First-In-Class Inhibitors Targeting the Interaction between Bacterial RNA Polymerase and Sigma Initiation Factor Affect the Viability and Toxin Release of Streptococcus pneumoniae


Abstract
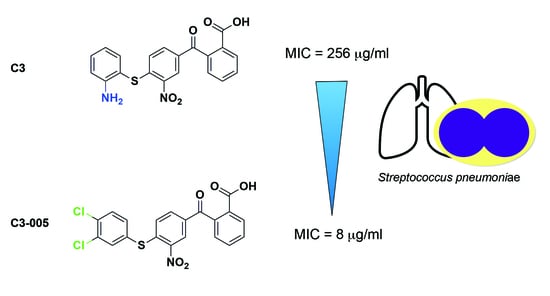
1. Introduction
2. Results and Discussion
2.1. Docking Study of C3 and Its Antimicrobial Activity
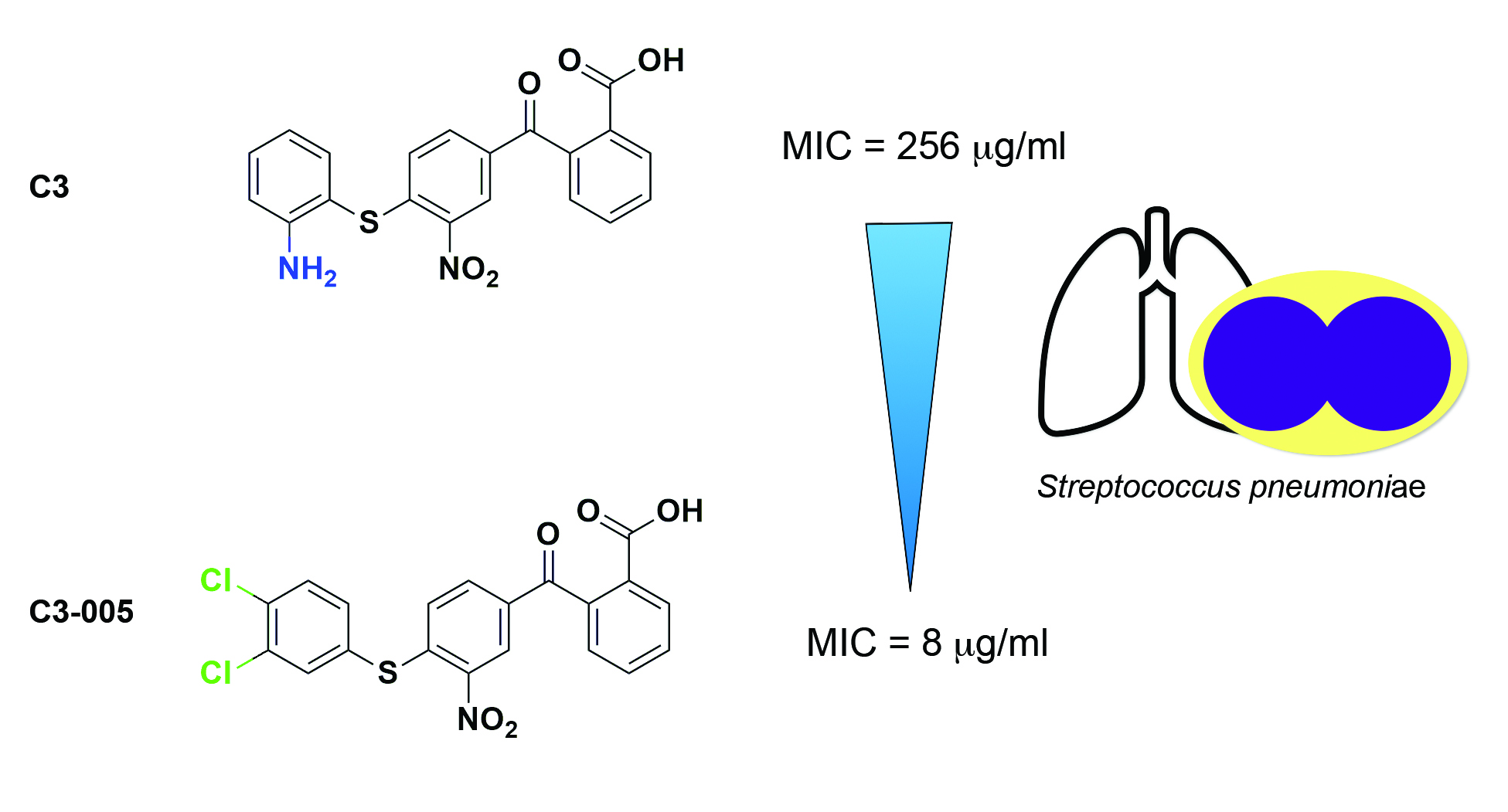
2.2. Molecular Mechanism of C3 by Inhibiting the Protein-Protein Interaction between RNAP CH-σ
2.3. Antimicrobial and Inhibitory Activities of C3 Derivatives
2.4. Antimicrobial Activity of C3-005 against Representative Gram-Positive Bacterial Pathogens
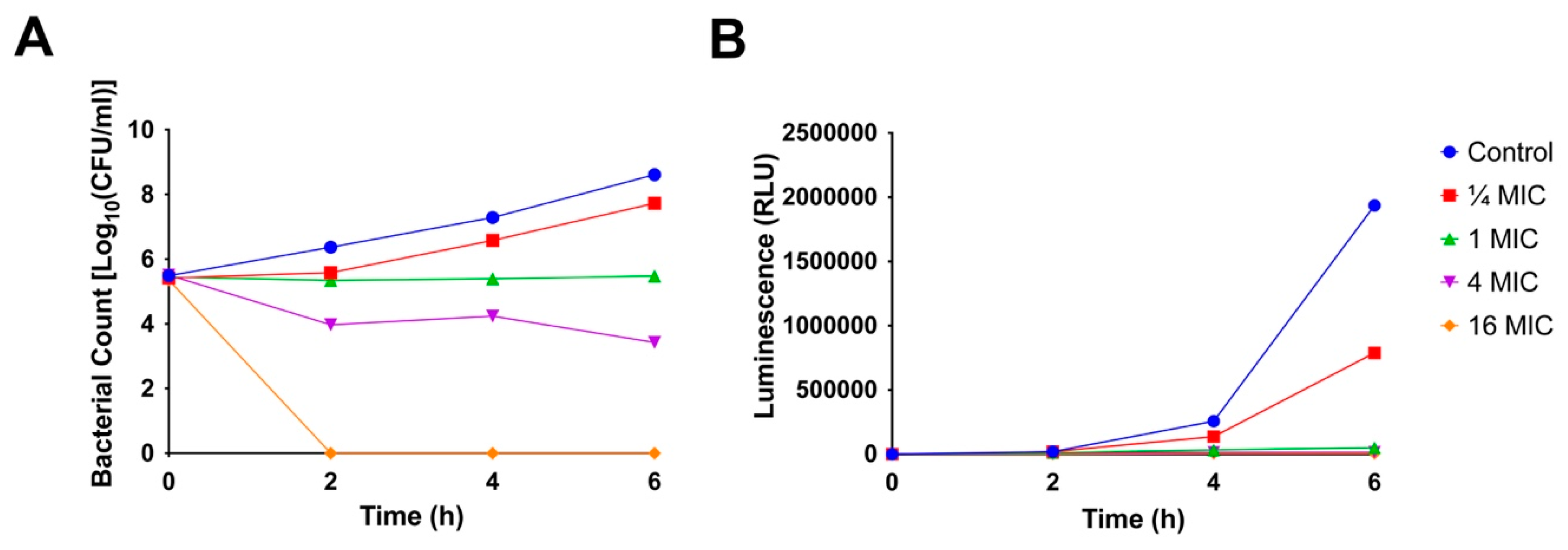
2.5. Time-Kill Kinetics
2.6. ATP Production
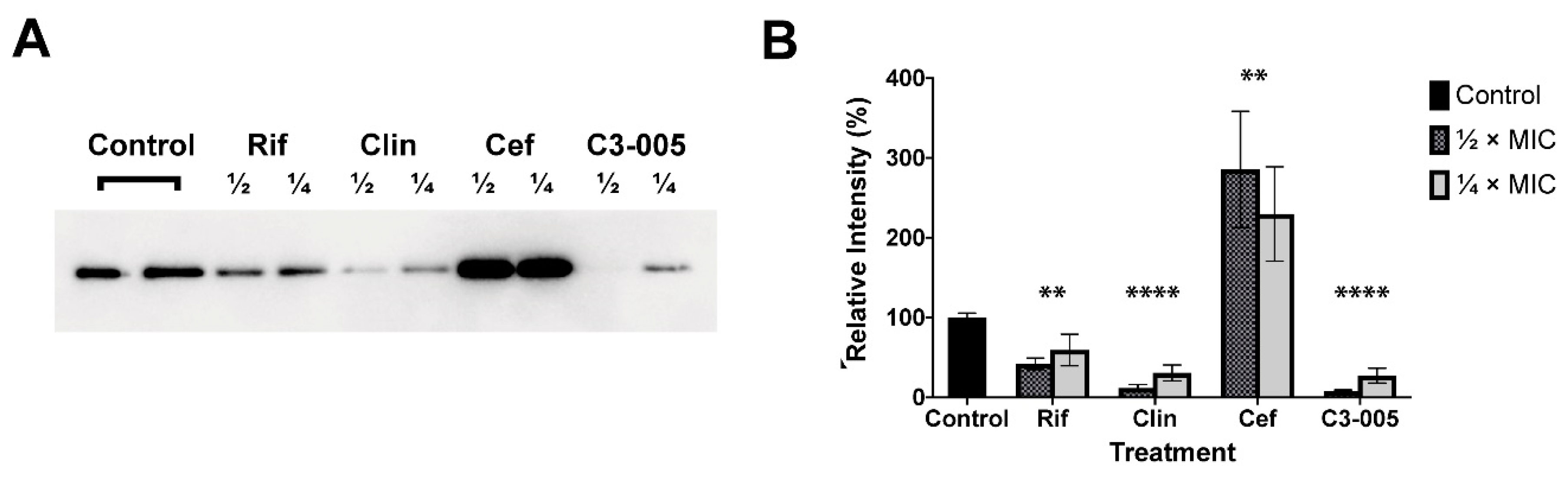
2.7. S. pneumoniae Toxin Secretion
2.8. Cytotoxicity of C3-005
3. Materials and Methods
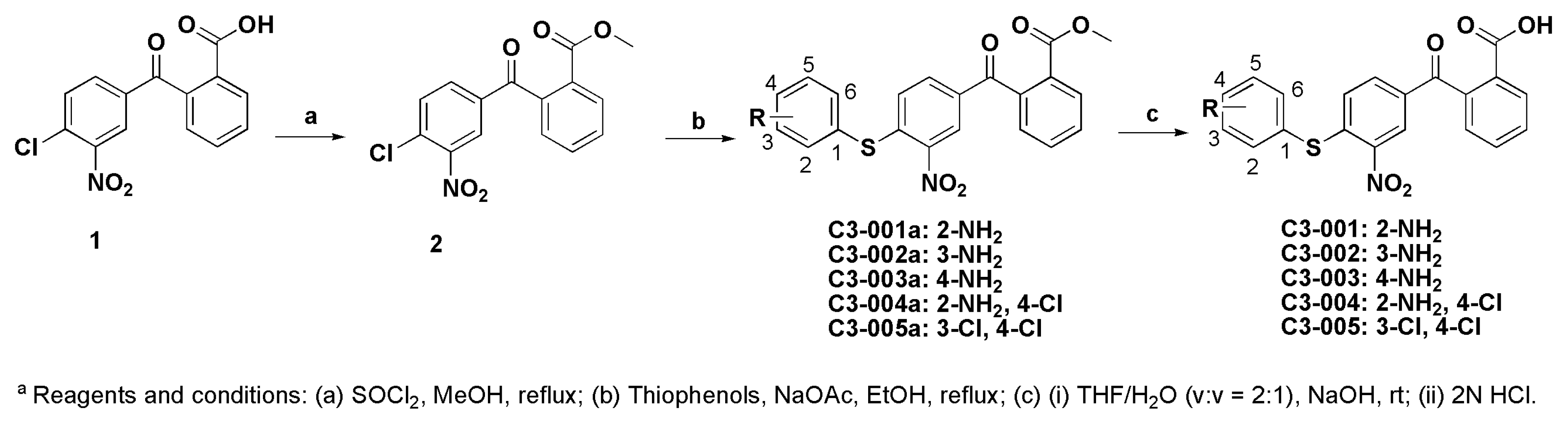
3.1. Chemistry
3.1.1. Synthesis of Methyl 2-(4-chloro-3-nitrobenzoyl)benzoate (2)
3.1.2. General Procedure for the Synthesis of Compound C3-001a and its Derivatives
3.1.3. General Procedure for the Synthesis of Compound C3-001 and Its Derivatives
3.2. Biology
3.2.1. Bacterial Strains and Antibiotics
3.2.2. Determination of Minimum Inhibitory Concentration (MIC)
3.2.3. Protein-Protein Interaction Inhibition Assay
3.2.4. Time-Kill Kinetics
3.2.5. Assessment of ATP Production
3.2.6. S. pneumoniae Toxin Secretion
3.2.7. Western Blot
3.2.8. Cytotoxicity Assay
3.3. Molecular Modelling
3.4. Data and Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Tacconelli, E.; Pezzani, M.D. Public health burden of antimicrobial resistance in Europe. Lancet Infect. Dis. 2019, 19, 4–6. [Google Scholar] [CrossRef]
