
In Vitro Validation of Phosphorodiamidate Morpholino Oligomers

 , ,
, , 
Abstract
:1. Introduction
2. Results
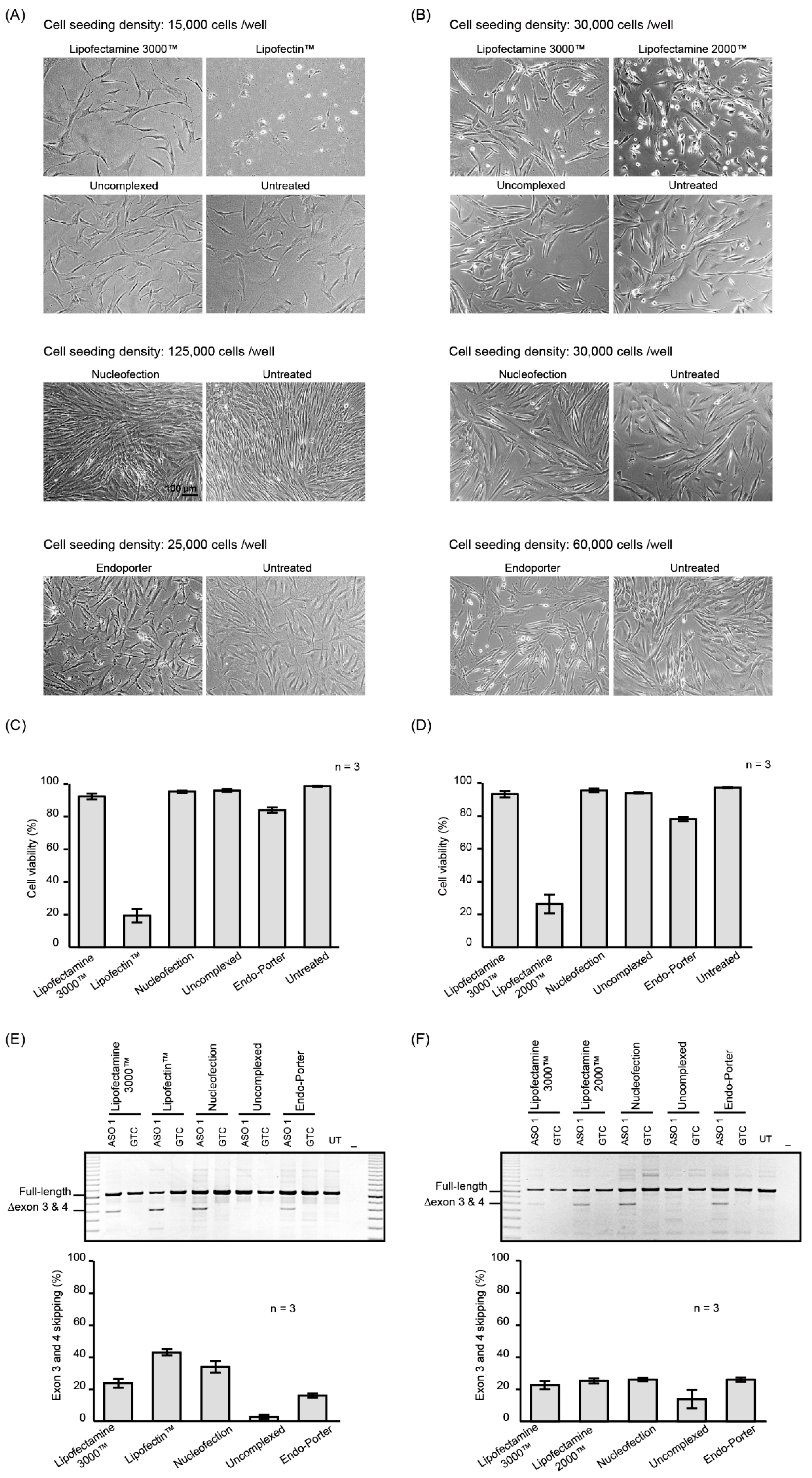
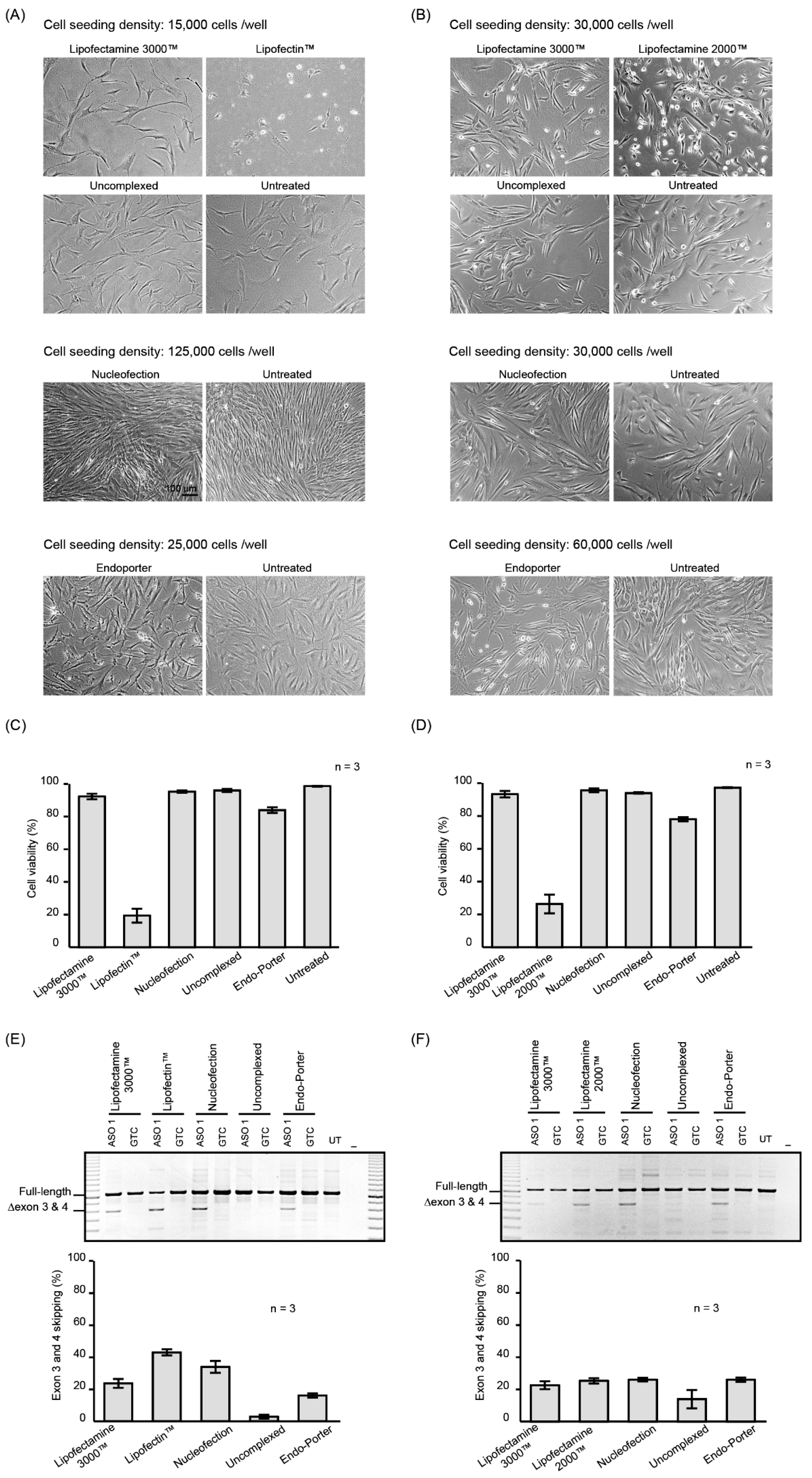
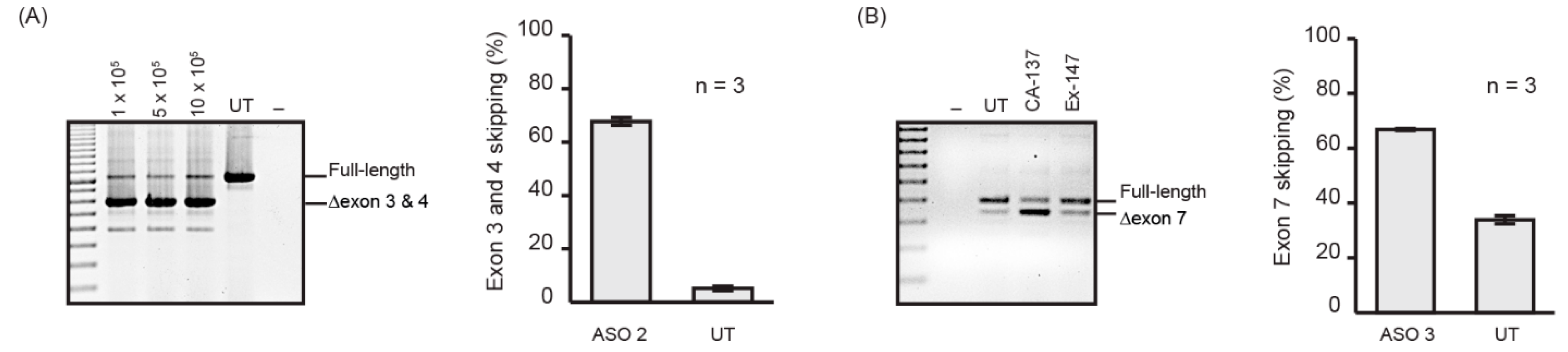
2.1. PMO Delivery into Human Dermal Fibroblasts
2.2. Nucleofection Programs for PMO Delivery into Different Cell Lines
3. Discussion
4. Materials and Methods
4.1. PMO Nomenclature
4.2. PMOs
4.3. Cell Culture
4.4. Isolation of Splenocytes
4.5. Transfection
4.6. Nucleofection
4.7. Microscopy
4.8. RT-PCR
4.9. Protein Stress Array
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Stephenson, M.L.; Zamecnik, P.C. Inhibition of rous sarcoma viral rna translation by a specific oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Zamecnik, P.C.; Stephenson, M.L. Inhibition of rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Dias, N.; Stein, C. Antisense oligonucleotides: Basic concepts and mechanisms. Mol. Cancer Ther. 2002, 1, 347–355. [Google Scholar] [PubMed]
- Chery, J.; Näär, A. Rna therapeutics: Rnai and antisense mechanisms and clinical applications. Postdoc J. 2016, 4, 35. [Google Scholar] [CrossRef] [PubMed]
- Summerton, J.; Weller, D. Morpholino antisense oligomers: Design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997, 7, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Bauman, J.; Jearawiriyapaisarn, N.; Kole, R. Therapeutic potential of splice-switching oligonucleotides. Oligonucleotides 2009, 19, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Craig, K.; Abrams, M.; Amiji, M. Recent preclinical and clinical advances in oligonucleotide conjugates. Expert Opin. Drug Deliv. 2018, 15, 629–640. [Google Scholar] [CrossRef]
