
Recent Advances in the Synthesis of 2H-Pyrans

 ,
, 
Abstract

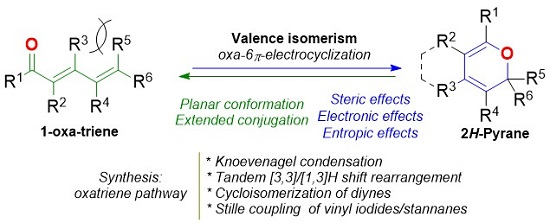
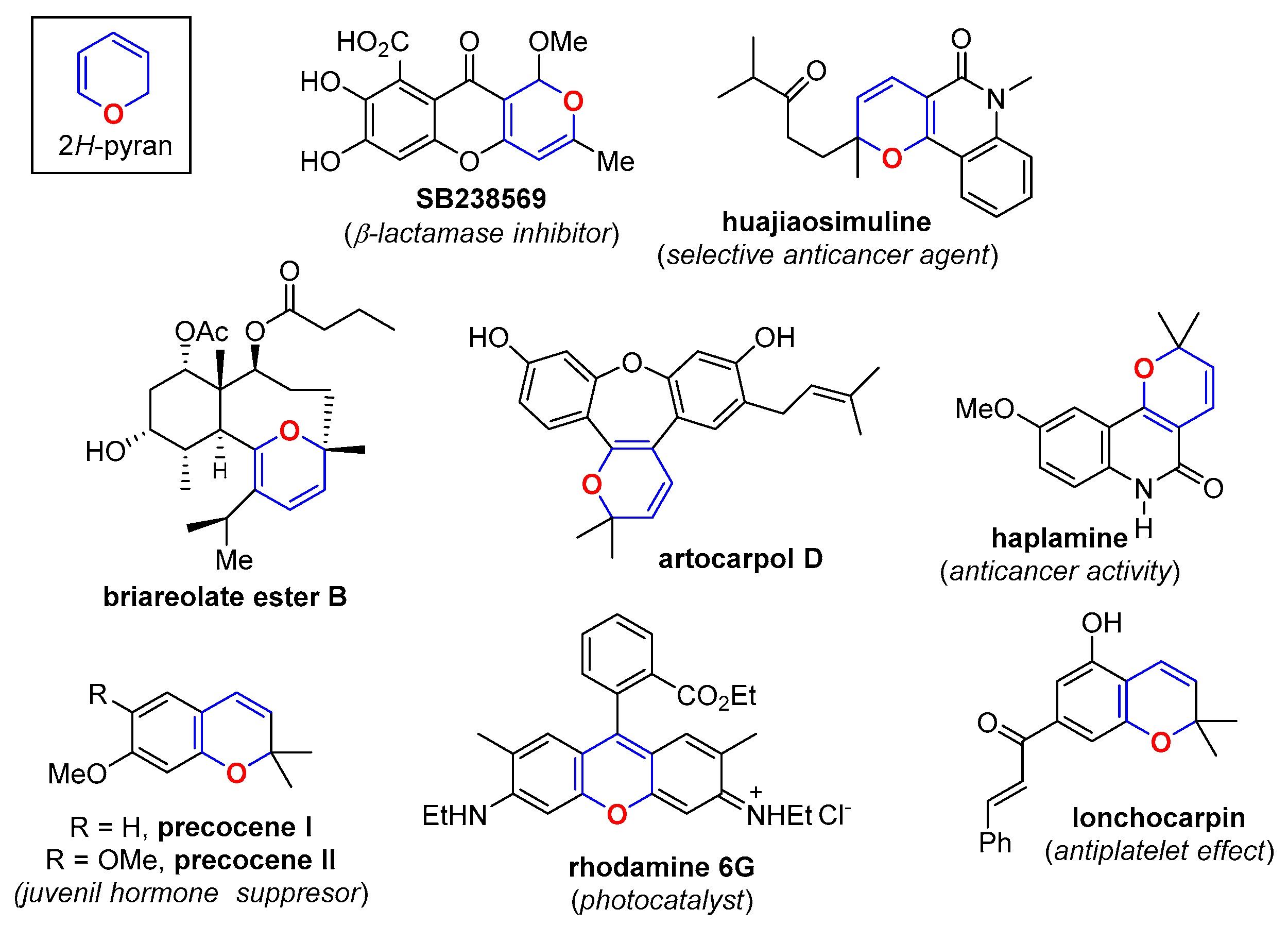
1. Introduction
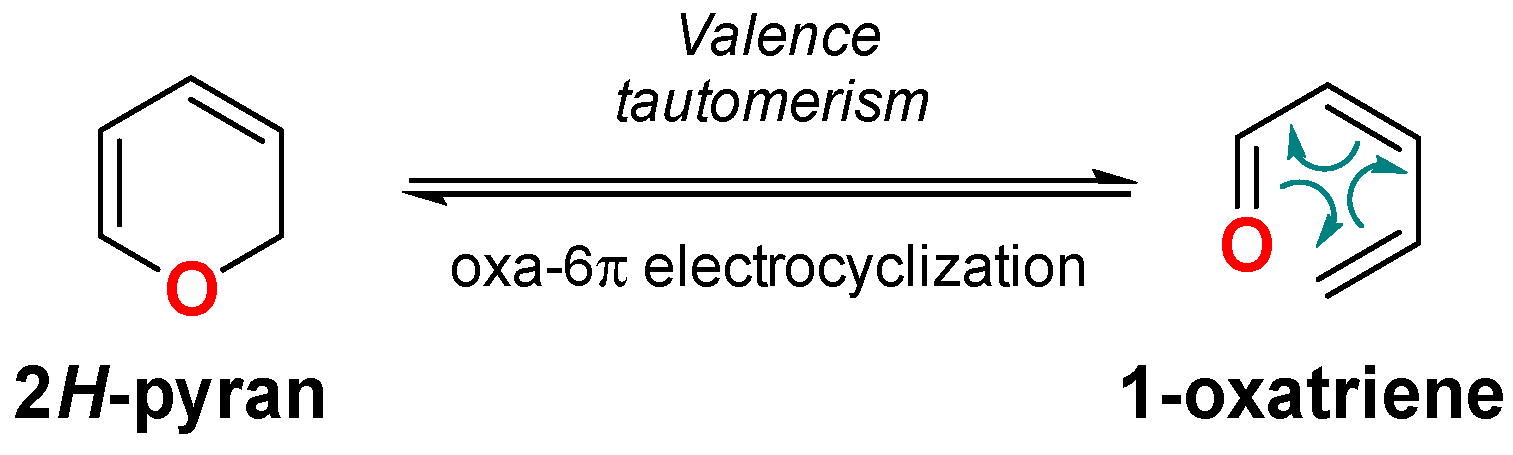
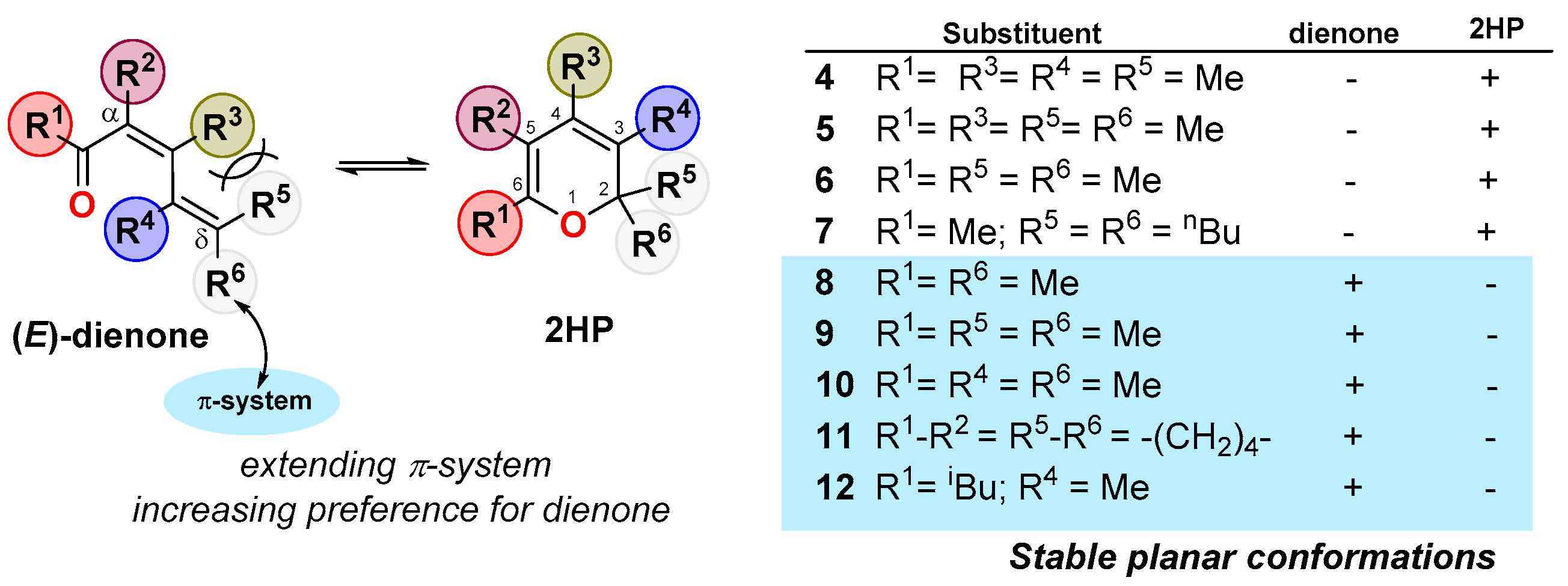
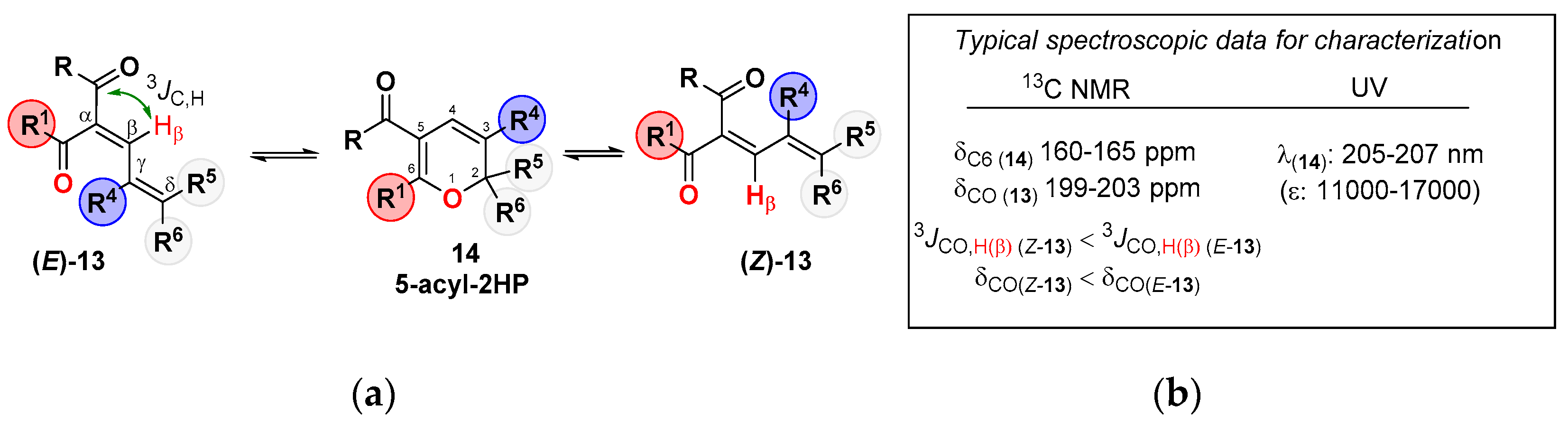
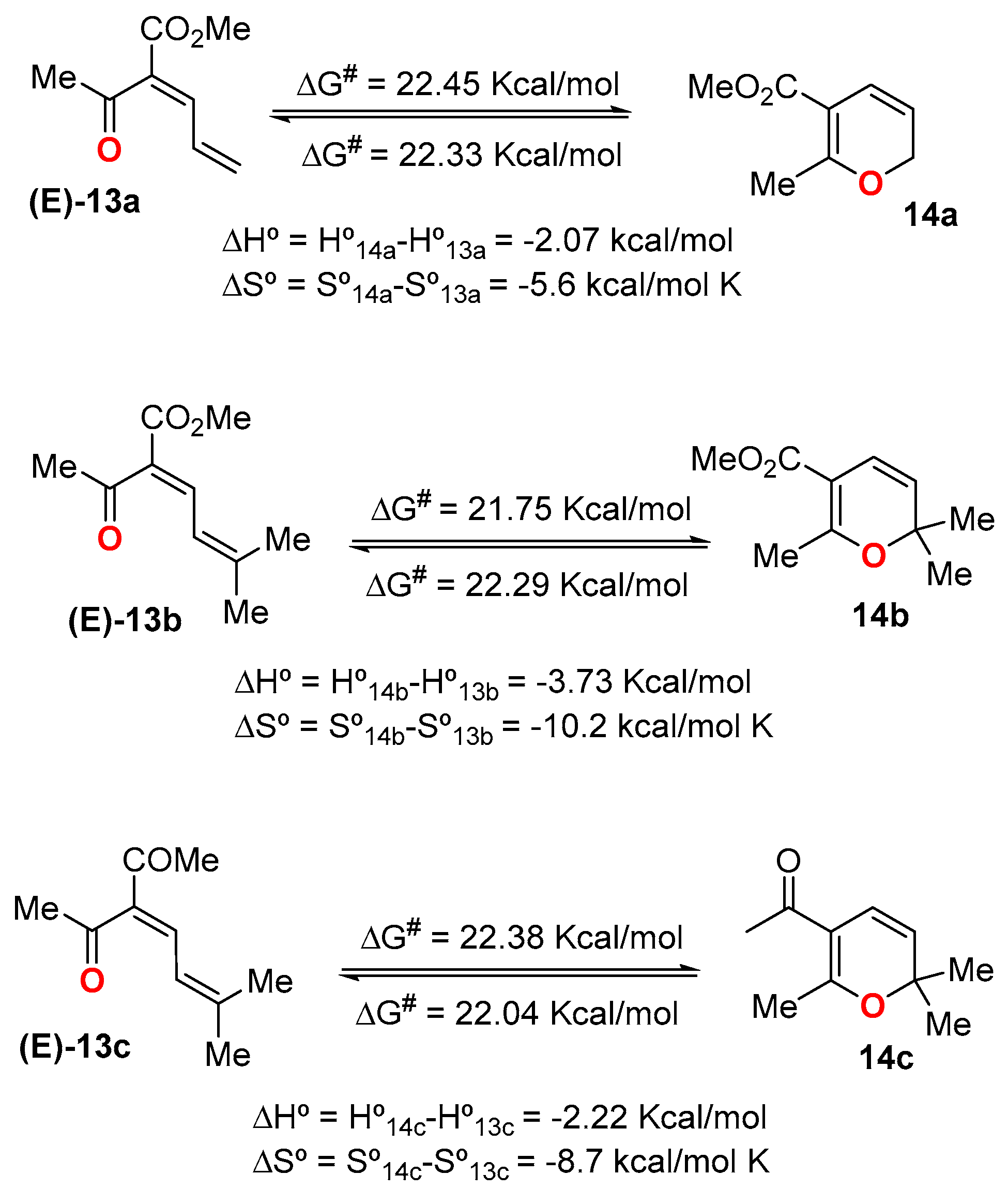
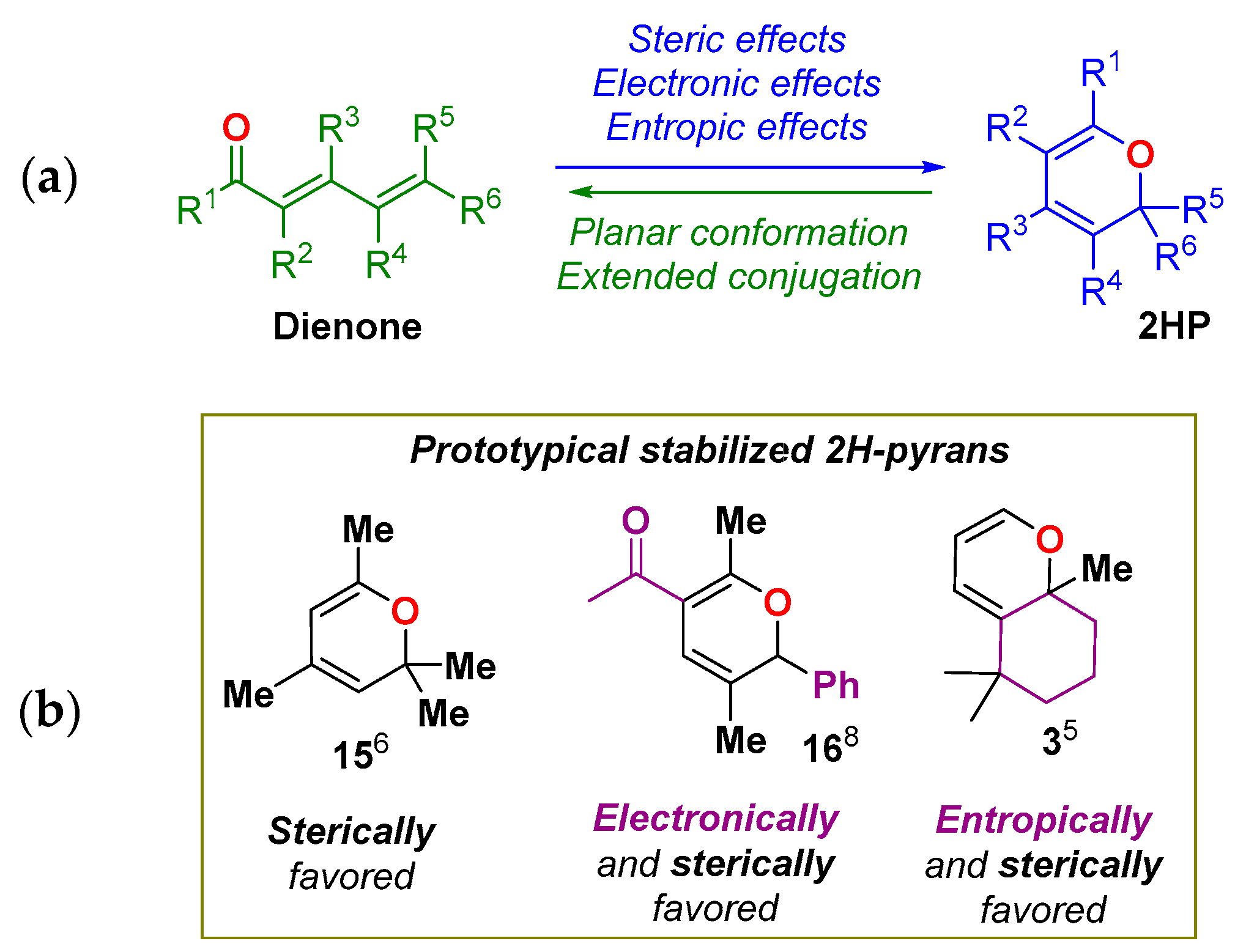
2. Dienone/2HP Equilibrium
- The successive substitution at position C2 in the ring (Cδ on dienone) leads to an increase in the content of 2HP (steric strain on the dienone) (compare entries 1–3 and 7–8).
- The elongation of the conjugated system results in an increase in the content of dienone (resonance delocalization) (compare entries 15, 25 and 17, 26).
- Substitution at the C2-position of the ring (Cδ on dienone) with two methyl groups strongly shifts the equilibrium toward the 2HP (entries 17, 19). In this case, it is possible to observe only the 2HP (compare entries 7, 17 with 11, 19).
- Aprotic polar solvent shifts the equilibrium toward the formation of the dienone.
- Although the electronic effect of the acyl group at the C5-position of the ring (Cα in the dienone) is masked in Table 1, other studies have shown that the presence of an electron-withdrawing substituent(s) at the ring, preferentially at this C5-position, favors the 2HP [23,24,25]. Table 1 shows that although this effect could be operative in α-acyl-dienones 13, it can be completely surpassed by other structural/electronic effects (Table 1, entries 11–14).