- Cherazard, R.; Epstein, M.; Doan, T.L.; Salim, T.; Bharti, S.; Smith, M.A. Antimicrobial Resistant Streptococcus pneumoniae: Prevalence, Mechanisms, and Clinical Implications. Am. J. Therapeutics 2017, 24, e361–e369. [Google Scholar] [CrossRef] [PubMed]
- Jedrzejas, M.J. Pneumococcal virulence factors: Structure and function. Microbiol. Mol. Biol. Rev. 2001, 65, 187–207. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.; Steel, H.C.; Cockeran, R.; Smith, A.M.; von Gottberg, A.; de Gouveia, L.; Brink, A.; Klugman, K.P.; Mitchell, T.J.; Feldman, C. Clarithromycin alone and in combination with ceftriaxone inhibits the production of pneumolysin by both macrolide-susceptible and macrolide-resistant strains of Streptococcus pneumoniae. J. Antimicrob. Chemother. 2007, 59, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Yang, X.; Lewis, P.J. Bacterial transcription as a target for antibacterial drug development. Microbiol. Mol. Biol. Rev. 2016, 80, 139–160. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.S. Structural biology of bacterial RNA polymerase. Biomolecules 2015, 5, 848–864. [Google Scholar] [CrossRef] [PubMed]
- Werner, F.; Grohmann, D. Evolution of multisubunit RNA polymerases in the three domains of life. Nat. Rev. Microbiol. 2011, 9, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, L.; Zhang, Y.; Lin, Z. Bacterial sigma factors as targets for engineered or synthetic transcriptional control. Front. Bioeng. Biotechnol. 2014, 2, 33. [Google Scholar] [CrossRef] [PubMed]
- Hinsberger, S.; Hüsecken, K.; Groh, M.; Negri, M.; Haupenthal, J.; Hartmann, R.W. Discovery of novel bacterial RNA polymerase inhibitors: Pharmacophore-based virtual screening and hit optimization. J. Med. Chem. 2013, 56, 8332–8338. [Google Scholar] [CrossRef]