- Aartsma-Rus, A. Fda approval of nusinersen for spinal muscular atrophy makes 2016 the year of splice modulating oligonucleotides. Nucleic Acid Ther. 2017, 27, 67–69. [Google Scholar] [CrossRef]
- Mendell, J.R.; Sahenk, Z.; Rodino-Klapac, L.R. Clinical trials of exon skipping in duchenne muscular dystrophy. Expert Opin. Orphan Drugs 2017, 5, 683–690. [Google Scholar] [CrossRef]
- Rinaldi, C.; Wood, M.J. Antisense oligonucleotides: The next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 2018, 14, 9. [Google Scholar] [CrossRef]
- Godfrey, C.; Desviat, L.R.; Smedsrød, B.; Piétri-Rouxel, F.; Denti, M.A.; Disterer, P.; Lorain, S.; Nogales-Gadea, G.; Sardone, V.; Anwar, R. Delivery is key: Lessons learnt from developing splice-switching antisense therapies. EMBO Mol. Med. 2017, 9, 545–557. [Google Scholar] [CrossRef]
- Summerton, J.; Stein, D.; Huang, S.B.; Matthews, P.; Weller, D.; Partridge, M. Morpholino and phosphorothioate antisense oligomers compared in cell-free and in-cell systems. Antisense Nucleic Acid Drug Dev. 1997, 7, 63–70. [Google Scholar] [CrossRef]
- Summerton, J.E. Morpholino, sirna, and s-DNA compared: Impact of structure and mechanism of action on off-target effects and sequence specificity. Curr. Top. Med. Chem. 2007, 7, 651–660. [Google Scholar] [CrossRef]
- Adams, A.M.; Harding, P.L.; Iversen, P.L.; Coleman, C.; Fletcher, S.; Wilton, S.D. Antisense oligonucleotide induced exon skipping and the dystrophin gene transcript: Cocktails and chemistries. BMC Mol. Biol. 2007, 8, 57. [Google Scholar] [CrossRef]
- Miyatake, S.; Mizobe, Y.; Tsoumpra, M.K.; Lim, K.R.Q.; Hara, Y.; Shabanpoor, F.; Yokota, T.; Takeda, S.I.; Aoki, Y. Scavenger receptor class a1 mediates uptake of morpholino antisense oligonucleotide into dystrophic skeletal muscle. Mol. Ther. Nucleic Acids 2019, 14, 520–535. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Kaman, W.; Bremmer-Bout, M.; Janson, A.; Den Dunnen, J.; van Ommen, G.B.; Van Deutekom, J. Comparative analysis of antisense oligonucleotide analogs for targeted dmd exon 46 skipping in muscle cells. Gene Ther. 2004, 11, 1391. [Google Scholar] [CrossRef]
- Heemskerk, H.A.; de Winter, C.L.; de Kimpe, S.J.; van Kuik-Romeijn, P.; Heuvelmans, N.; Platenburg, G.J.; van Ommen, G.J.B.; van Deutekom, J.C.; Aartsma-Rus, A. In Vivo comparison of 2′-o-methyl phosphorothioate and morpholino antisense oligonucleotides for duchenne muscular dystrophy exon skipping. J. Gene Med. 2009, 11, 257–266. [Google Scholar] [CrossRef]
- McClorey, G.; Moulton, H.; Iversen, P.; Fletcher, S.; Wilton, S. Antisense oligonucleotide-induced exon skipping restores dystrophin expression in vitro in a canine model of dmd. Gene Ther. 2006, 13, 1373–1381. [Google Scholar] [CrossRef]
- Partridge, M.; Vincent, A.; Matthews, P.; Puma, J.; Stein, D.; Summerton, J. A simple method for delivering morpholino antisense oligos into the cytoplasm of cells. Antisense Nucleic Acid Drug Dev. 1996, 6, 169–175. [Google Scholar] [CrossRef]
- Gebski, B.L.; Mann, C.J.; Fletcher, S.; Wilton, S.D. Morpholino antisense oligonucleotide induced dystrophin exon 23 skipping in mdx mouse muscle. Hum. Mol. Genet. 2003, 12, 1801–1811. [Google Scholar] [CrossRef]
- Summerton, J.E. Endo-porter: A novel reagent for safe, effective delivery of substances into cells. Ann. N. Y. Acad. Sci. 2005, 1058, 62–75. [Google Scholar] [CrossRef]
- Brees, C.; Fransen, M. A cost-effective approach to microporate mammalian cells with the neon transfection system. Anal. Biochem. 2014, 466, 49–50. [Google Scholar] [CrossRef]
- Singh, N.N.; Luo, D.; Singh, R.N. Pre-mrna splicing modulation by antisense oligonucleotides. In Exon Skipping and Inclusion Therapies; Springer: New York, NY, USA, 2018; pp. 415–437. [Google Scholar]