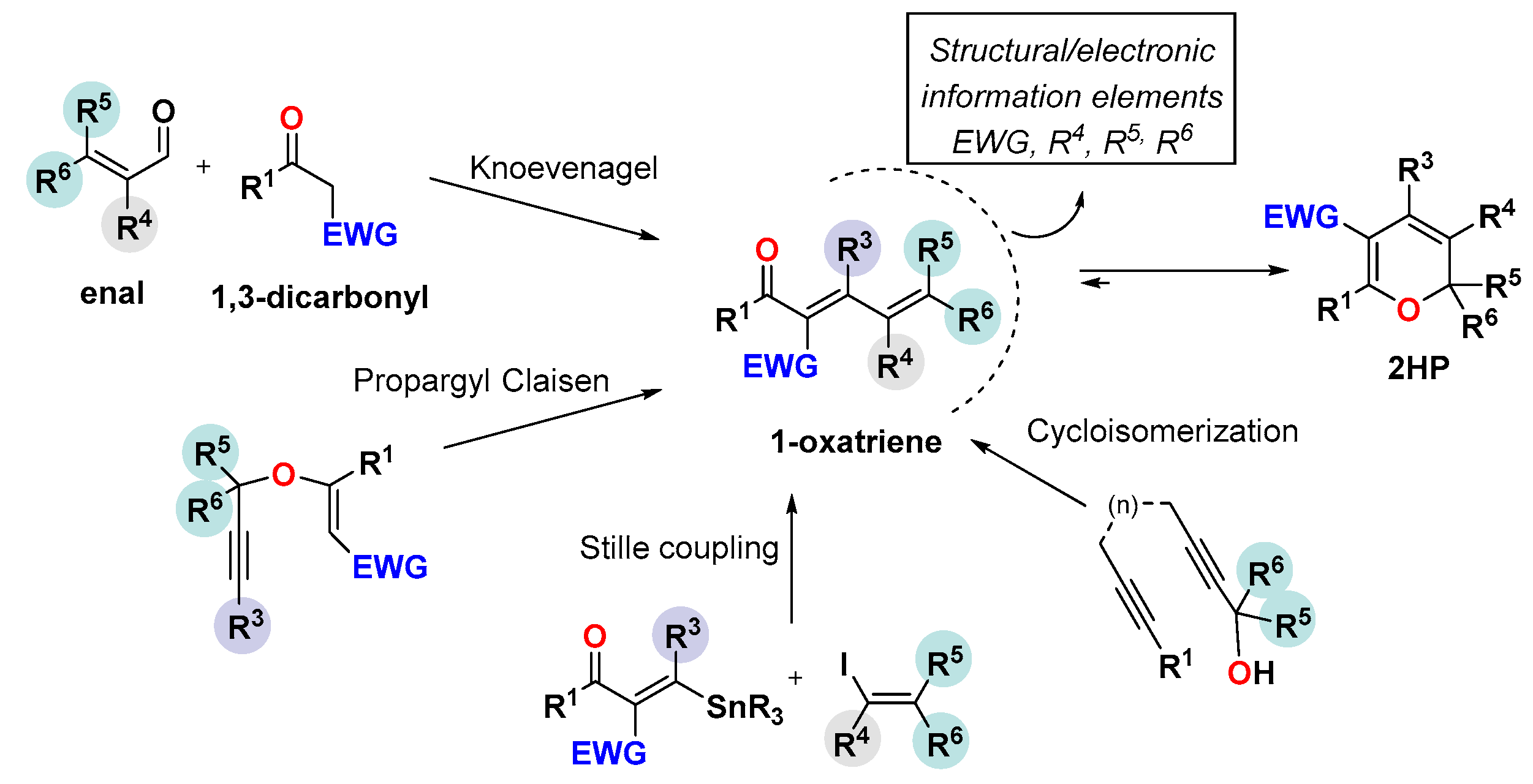
3. Synthesis of the 2HP Core
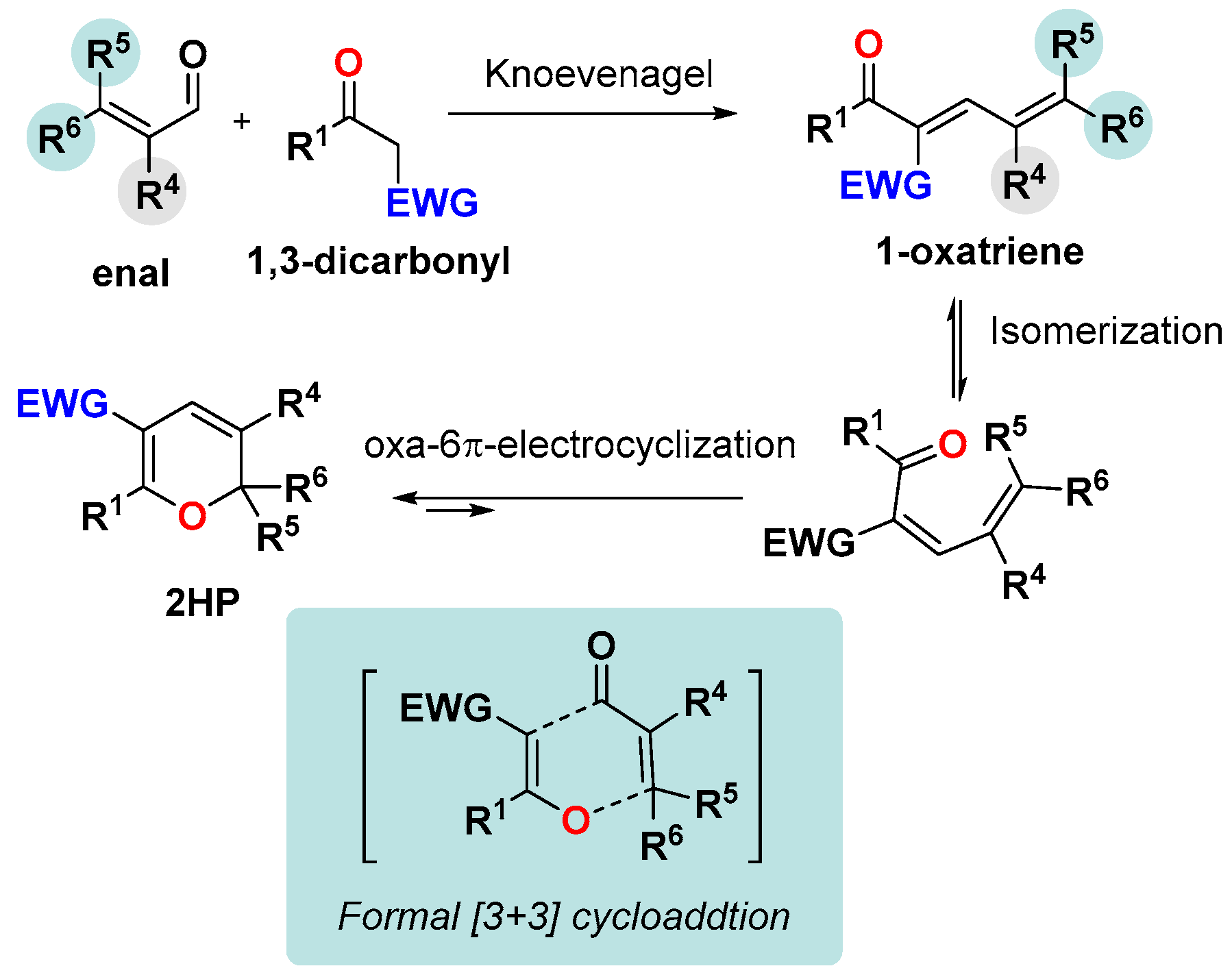
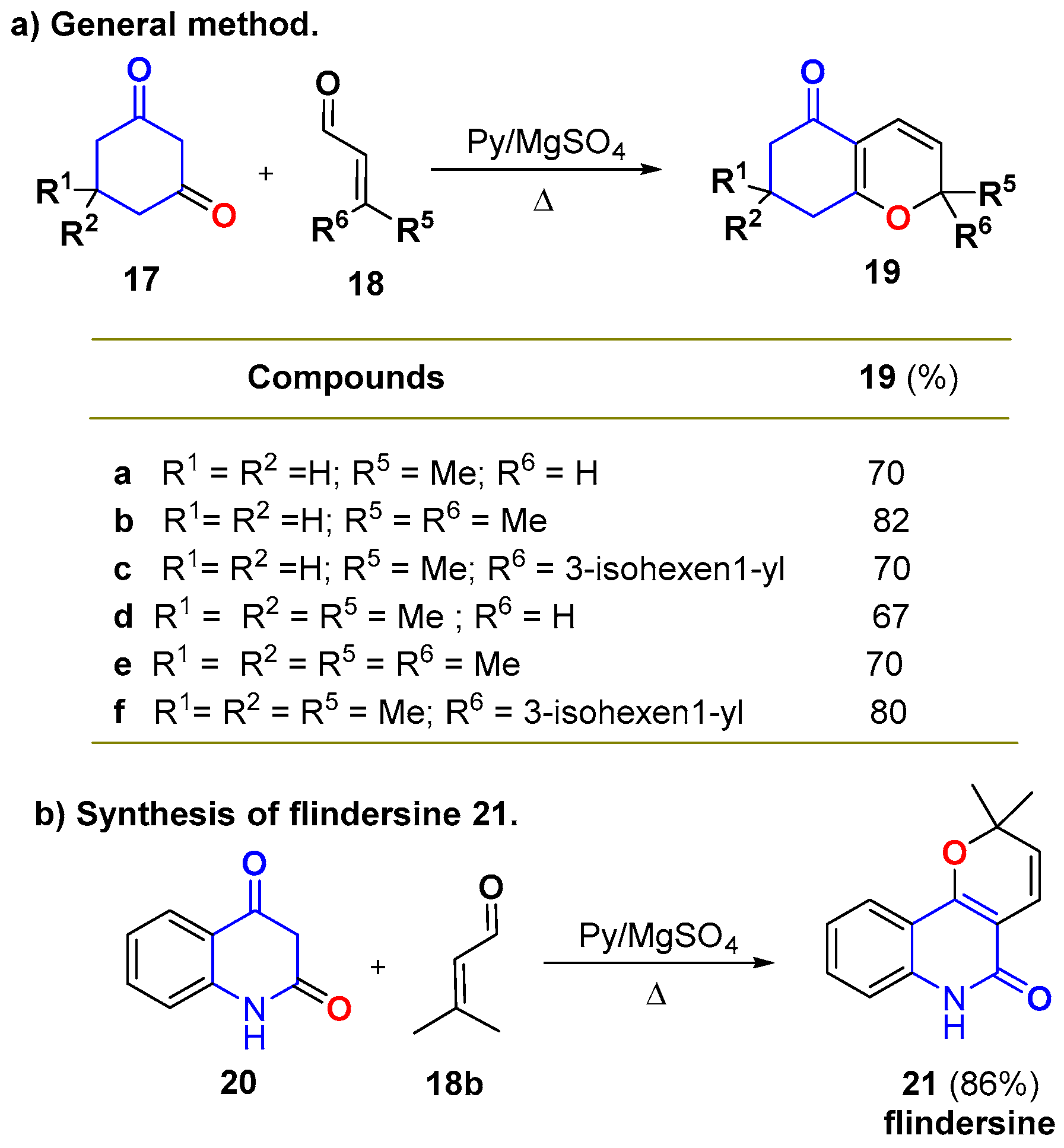
3.1. The Knoevenagel/Electrocyclization Protocol
3.2. From Other Heterocycles
3.3. From Allenolates
3.4. From Alkynes
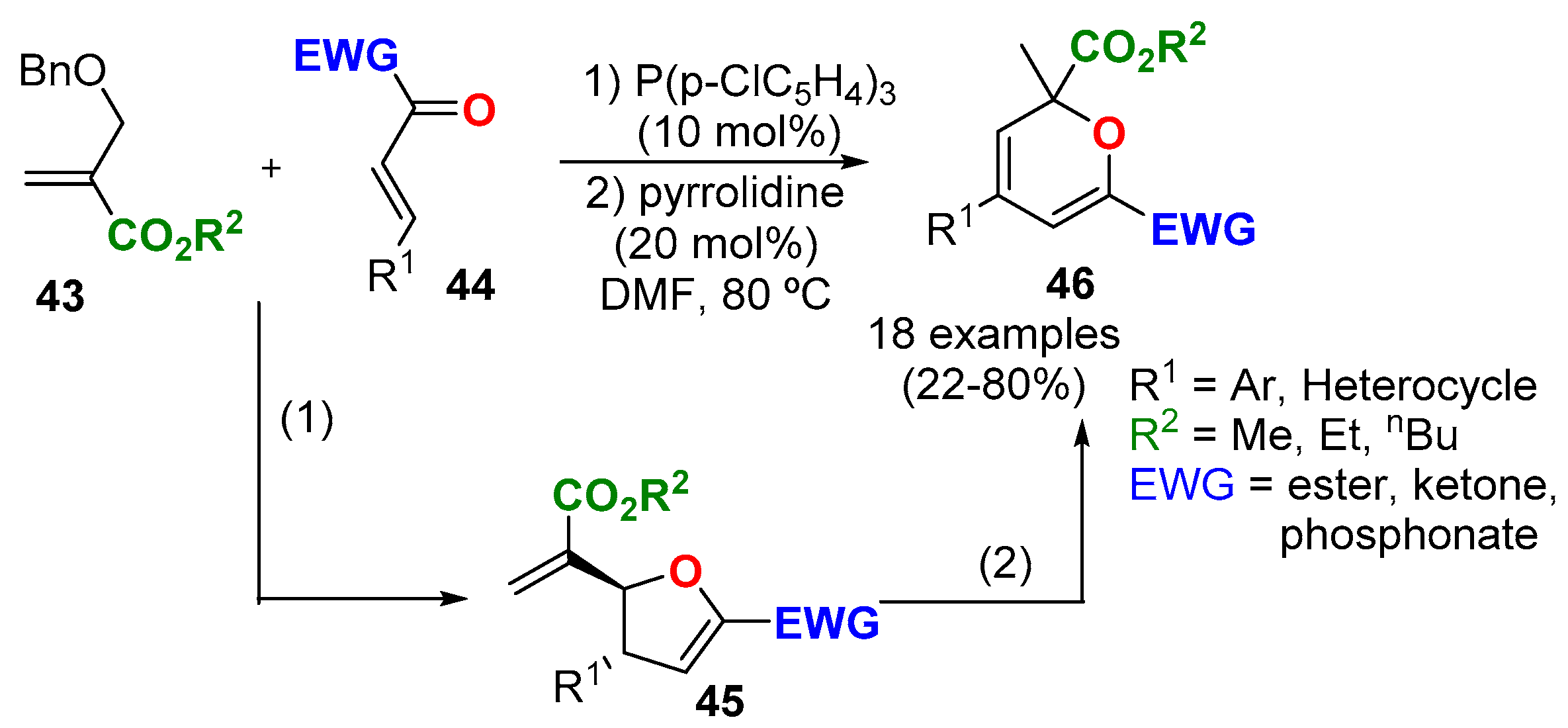
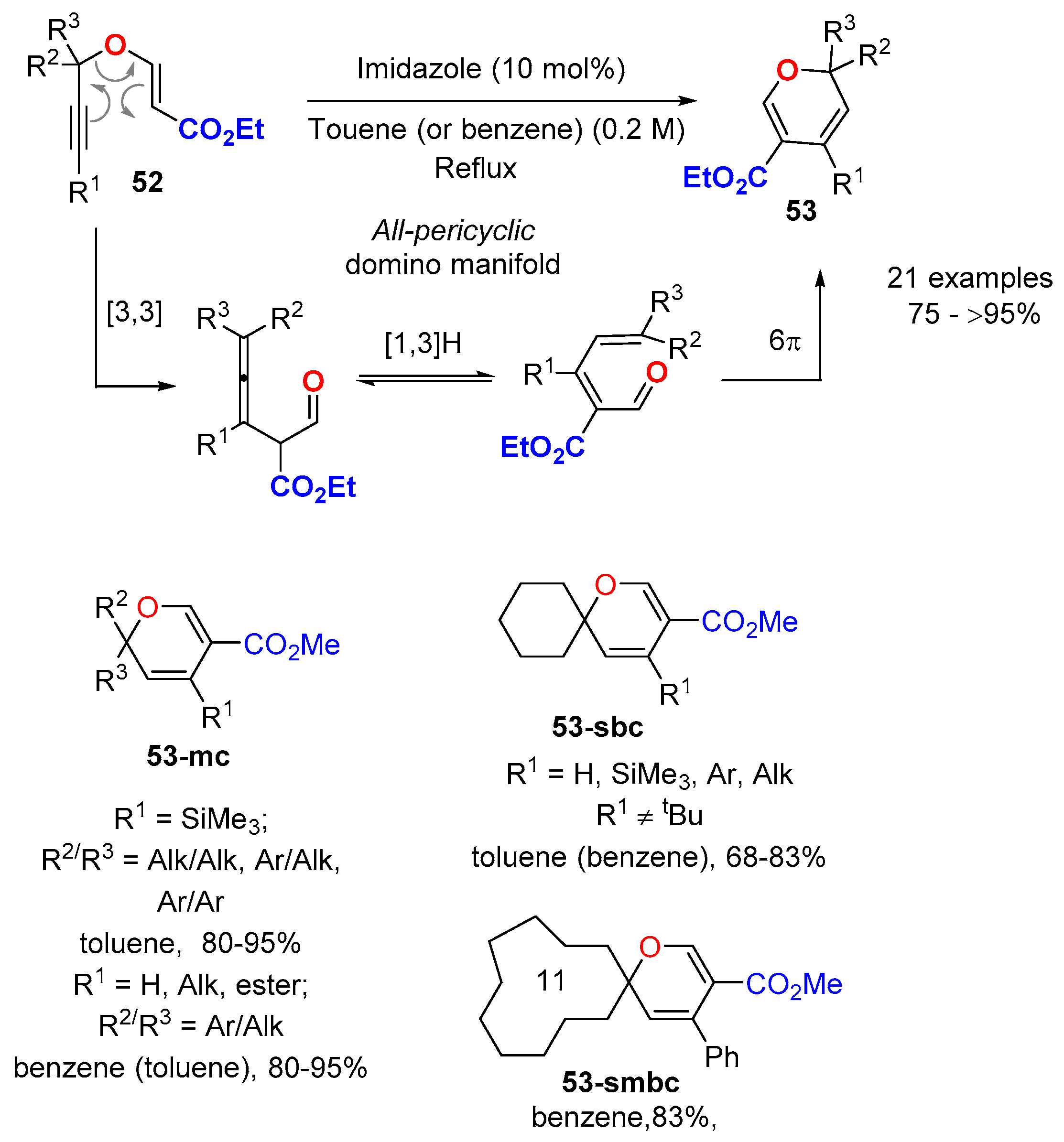
3.4.1. From Propargyl Vinyl Ethers
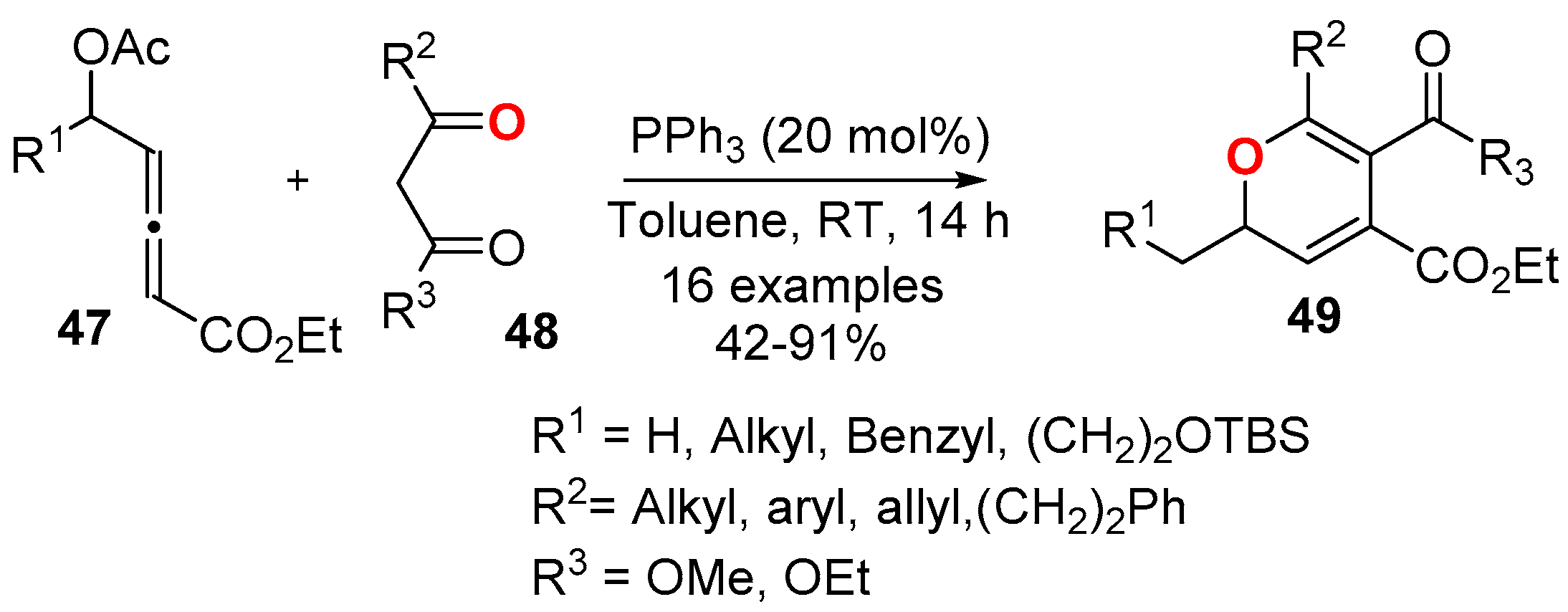
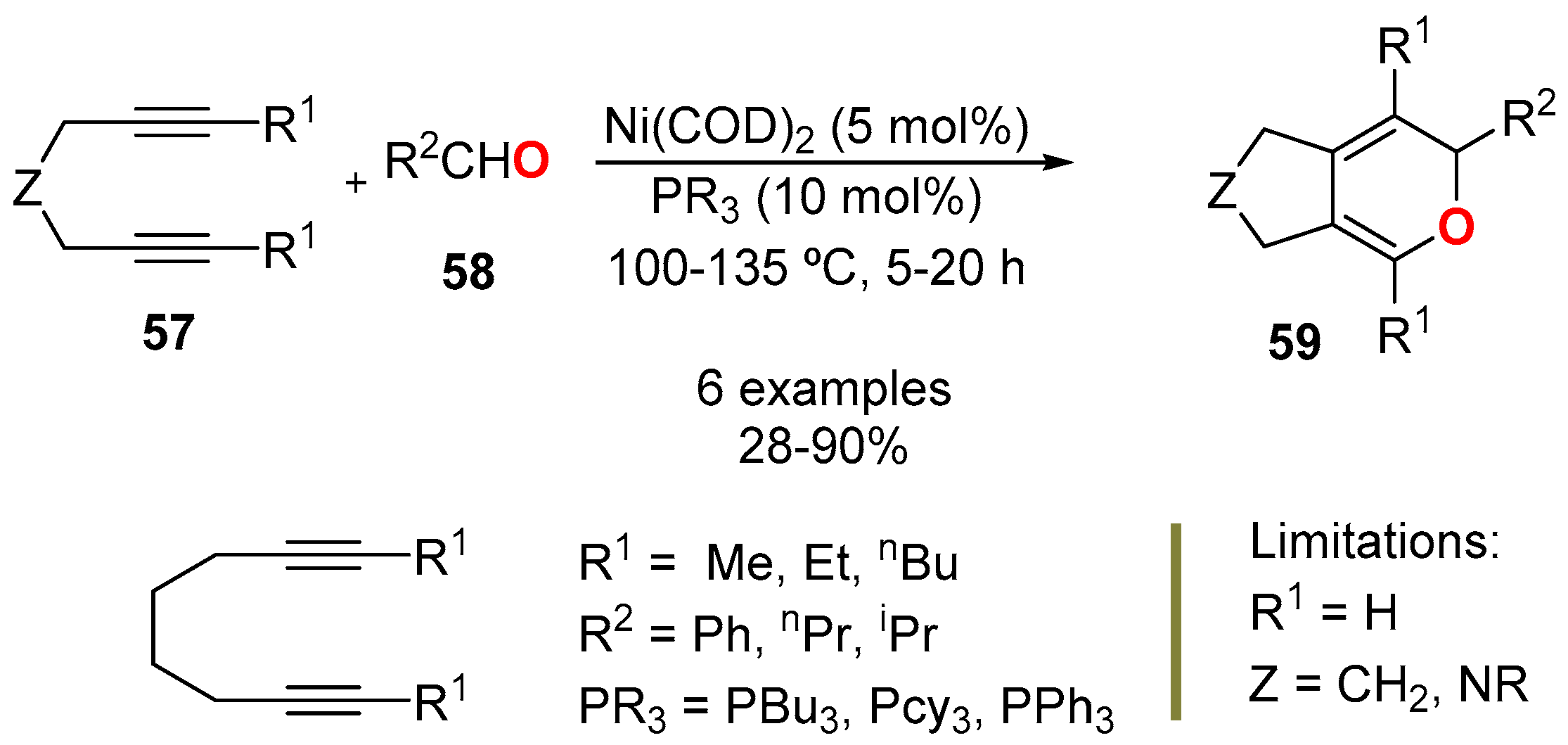
3.4.2. From Diynes
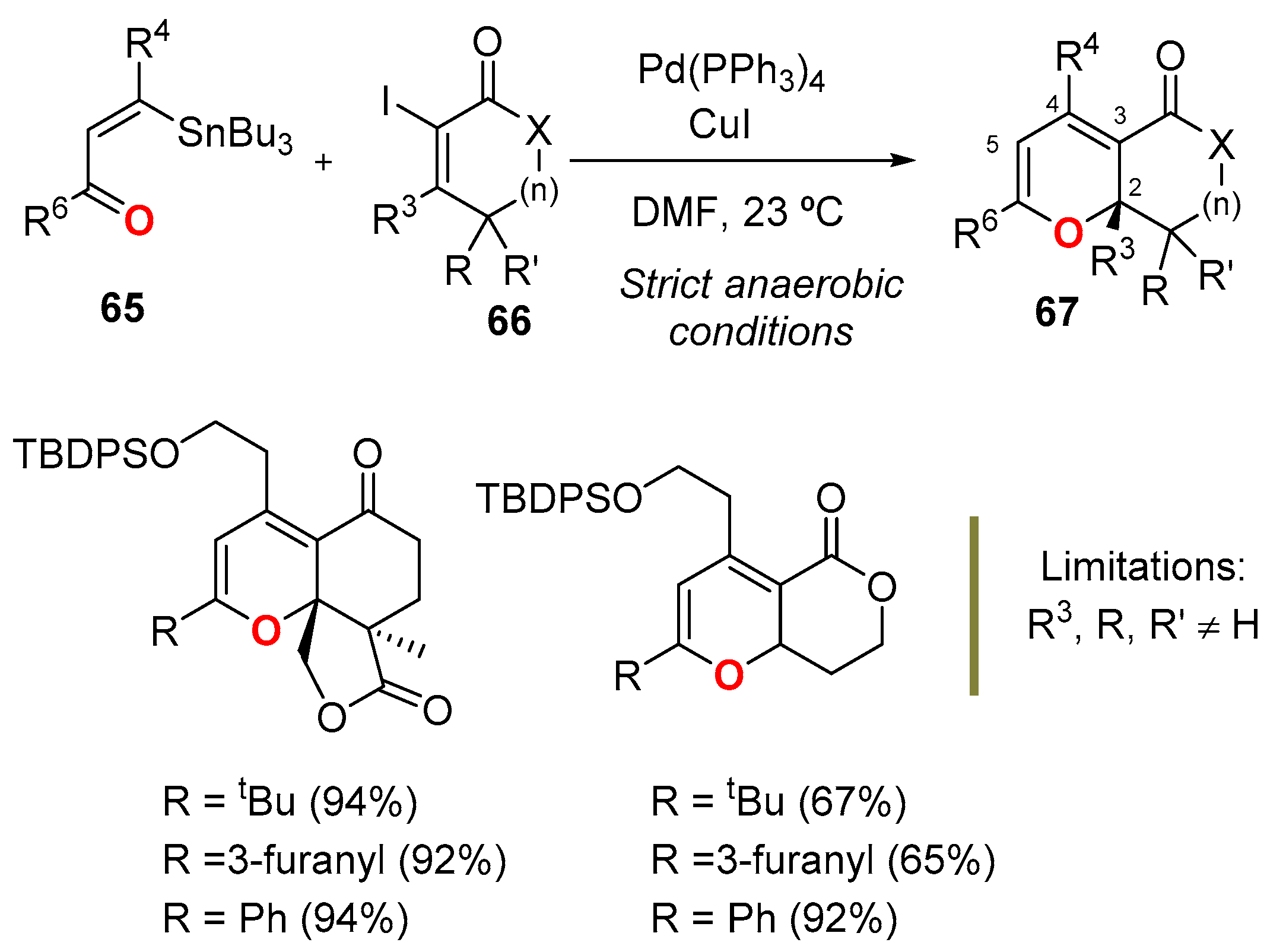
3.4.3. From Alkenes: Tandem Stille-Oxa-Electrocyclization Reaction
4. Summary and Conclusions
Acknowledgments
Conflicts of Interest
References
- Fravel, B.W. Pyrans and their Benzo Derivatives: Applications. In Comprehensive Heterocyclic Chemistry III; Katritzky, A.R., Ramsdem, C.A., Scriven, E.F.V., Taylor, R.J.K., Eds.; Elsevier: Oxford, UK, 2008; Volume 7, pp. 701–726. [Google Scholar]
- Hsung, R.P.; Kurdyumov, A.V.; Sydorenko, N. A formal [3 + 3] cycloaddition approach to Natural-Product synthesis. Eur. J. Org. Chem. 2005, 2005, 23–44. [Google Scholar] [CrossRef]
- Beaudry, C.M.; Malerich, J.P.; Trauner, D. Biosynthetic and biomimetic electrocyclizations. Chem. Rev. 2005, 105, 4757–4778. [Google Scholar] [CrossRef]
- Drygina, O.V.; Garnovskii, A.D.; Kazantsev, A.V. 2H-pyrans (review). Chem. Heterocycl. Compd. 1985, 21, 239–253. [Google Scholar] [CrossRef]
- Brimble, M.A.; Gibson, J.S.; Sperry, J. Pyrans and their Benzo Derivatives: Synthesis. In Comprehensive Heterocycles Chemistry III; Katritzky, A.R., Ramsdem, C.A., Scriven, E.F.V., Taylor, R.J.K., Eds.; Elsevier: Oxford, UK, 2008; Volume 5, pp. 419–699. [Google Scholar]
- Hepworth, J.D.; Gabbutt, C.D.; Heron, B.M. Pyrans and their Benzo Derivatives: Synthesis. In Comprehensive Heterocycles Chemistry II; Katritzky, A.R., Rees, C.W., Scriven, R.J.K., Eds.; Elsevier: Oxford, UK, 1996; Volume 7, pp. 301–468. [Google Scholar]
- Hepworth, J.D. Pyrans and Fused Pyrans: (iii) Synthesis and Applications. In Comprehensive Heterocycles Chemistry; Katritzky, A.R., Rees, C.W., Eds.; Elsevier: Oxford, UK, 1984; Volume 3, pp. 737–883. [Google Scholar]
- Hepworth, J.D.; Heron, B.M. Six-membered ring systems: With O and/or S atoms. In Progress in Heterocyclic Chemistry; Gribble, G.W., Joule, J.A., Eds.; Elsevier: Oxford, UK, 2009; Volume 20, pp. 365–398. [Google Scholar]
- Kuthan, J.; Šebek, P.; Böhm, S. New Developments in the Chemistry of Pyrans. Adv. Heterocyclic. Chem. 1995, 62, 19–135. [Google Scholar]
- Pratap, R.; Ram, V.J. Natural and Synthetic Chromenes, Fused Chromenes, and Versatility of Dihydrobenzo[h]chromenes in Organic Synthesis. Chem. Rev. 2014, 114, 10476–10526. [Google Scholar] [CrossRef]