- André, E.; Bastide, L.; Villain-Guillot, P.; Latouche, J.; Rouby, J.; Leonetti, J.P. A multiwell assay to isolate compounds inhibiting the assembly of the prokaryotic RNA polymerase. Assay Drug Dev. Technol. 2004, 2, 629–635. [Google Scholar] [CrossRef]
- Mariner, K.R.; Trowbridge, R.; Agarwal, A.K.; Miller, K.; O’Neill, A.J.; Fishwick, C.W.G.; Chopra, I. Furanyl-rhodanines are unattractive drug candidates for development as inhibitors of bacterial RNA polymerase. Antimicrob. Agents Chemother. 2010, 54, 4506–4509. [Google Scholar] [CrossRef]
- Martínez-Lumbreras, S.; Alfano, C.; Evans, N.J.; Collins, K.M.; Flanagan, K.A.; Atkinson, R.A.; Krysztofinska, E.M.; Vydyanath, A.; Jackter, J.; Fixon-Owoo, S.; et al. Structural and Functional Insights into Bacillus subtilis Sigma Factor Inhibitor, CsfB. Structure 2018, 26, 640–648.e5. [Google Scholar] [CrossRef]
- Ma, C.; Yang, X.; Kandemir, H.; Mielczarek, M.; Johnston, E.B.; Griffith, R.; Kumar, N.; Lewis, P.J. Inhibitors of bacterial transcription initiation complex formation. ACS Chem. Biol. 2013, 8, 1972–1980. [Google Scholar] [CrossRef]
- Ma, C.; Yang, X.; Lewis, P.J. Bacterial Transcription Inhibitor of RNA Polymerase Holoenzyme Formation by Structure-Based Drug Design: From in Silico Screening to Validation. ACS Infect. Dis. 2016, 2, 39–46. [Google Scholar] [CrossRef]
- CLSI, Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically, 11th ed.; (M07Ed11); CLSI: Wayne, PA, USA, 2018. [Google Scholar]
- Tsang, T.F.; Qiu, Y.; Lin, L.; Ye, J.; Ma, C.; Yang, X. Simple method for studying in vitro protein-protein interactions based on protein complementation and its application in drug screening targeting bacterial transcription. ACS Infect. Dis. 2019, 5, 521–527. [Google Scholar] [CrossRef]