- Wilton, S.D.; Fletcher, S.; McClorey, G. Antisense oligonucleotides for inducing exon skipping and methods of use thereof. Patent Number 1766010, 28 March 2007. [Google Scholar]
- Fletcher, S.; Bellgard, M.; Price, L.; Akkari, A.; Wilton, S. Translational development of splice-modifying antisense oligomers. Expert Opin. Biol. Ther. 2017, 17, 15–30. [Google Scholar] [CrossRef]
- Arechavala-Gomeza, V.; Graham, I.; Popplewell, L.; Adams, A.; Aartsma-Rus, A.; Kinali, M.; Morgan, J.; Van Deutekom, J.; Wilton, S.; Dickson, G. Comparative analysis of antisense oligonucleotide sequences for targeted skipping of exon 51 during dystrophin pre-mrna splicing in human muscle. Hum. Gene Ther. 2007, 18, 798–810. [Google Scholar] [CrossRef]
- Charleston, J.; Schnell, F.; Dworzak, J.; Donoghue, C.; Lewis, S.; Rodino-Klapac, L.; Sahenk, Z.; Shanks, C.; Voss, J.; DeAlwis, U. Long-term treatment with eteplirsen promotes exon 51 skipping and novel dystrophin protein production in duchenne muscular dystrophy patients. Neuromuscul. Disord. 2016, 26, S153. [Google Scholar] [CrossRef]
- Mendell, J.R.; Goemans, N.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Shao, J.; Kaye, E.M.; Mercuri, E.; Group, E.S.; Network, T.F.D.I.; et al. Longitudinal effect of eteplirsen versus historical control on ambulation in d uchenne muscular dystrophy. Ann. Neurol. 2016, 79, 257–271. [Google Scholar] [CrossRef]
- Mendell, J.R.; Rodino-Klapac, L.R.; Sahenk, Z.; Roush, K.; Bird, L.; Lowes, L.P.; Alfano, L.; Gomez, A.M.; Lewis, S.; Kota, J. Eteplirsen for the treatment of duchenne muscular dystrophy. Ann. Neurol. 2013, 74, 637–647. [Google Scholar] [CrossRef]