- Majumdar, N.; Paul, N.P.; Mandal, S.; de Bruin, B.; Wulff, W.D. Catalytic Synthesis of 2H-Chromenes. ACS Catal. 2015, 5, 2329–2366. [Google Scholar] [CrossRef]
- Ellis, G.P.; Lockhart, I.M. The Chemistry of Heterocyclic Compounds: Chromenes, Chromanones, and Chromones; Ellis, G.P., Ed.; Wiley-VCH: New York, NY, USA, 2009; Volume 31, pp. 1–1196. [Google Scholar]
- Ellis, G.P. Pyrans and fused pyrans: (ii) reactivity. In Comprehensive Heterocycles Chemistry; Katritzky, A.R., Rees, C.W., Eds.; Elsevier: Oxford, UK, 1984; Volume 3, pp. 647–736. [Google Scholar]
- Schweizer, E.E.; Meeder-Nycz, O. Chromenes, Chromanes, Chromones; Ellis, G.P., Ed.; Wiley-Interscience: New York, NY, USA, 1977; Volume 31, pp. 11–139. [Google Scholar]
- Hepworth, J.D.; Heron, B.M. Synthesis and photochromic properties of naphthopyrans. In Progress in Heterocyclic Chemistry; Gribble, G.W., Joule, J.A., Eds.; Elsevier: Oxford, UK, 2005; Volume 17, pp. 33–62. [Google Scholar]
- Vogel, E. Valence Isomerizations in compounds with strained rings. Angew. Chem. Int. Ed. Engl. 1963, 2, 1–11. [Google Scholar] [CrossRef]
- Rodriguez-Otero, J. Study of the electrocyclization of (Z)-hexa-1,3,5-triene and its heterosubstituted analogues based on ab initio and DFT calculations. J. Org. Chem. 1999, 64, 6842–6848. [Google Scholar] [CrossRef]
- Marvell, E.N.; Caple, G.; Gosink, T.A.; Zimmer, G. Valence isomerization of a cis-dienone to an α-pyran. J. Am. Chem. Soc. 1966, 88, 619–620. [Google Scholar] [CrossRef]
- Marvell, E.N.; Gosink, T.A. Valence isomerization of 2,4,6-trimethyl-2H-pyran. J. Org. Chem. 1972, 37, 3036–3037. [Google Scholar] [CrossRef]
- Lillya, C.P.; Kluge, A.F. Molecular spectra and conformations of conjugated dienones. J. Org. Chem. 1971, 36, 1977–1988. [Google Scholar] [CrossRef]
- Gosink, T.A. Valence isomers. Substituent effects on the equilibrium between 2H-pyrans and cis dienones. J. Org. Chem. 1974, 39, 1942–1943. [Google Scholar] [CrossRef]
- Krasnaya, Z.A. Dienone ⇄ 2H-pyran valence isomerization. Chem. Heterocycl. Comp. 1999, 35, 1255–1271. [Google Scholar] [CrossRef]
- Adams, R.D.; Chen, L. Coupling of alkynes to carbon monoxide at a dimanganese center. A new route to carboxylate-functionalized pyrans. J. Am. Chem. Soc. 1994, 116, 4467–4468. [Google Scholar] [CrossRef]
- Fan, M.; Yan, Z.; Liu, W.; Liang, Y. DABCO-catalyzed reaction of α-halo carbonyl compounds with dimethyl acetylenedicarboxylate: A novel method for the preparation of polysubstituted furans and highly functionalized 2H-pyrans. J. Org. Chem. 2005, 70, 8204–8207. [Google Scholar] [CrossRef] [PubMed]
- Qing, F.L.; Gao, W.Z. The first synthesis of 4-trifluoromethyl-2H-pyrans by palladium-catalyzed cyclization of (E)-3-alkynyl-3-trifluoromethyl allylic alcohols. Tetrahedron Lett. 2000, 41, 7727–7730. [Google Scholar] [CrossRef]