- Watanabe, S.; Takemoto, N.; Ogura, K.; Miyoshi-Akiyama, T. Severe invasive streptococcal infection by Streptococcus pyogenes and Streptococcus dysgalactiae subsp. equisimilis. Microbiol. Immunol. 2016, 60, 1–9. [Google Scholar] [CrossRef]
- Raabe, V.N.; Shane, A.L. Group B Streptococcus (Streptococcus agalactiae). Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef]
- Balemans, W.; Vranckx, L.; Lounis, N.; Pop, O.; Guillemont, J.; Vergauwen, K.; Mol, S.; Gilissen, R.; Motte, M.; Lançois, D.; et al. Novel antibiotics targeting respiratory ATP synthesis in gram-positive pathogenic bacteria. Antimicrob. Agents Chemother. 2012, 56, 4131–4139. [Google Scholar] [CrossRef]
- Lobritz, M.A.; Belenky, P.; Porter, C.B.M.; Gutierrez, A.; Yang, J.H.; Schwarz, E.G.; Dwyer, D.J.; Khalil, A.S.; Collins, J.J. Antibiotic efficacy is linked to bacterial cellular respiration. Proc. Natl. Acad. Sci. USA 2015, 112, 8173–8180. [Google Scholar] [CrossRef]
- Yang, J.H.; Bening, S.C.; Collins, J.J. Antibiotic efficacy—Context matters. Curr. Opin. Microbiol. 2017, 39, 73–80. [Google Scholar] [CrossRef]
- Spreer, A.; Von Rüden, C.; Mitchell, T.J.; Eiffert, H.; Nau, R. Influence of subinhibitory concentrations of protein-synthesis-inhibiting antibiotics on production and release of the pneumococcal virulence factor pneumolysin in vitro. Chemotherapy 2007, 53, 327–331. [Google Scholar] [CrossRef]
- Brown, L.A.; Mitchell, A.M.; Mitchell, T.J. Streptococcus pneumoniae and lytic antibiotic therapy: Are we adding insult to injury during invasive pneumococcal disease and sepsis? J. Med. Microbiol. 2017, 66, 1253–1256. [Google Scholar] [CrossRef]