- Wilton, S.D.; Fletcher, S.; Aung-Htut, M. Multiple sclerosis treatment. Patent Number 20180104273, 19 April 2018. [Google Scholar]
- Flynn, L.L.; Mitrpant, C.; Pitout, I.L.; Fletcher, S.; Wilton, S.D. Antisense oligonucleotide-mediated terminal intron retention of the smn2 transcript. Mol. Ther. Nucleic Acids 2018, 11, 91–102. [Google Scholar] [CrossRef]
- Collett, G.P.; Redman, C.W.; Sargent, I.L.; Vatish, M. Endoplasmic reticulum stress stimulates the release of extracellular vesicles carrying danger-associated molecular pattern (damp) molecules. Oncotarget 2018, 9, 6707–6717. [Google Scholar] [CrossRef]
- Mann, C.J.; Honeyman, K.; McClorey, G.; Fletcher, S.; Wilton, S.D. Improved antisense oligonucleotide induced exon skipping in the mdx mouse model of muscular dystrophy. J. Gene Med. 2002, 4, 644–654. [Google Scholar] [CrossRef]
- Rando, T.A.; Blau, H.M. Primary mouse myoblast purification, characterization, and transplantation for cell-mediated gene therapy. J. Cell Biol. 1994, 125, 1275–1287. [Google Scholar] [CrossRef]
- Harding, P.; Fall, A.; Honeyman, K.; Fletcher, S.; Wilton, S. The influence of antisense oligonucleotide length on dystrophin exon skipping. Mol. Ther. 2007, 15, 157–166. [Google Scholar] [CrossRef]
- Greer, K.L.; Lochmüller, H.; Flanigan, K.; Fletcher, S.; Wilton, S.D. Targeted exon skipping to correct exon duplications in the dystrophin gene. Mol. Ther. Nucleic Acids 2014, 3, e155. [Google Scholar] [CrossRef]
Sample Availability: Samples of 2′-OMe ASOs are available on request from the authors. However, PMOs can be purchased from Gene Tools. |


{kind=link}
{kind=link}
| Cell Type | Name | P3 Program | Cell Number |
|---|---|---|---|
| Human dermal fibroblasts | HDF | CA-137 | 2.5−10 × 105 |
| Human skeletal muscle myoblasts | HSkM | CM-138 | 2.5 × 105 |