- Rauser, M.; Schroeder, S.; Niggemann, M. Cooperative catalysis: Calcium and camphorsulfonic acid catalyzed cycloisomerization of diynols. Chem. Eur. J. 2015, 21, 15929–15933. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, T.; Kiyoi, T.; Miyane, T.; Saegusa, T. Nickel(0)-catalyzed reaction of diynes with aldehydes. J. Am. Chem. Soc. 1988, 110, 8570–8572. [Google Scholar] [CrossRef]
- Voskressensky, L.G.; Festa, A.A.; Varlamov, A.V. Domino reactions based on Knoevenagel condensation in the synthesis of heterocyclic compounds. Recent advances. Tetrahedron 2014, 70, 551–572. [Google Scholar] [CrossRef]
- Tietze, L.F.; Beifuss, U. The Knoevenagel reaction. In Comprehensive Organic Synthesis; Trost, B.M., Ed.; Pergamon Press: Oxford, UK, 1992; Volume 2, pp. 341–394. [Google Scholar]
- de Groot, A.; Jansen, B.J.M. A simple synthesis of 2H-pyrans; a one-step synthesis of flindersine. Tetrahedron Lett. 1975, 39, 3407–3410. [Google Scholar] [CrossRef]
- Lelais, G.; MacMillan, D.W.C. Modern Strategies in organic catalysis: The advent and development of iminium activation. Aldrichimica Acta 2006, 39, 79–87. [Google Scholar]
- Erkkila, A.; Majander, I.; Pihko, P.M. Iminium catalysis. Chem. Rev. 2007, 107, 5416–5470. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.C.; Wang, J.; Cole, K.P.; McLaughlin, M.J.; Morgan, C.D.; Douglas, C.J.; Hsung, R.P.; Coverdale, H.A.; Gerasyuto, A.I.; Hahn, J.M.; et al. A formal [3 + 3] cycloaddition reaction. improved reactivity using α,β-unsaturated iminium salts and evidence for reversibility of 6π-electron electrocyclic ring closure of 1-oxatrienes. J. Org. Chem. 2003, 68, 1729–1735. [Google Scholar] [CrossRef] [PubMed]
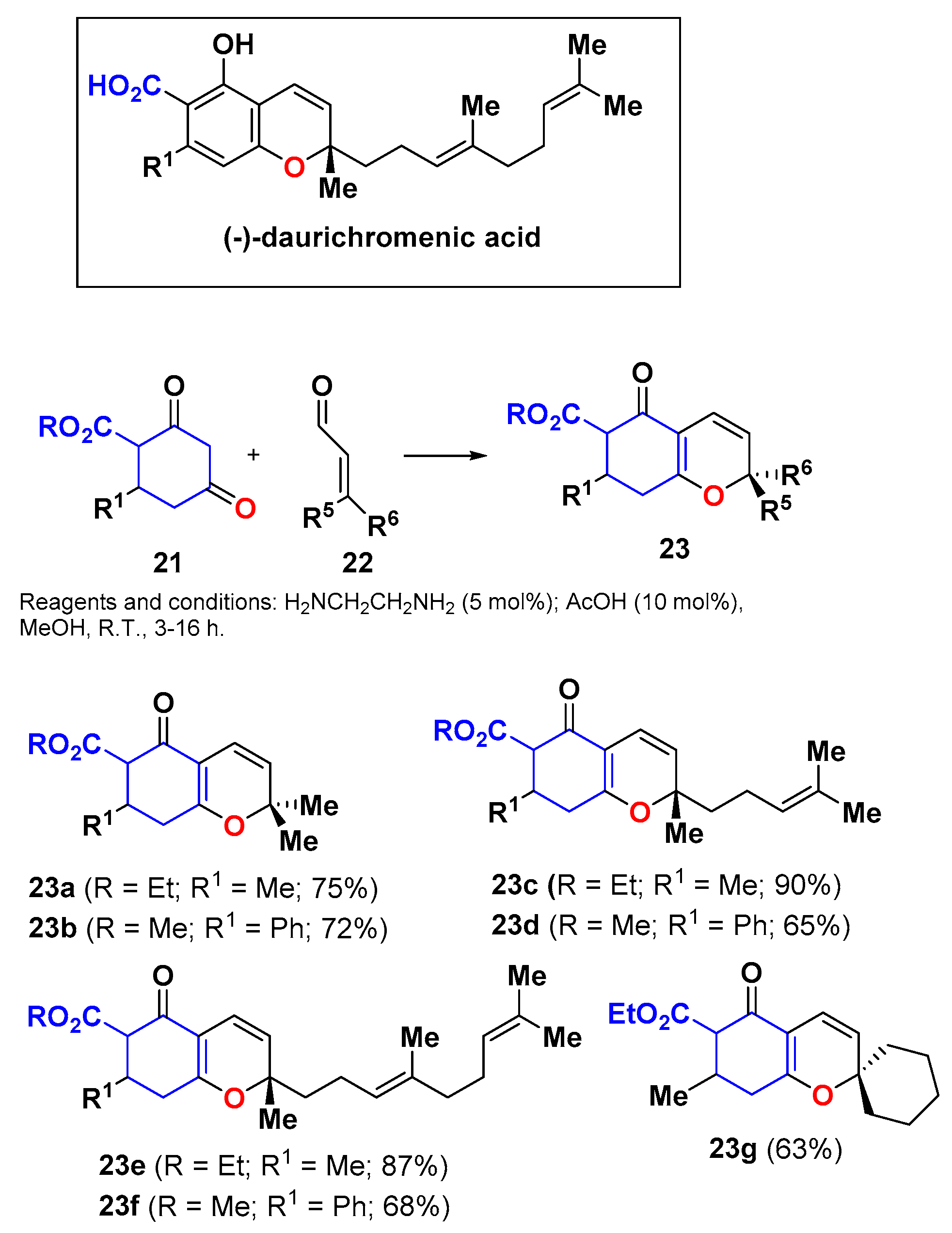
- Hu, H.; Harrison, T.J.; Wilson, P.D. A modular and concise total synthesis of (±)-daurichromenic acid and analogues. J. Org. Chem. 2004, 69, 3782–3786. [Google Scholar] [CrossRef] [PubMed]
- Kurdyumov, A.V.; Lin, N.; Hsung, R.P.; Gullickson, G.C.; Cole, K.P.; Sydorenko, N.; Swidorski, J.J. A Lewis acid-catalyzed formal [3 + 3] cycloaddition of α,β-unsaturated aldehydes with 4-hydroxy-2-pyrone, diketones, and vinylogous esters. Org. Lett. 2006, 8, 191–193. [Google Scholar] [CrossRef] [PubMed]
- Hubert, C.; Moreau, J.; Batany, J.; Duboc, A.; Hurvois, J.P.; Renaud, J.L. Brønsted acid-catalyzed synthesis of pyrans via a formal [3 + 3] cycloaddition. Adv. Synth. Catal. 2008, 350, 40–42. [Google Scholar] [CrossRef]
- Lee, Y.R.; Kim, D.H.; Shim, J.J.; Kim, S.K.; Park, J.H.; Cha, J.S.; Lee, C.S. One-pot synthesis of 2H-pyrans by indium(iii) chloride-catalyzed reactions. Efficient synthesis of pyranocoumarins, pyranophenalenones, and pyranoquinolinones. Bull. Korean Chem. Soc. 2002, 23, 998–1002. [Google Scholar] [CrossRef]