- Spreer, A.; Kerstan, H.; Böttcher, T.; Gerber, J.; Siemer, A.; Zysk, G.; Mitchell, T.J.; Eiffert, H.; Nau, R. Reduced release of pneumolysin by Streptococcus pneumoniae in vitro and in vivo after treatment with nonbacteriolytic antibiotics in comparison to ceftriaxone. Antimicrob. Agents Chemother. 2003, 47, 2649–2654. [Google Scholar] [CrossRef]
- Yang, X.; Luo, M.J.; Yeung, A.C.M.; Lewis, P.J.; Chan, P.K.S.; Ip, M.; Ma, C. First-In-Class Inhibitor of Ribosomal RNA Synthesis with Antimicrobial Activity against Staphylococcus aureus. Biochemistry 2017, 56, 5049–5052. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Bae, B.; Davis, E.; Brown, D.; Campbell, E.A.; Wigneshweraraj, S.; Darst, S.A. Phage T7 Gp2 inhibition of Escherichia coli RNA polymerase involves misappropriation of σ70 domain 1.1. Proc. Natl. Acad. Sci. USA 2013, 110, 19772–19777. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds C3-001–005 are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | EFAE | SAURa | SAURb | SPNE | KPNE | ABAU | PAER | ECLO | ECOL |
|---|---|---|---|---|---|---|---|---|---|
| C3 | >256 | >256 | >256 | 256 | >256 | >256 | >256 | >256 | >256 |
| C3-002 | 256 | 256 | 256 | 64 | >256 | >256 | >256 | >256 | >256 |
| C3-003 | >256 | >256 | >256 | 128 | >256 | >256 | >256 | >256 | >256 |
| C3-004 | 256 | 128 | 256 | 64 | >256 | >256 | >256 | >256 | >256 |
| C3-005 | 32 | 16 | 16 | 8 | >256 | >256 | >256 | >256 | >256 |
| VAN | 1 | 1 | 0.5 | 0.25 | >64 | >64 | >64 | >64 | >64 |
| RIF | 4 | 0.063 | 0.063 | 0.063 | 32 | 4 | 32 | ≥64 | 64 |
| Compound | R | MIC a (μg/mL) | % Inhibition b | ClogP |
|---|---|---|---|---|
| C3-001 | 2-NH2 | 256 | 62.2 ± 2.6 | 3.39 |
| C3-002 | 3-NH2 | 64 | 81.3 ± 4.9 | 3.39 |
| C3-003 | 4-NH2 | 128 | 69.1 ± 10.7 | 3.39 |
| C3-004 | 2-NH2, 4-Cl | 64 | 60.5 ± 16.5 | 4.39 |