| Human T lymphocytes | Jurkat | CL-120 | 2-5 × 105 |
| Human Glial (Oligodendrocytic) Hybrid Cell Line | MO3.13 | CA-138 | 2.5 × 105 |
| DR-114 | 2.5 × 105 | ||
| EM-110 | 2.5 × 105 | ||
| Human neuroblastoma | SH-SY5Y | CA-137 | 2.5−10 × 105 |
| Human hepatocarcinoma | Huh7 | CA-137 | 2.5 × 105 |
| Human hepatocarcinoma | HepG2 | EH-100 | 2.5 × 105 |
| Murine primary splenocytes | DN-100 | 10 × 105 | |
| Murine myoblast | H2Kmdx | CM-138 | 2.5 × 105 |
| Name | ASO Nomenclature | Sequence (5′ to 3′) | Target Protein |
|---|---|---|---|
| ASO 1 | ITGA4 H3A (+ 30 + 49) | TCTCTCTCTTCCAAACAAGT | Integrin alpha 4 |
| ASO 2 | ITGA4 H3A (+ 41 + 65) | CCCCAACCACTGATTGTCTCTCTCT | Integrin alpha 4 |
| ASO 3 | Smn M7A (+ 7 + 36) | TGAGCACTTTCCTTCTTTTTTATTTTGTCT | Survival motor neuron |
| GTC | GTC-Gene Tools Control | CCTCTTACCTCAGTTACAATTTATA | Beta-globin chain |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aung-Htut, M.T.; McIntosh, C.S.; West, K.A.; Fletcher, S.; Wilton, S.D. In Vitro Validation of Phosphorodiamidate Morpholino Oligomers. Molecules 2019, 24, 2922. https://doi.org/10.3390/molecules24162922
Aung-Htut MT, McIntosh CS, West KA, Fletcher S, Wilton SD. In Vitro Validation of Phosphorodiamidate Morpholino Oligomers. Molecules. 2019; 24(16):2922. https://doi.org/10.3390/molecules24162922
Chicago/Turabian StyleAung-Htut, May T., Craig S. McIntosh, Kristin A. West, Sue Fletcher, and Steve D. Wilton. 2019. "In Vitro Validation of Phosphorodiamidate Morpholino Oligomers" Molecules 24, no. 16: 2922. https://doi.org/10.3390/molecules24162922
APA StyleAung-Htut, M. T., McIntosh, C. S., West, K. A., Fletcher, S., & Wilton, S. D. (2019). In Vitro Validation of Phosphorodiamidate Morpholino Oligomers. Molecules, 24(16), 2922. https://doi.org/10.3390/molecules24162922