- Jung, E.J.; Lee, Y.R.; Lee, H.J. Iodine-catalyzed one-pot synthesis of 2H-pyrans by domino Knoevenagel/6π-electrocylization. Bull. Korean Chem. Soc. 2009, 30, 2833–2836. [Google Scholar]
- Burke, M.D.; Schreiber, S.L. A planning strategy for diversity-oriented synthesis. Angew. Chem. Int. Ed. 2004, 43, 46–58. [Google Scholar] [CrossRef] [PubMed]
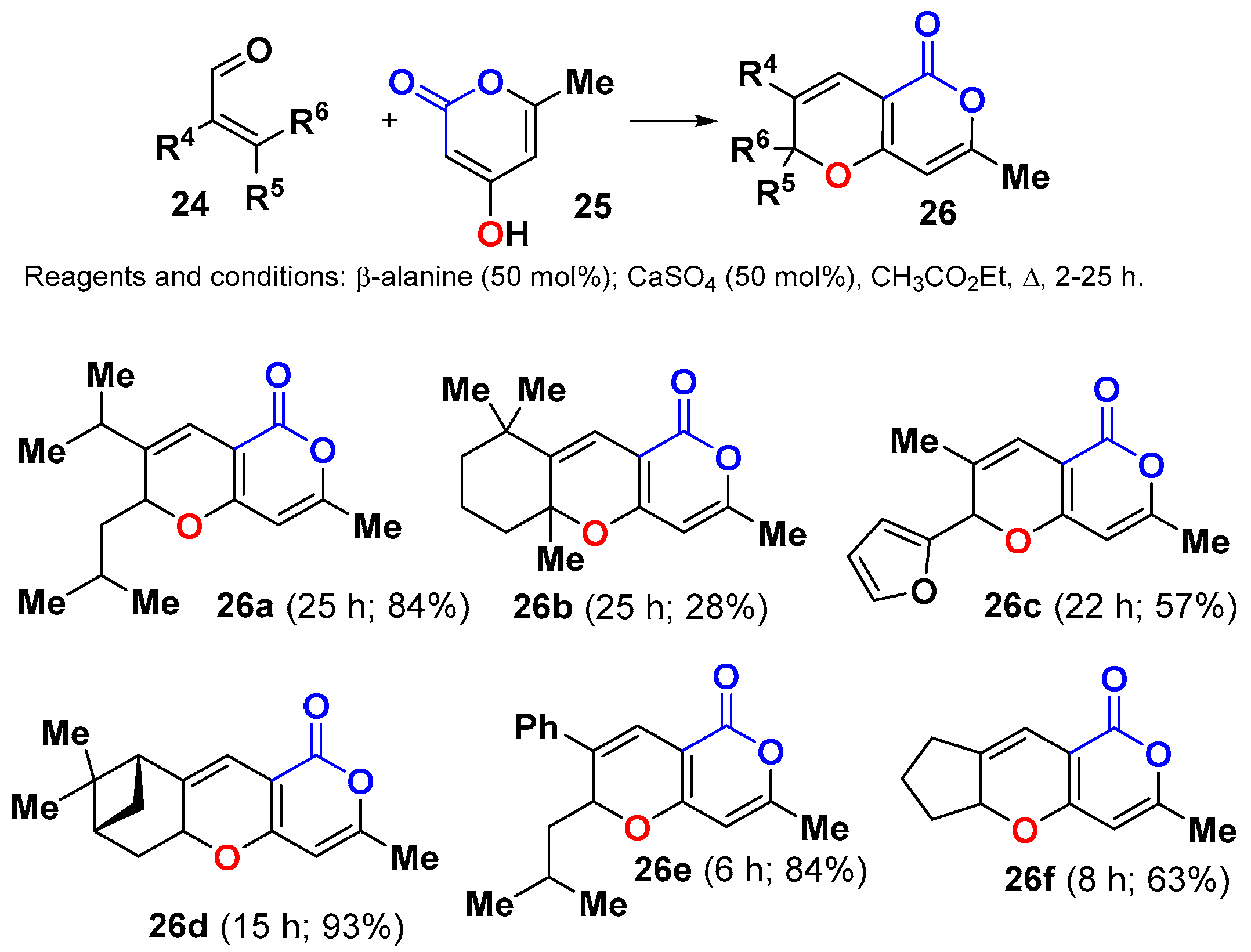
- Leutbecher, H.; Williams, L.A.D.; Rösner, H.; Beifuss, U. Efficient synthesis of substituted 7-methyl-2H,5H-pyrano[4,3-b]pyran-5-ones and evaluation of their in vitro antiproliferative/cytotoxic activities. Bioorg. Med. Chem. Lett. 2007, 17, 978–982. [Google Scholar] [CrossRef] [PubMed]
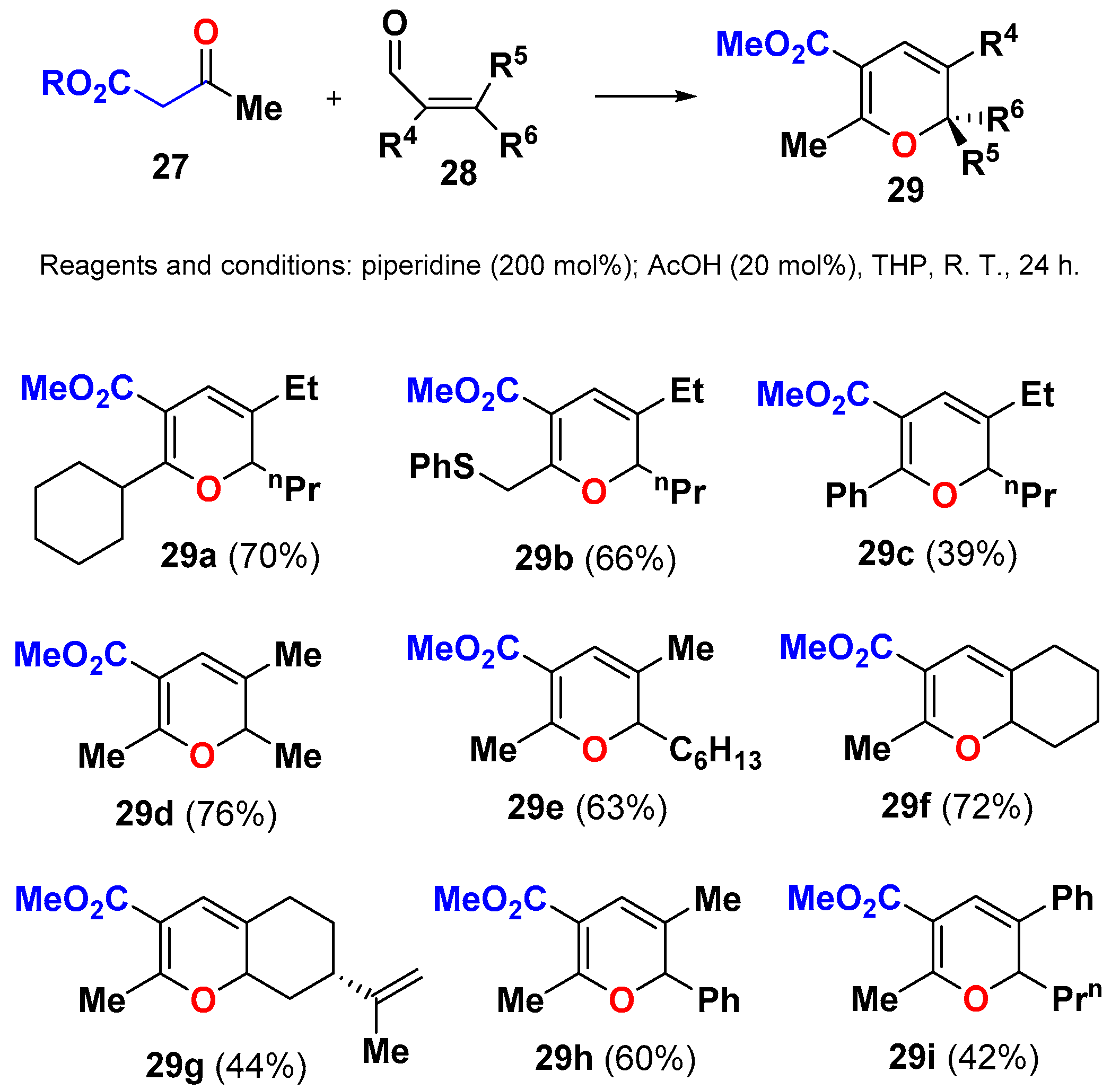
- Peng, W.; Hirabaru, T.; Kawafuchi, H.; Inokuchi, T. Substituent-controlled electrocyclization of 2,4-dienones: Synthesis of 2,3,6-trisubstituted 2H-pyran-5-carboxylates and their transformations. Eur. J. Org. Chem. 2011, 2011, 5469–5474. [Google Scholar] [CrossRef]
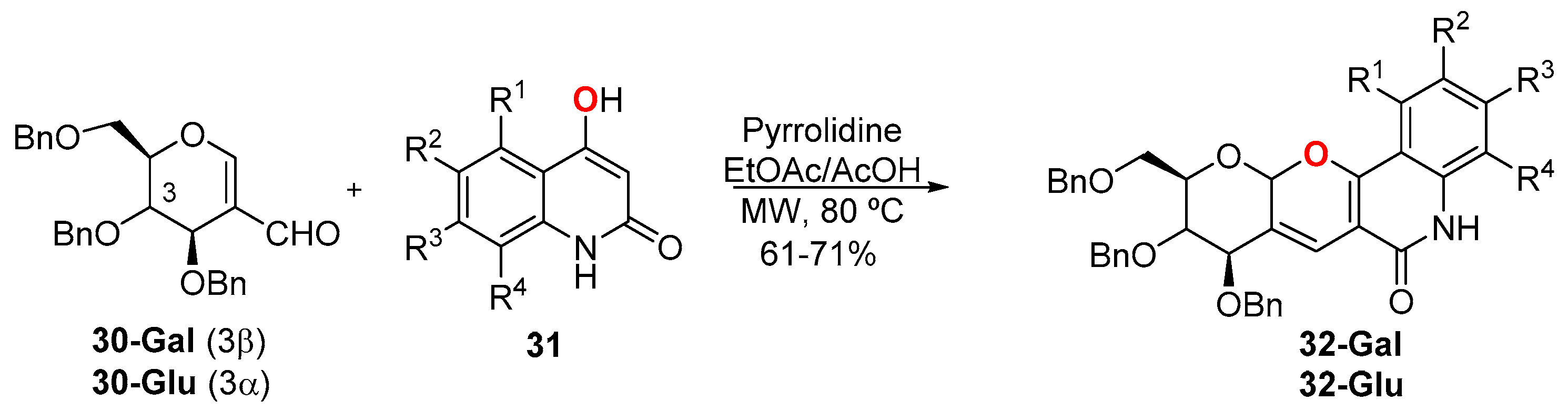
- Kumari, P.; Narayana, C.; Dubey, S.; Gupta, A.; Sagar, R. Stereoselective synthesis of natural product inspired carbohydrate fused pyrano[3,2-c]quinolones as antiproliferative agents. Org. Biomol. Chem. 2018, 16, 2049–2059. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.J.; Park, B.H.; Lee, Y.R. Environmentally benign, one-pot synthesis of pyrans by domino Knoevenagel/6π-electrocyclization in water and application to natural products. Green Chem. 2010, 12, 2003–2011. [Google Scholar] [CrossRef]
- Narayan, S.; Muldoon, J.; Finn, M.G.; Fokin, V.V.; Kolb, H.C.; Sharpless, K.B. “On water”: Unique reactivity of organic compounds in aqueous suspension. Angew. Chem. Int. Ed. 2005, 44, 3275–3279. [Google Scholar] [CrossRef] [PubMed]
- Riviera, M.J.; Mischine, M.P. Green one-pot synthesis of 2H-pyrans under solvent-free conditions catalyzed by ethylenediammonium diacetate. Synth. Commun. 2013, 43, 208–220. [Google Scholar] [CrossRef]
- Peña, J.; Moro, R.F.; Basabe, P.; Marcos, I.S.; Díez, D. Solvent free L-proline-catalysed domino Knoevenagel/6π-electrocyclization for the synthesis of highly functionalised 2H-pyrans. RSC Adv. 2012, 2, 8041–8049. [Google Scholar] [CrossRef]
- Chang, L.; Plevová, K.; Thorimbert, S.; Dechoux, L. Preparation of substituted 2H-pyrans via a cascade reaction from methyl coumalate and activated methylene nucleophiles. J. Org. Chem. 2017, 82, 5499–5505. [Google Scholar] [CrossRef]
- Xie, P.; Yang, J.; Zheng, J.; Huang, Y. Sequential catalyst phosphine/secondary amine promoted [1 + 4]/rearrangement domino reaction for the construction of (2H)-pyrans and 2-oxabicyclo[2,2,2]oct-5-ene skeletons. Eur. J. Org. Chem. 2014, 2014, 1189–1194. [Google Scholar] [CrossRef]
- Hu, J.; Dong, W.; Wu, X.Y.; Tong, X. PPh3-catalyzed [3 + 3] annulations of 5-acetoxypenta-2,3-dienoate with 1C,3O-bisnucleophiles: Facile entry to stable monocyclic 2H-pyrans. Org. Lett. 2012, 14, 5530–5533. [Google Scholar] [CrossRef]