| C3-005 | 3-Cl, 4-Cl | 8 | 81.7 ± 0.9 | 5.94 |
| Vancomycin | N.A. | 0.25 | N.D. | N.D. |
| Rifampicin | N.A. | 0.0625 | N.D. | N.D. |
| Compound | MIC (μg/mL) | |||
|---|---|---|---|---|
| SPYO | SAGA | SEPI | SSAP | |
| C3-005 | 16 | 16 | 16 | 32 |
| VAN | 0.5 | 1 | 2 | 1 |
| RIF | 0.031 | 0.031 | ≤0.063 | ≤0.063 |
| Compound | CC50 (μM) | |
|---|---|---|
| HepG2 | A549 | |
| C3-005 | 63.82 ± 9.32 | 76.65 ± 11.37 |
| cisplatin | 6.81 ± 0.61 | 7.70 ± 0.58 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, J.; Chu, A.J.; Lin, L.; Yang, X.; Ma, C. First-In-Class Inhibitors Targeting the Interaction between Bacterial RNA Polymerase and Sigma Initiation Factor Affect the Viability and Toxin Release of Streptococcus pneumoniae. Molecules 2019, 24, 2902. https://doi.org/10.3390/molecules24162902
Ye J, Chu AJ, Lin L, Yang X, Ma C. First-In-Class Inhibitors Targeting the Interaction between Bacterial RNA Polymerase and Sigma Initiation Factor Affect the Viability and Toxin Release of Streptococcus pneumoniae. Molecules. 2019; 24(16):2902. https://doi.org/10.3390/molecules24162902
Chicago/Turabian StyleYe, Jiqing, Adrian Jun Chu, Lin Lin, Xiao Yang, and Cong Ma. 2019. "First-In-Class Inhibitors Targeting the Interaction between Bacterial RNA Polymerase and Sigma Initiation Factor Affect the Viability and Toxin Release of Streptococcus pneumoniae" Molecules 24, no. 16: 2902. https://doi.org/10.3390/molecules24162902
APA StyleYe, J., Chu, A. J., Lin, L., Yang, X., & Ma, C. (2019). First-In-Class Inhibitors Targeting the Interaction between Bacterial RNA Polymerase and Sigma Initiation Factor Affect the Viability and Toxin Release of Streptococcus pneumoniae. Molecules, 24(16), 2902. https://doi.org/10.3390/molecules24162902