- Menz, H.; Kirsch, S.F. Synthesis of stable 2H-pyran-5-carboxylates via a catalyzed propargyl-Claisen rearrangement/oxa-6π electrocyclization strategy. Org. Lett. 2006, 8, 4795–4797. [Google Scholar] [CrossRef]
- Tejedor, D.; Delgado-Hernández, S.; Diana-Rivero, R.; Díaz-Díaz, A.; García-Tellado, F. A domino strategy for the synthesis of 2H-pyrans from propargyl vinyl ethers. Eur. J. Org. Chem. 2019, 2019, 1784–1790. [Google Scholar] [CrossRef]
- Tejedor, D.; Díaz-Díaz, A.; Diana-Rivero, R.; Delgado-Hernández, S.; García-Tellado, F. Synthesis and utility of 2,2-dimethyl-2H-pyrans: Dienes for sequential Diels−Alder/Retro-Diels−Alder reactions. Org. Lett. 2018, 20, 7987–7990. [Google Scholar] [CrossRef] [PubMed]
- Tejedor, D.; Delgado-Hernández, S.; Peyrac, J.; González-Platas, J.; García-Tellado, F. Integrative pericyclic cascade: An atom economic, multi C-C bond-forming strategy for the construction of molecular complexity. Chem. Eur. J. 2017, 23, 10048–10052. [Google Scholar] [CrossRef] [PubMed]
- Tejedor, D.; Cotos, L.; Márquez-Arce, D.; Odriozola-Gimeno, M.; Torrent-Sucarrat, M.; Cossío, F.P.; García-Tellado, F. Microwave-assisted organocatalyzed rearrangement of propargyl vinyl ethers to salicylaldehyde derivatives: An experimental and theoretical study. Chem. Eur. J. 2015, 21, 18280–18289. [Google Scholar] [CrossRef] [PubMed]
- Chong, Q.; Wang, C.; Wang, D.; Wang, H.; Wu, F.; Xin, X.; Wan, B. DABCO-catalyzed synthesis of 3-bromo-/3-iodo-2H-pyrans from propargyl alcohols, dialkyl acetylene dicarboxylates, and N-bromo-/N-iodosuccinimides. Tetrahedron Lett. 2015, 56, 401–403. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Okude, Y.; Mori, S.; Shibuya, M. Combined experimental and computational study on ruthenium(II)-catalyzed reactions of diynes with aldehydes and N,N-dimethylformamide. J. Org. Chem. 2017, 82, 7964–7973. [Google Scholar] [CrossRef]
- Tambar, U.K.; Kano, V.; Zepernick, J.F.; Stoltz, B.M. The development and scope of a versatile tandem Stille-oxa-electrocyclization reaction. Tetrahedron Lett. 2007, 48, 345–350. [Google Scholar] [CrossRef]
- Tambar, U.K.; Kano, T.; Stoltz, B.M. Progress toward the total synthesis of saudin: Development of a tandem Stille-oxa-electrocyclization reaction. Org. Lett. 2005, 7, 2413–2416. [Google Scholar] [CrossRef][Green Version]
- Li, C.; Johnson, R.P.; Porco, J.A. Total synthesis of the quinone epoxide dimer (+)-torreyanic acid: Application of a biomimetic oxidation/electrocyclization/Diels-Alder dimerization cascade. J. Am. Chem. Soc. 2003, 125, 5095–5106. [Google Scholar] [CrossRef]
- Shoji, M.; Yamaguchi, J.; Kakeya, H.; Osada, H.; Hayashi, Y. Total synthesis of (+)-epoxyquinols A and B. Angew. Chem. Int. Ed. 2002, 41, 3192–3194. [Google Scholar] [CrossRef]
- Li, C.; Lobkovsky, E.; Porco, J.A. Total synthesis of (±)-torreyanic acid. J. Am. Chem. Soc. 2000, 122, 10484–10485. [Google Scholar] [CrossRef]

























{kind=link}
{kind=link}
{kind=link}
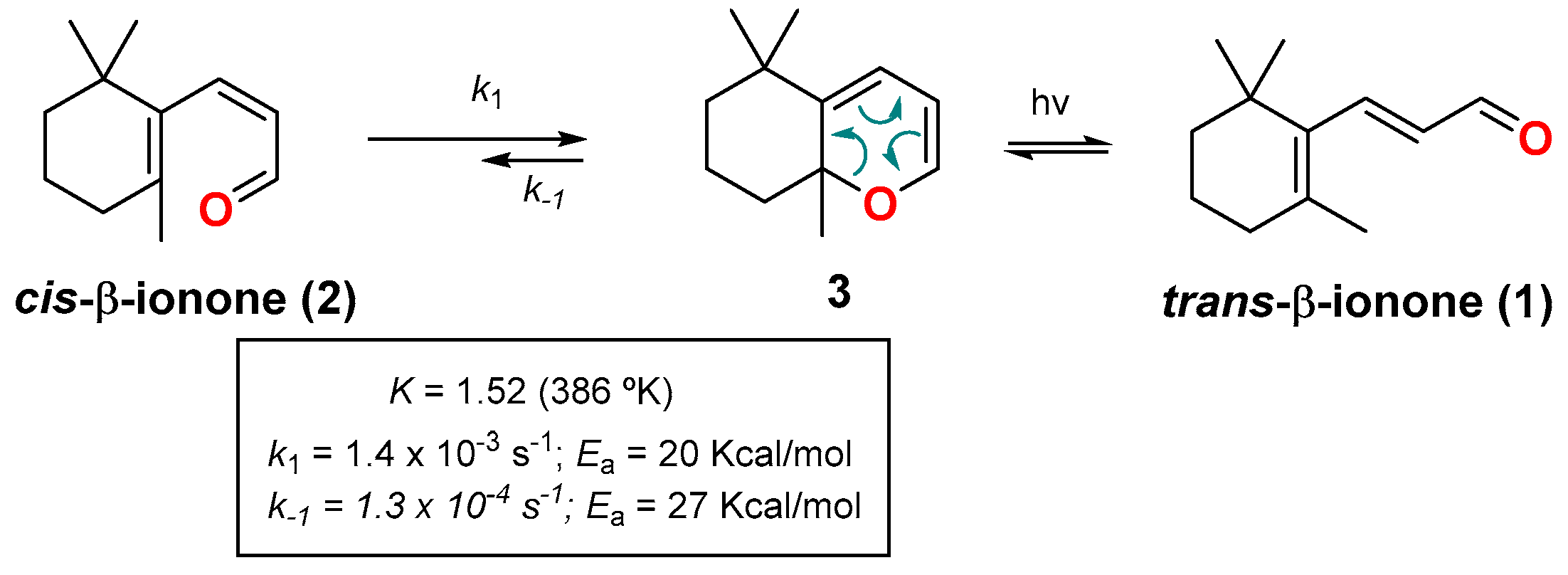{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
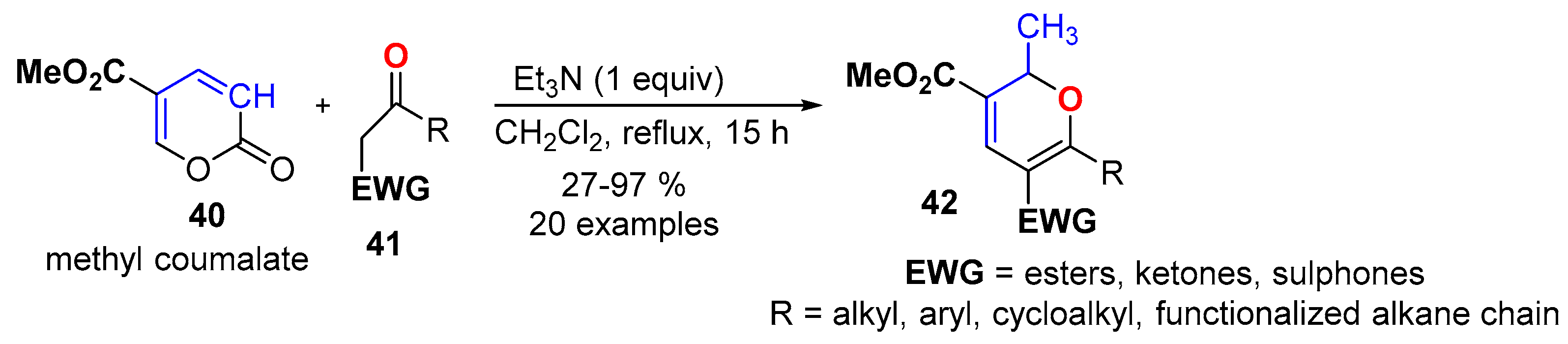{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
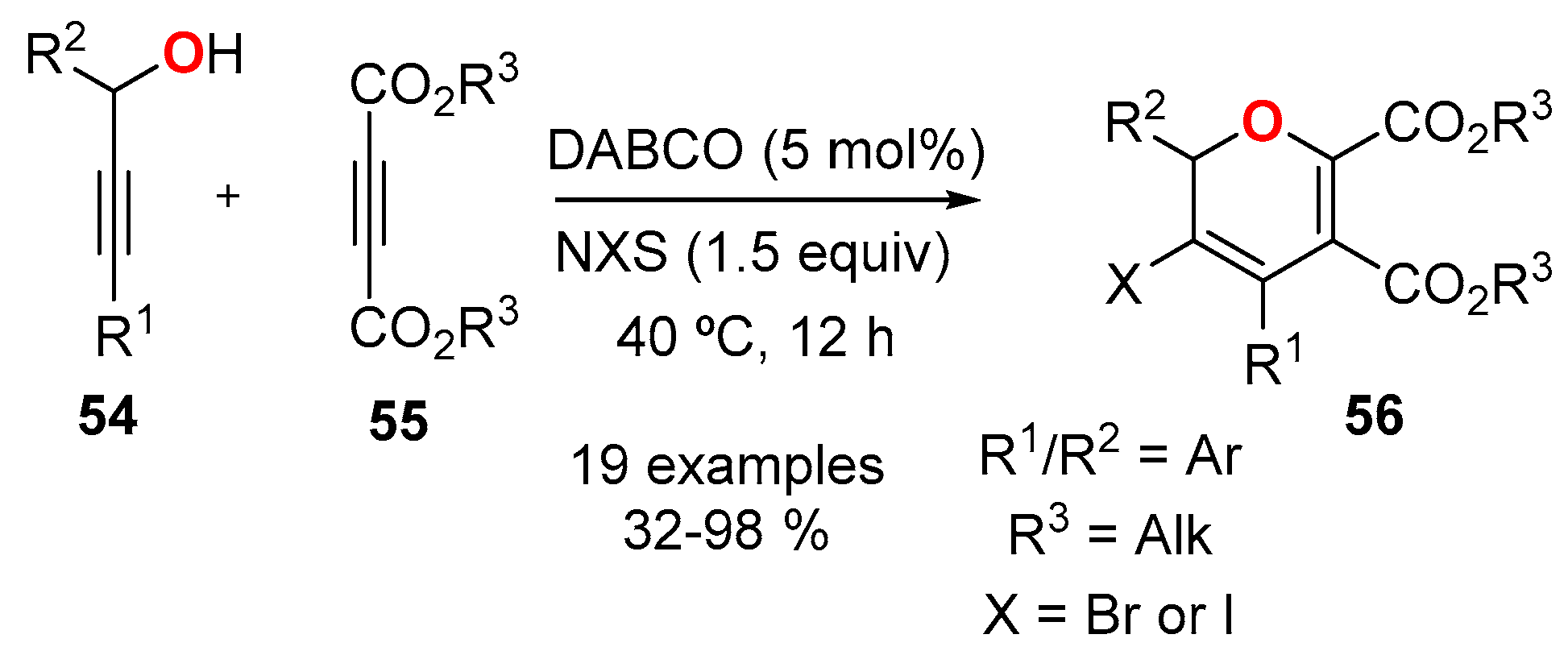{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | R | R1 | R4 | R5 | R6 | (E)-13 | 2HP 14 | (Z)-13 |
|---|---|---|---|---|---|---|---|---|
| 1 | EtO | Me | H | H | Me | 30 | 30 | 40 |
| 2 | EtO | Me | H | H | H | 45 | 30 | 25 |
| 3 | EtO | Me | H | Me | Me | 17 | 68 | 15 |
| 4 | MeO | Me | H | H | H | 43 | 37 | 30 |
| 5 | MeO | Me | H | H | Me | 40 | 40 | 20 |
| 6 | MeO | Me | H | Me | Me | 26 | 62 | 12 |
| 7 | Me | Me | H | H | Me | 72 | 28 | - |
| 8 | Me | Me | H | Me | Me | 64 | 36 | - |
| 9 | t-BuO | Me | H | Me | Me | 18 | 17 | 65 |
| 10 | EtO | Arb | H | Me | Me | 84 | 9 | 7 |
| 11 | EtO | Ph | H | H | Me | 67 | - | 33 |
| 12 | EtO | Ph | H | Me | Me | 86 | - | 14 |
| 13 | EtO | Me | H | H | Ph | 60 | - | 40 |
| 14 | Me | Me | H | H | Ph | 100 | - | - |
| 15 | MeO | Me | Me | Me | Me | - | 83 | 17 |
| 16 | MeO | Me | Me | H | Ph | - | 100 | - |
| 17 | Me | Me | Me | Me | Me | - | 100 | - |
| 18 | Me | Me | Me | H | Ph | - | 100 | - |
| 19 | EtO | Ph | Me | Me | Me | - | 100 | - |
| 20 | MeO | Me | H | H | c-C6H11 | 30 | 23 | 47 |
| 21 | MeO | Me | Me | H | c-C6H11 | - | 100 | - |
| 22 | MeO | Me | H | –(CH2)5– | 16 | 67 | 17 | |
| 23 | MeO | Me | Me | –(CH2)5– | - | 100 | - | |
| 24 | MeO | Me | H | –(CH2)4– | 47 | 31 | 22 | |
| 25 | MeO | Me | Me | H | HC=CMe2 | 75 | - | 25 |
| 26 | Me | Me | Me | H | HC=CMe2 | 100c | - | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tejedor, D.; Delgado-Hernández, S.; Diana-Rivero, R.; Díaz-Díaz, A.; García-Tellado, F. Recent Advances in the Synthesis of 2H-Pyrans. Molecules 2019, 24, 2904. https://doi.org/10.3390/molecules24162904
Tejedor D, Delgado-Hernández S, Diana-Rivero R, Díaz-Díaz A, García-Tellado F. Recent Advances in the Synthesis of 2H-Pyrans. Molecules. 2019; 24(16):2904. https://doi.org/10.3390/molecules24162904
Chicago/Turabian StyleTejedor, David, Samuel Delgado-Hernández, Raquel Diana-Rivero, Abián Díaz-Díaz, and Fernando García-Tellado. 2019. "Recent Advances in the Synthesis of 2H-Pyrans" Molecules 24, no. 16: 2904. https://doi.org/10.3390/molecules24162904
APA StyleTejedor, D., Delgado-Hernández, S., Diana-Rivero, R., Díaz-Díaz, A., & García-Tellado, F. (2019). Recent Advances in the Synthesis of 2H-Pyrans. Molecules, 24(16), 2904. https://doi.org/10.3390/molecules24162904