Insight into the PTP1B Inhibitory Activity of Arylbenzofurans: An In Vitro and In Silico Study

 , ,
, ,
Abstract
:1. Introduction
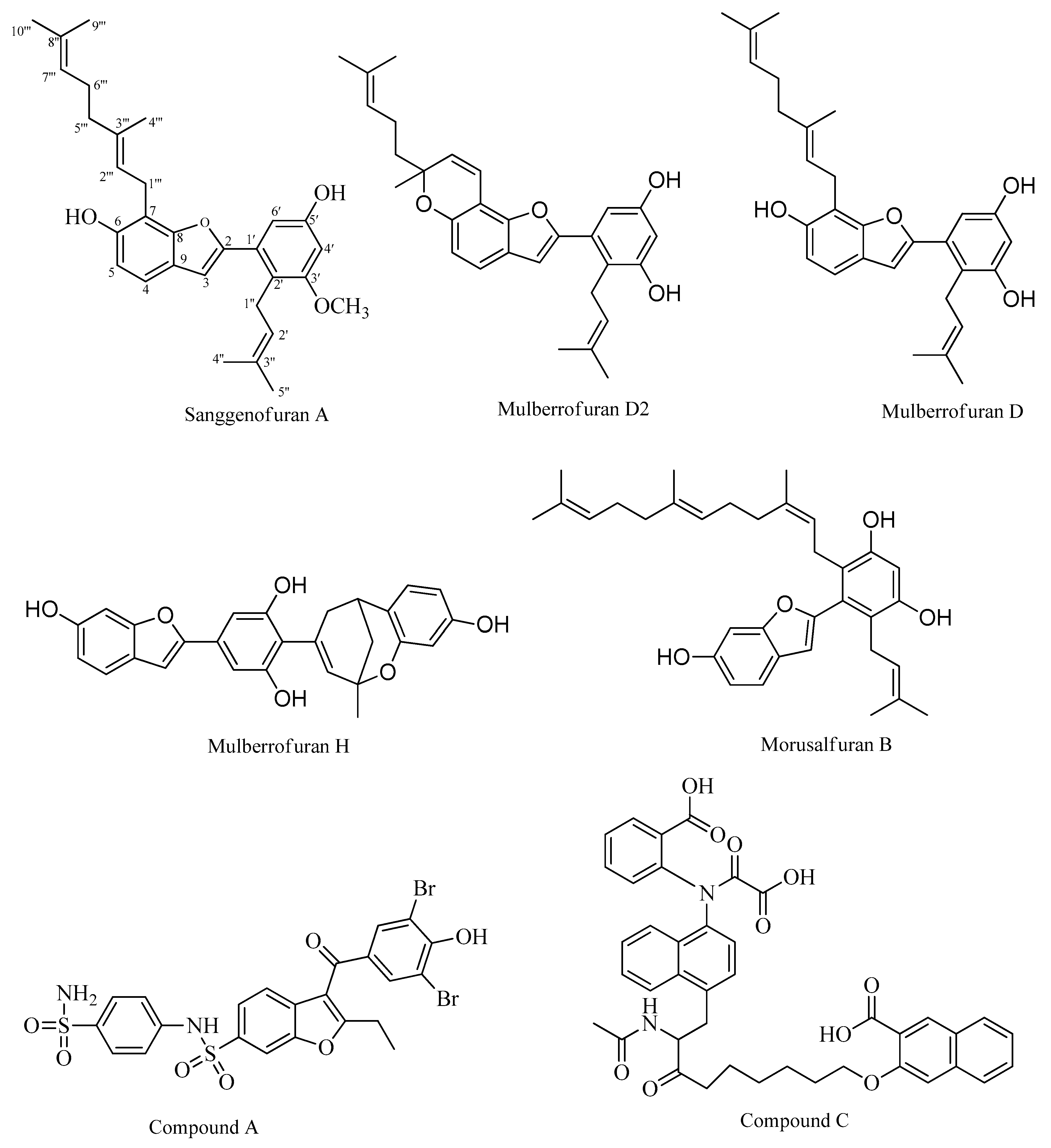
2. Results
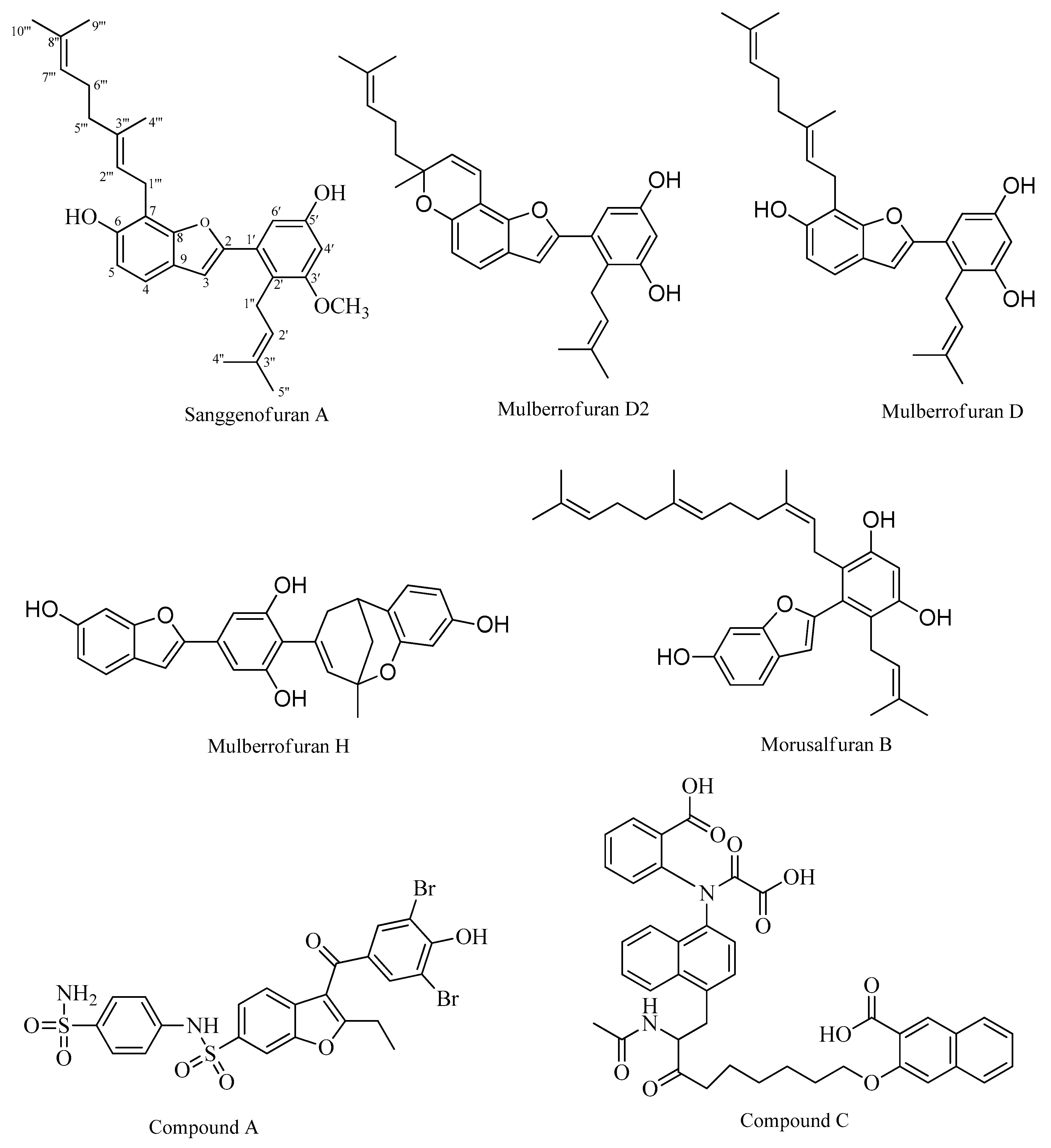
2.1. PTP1B Inhibitory Assays
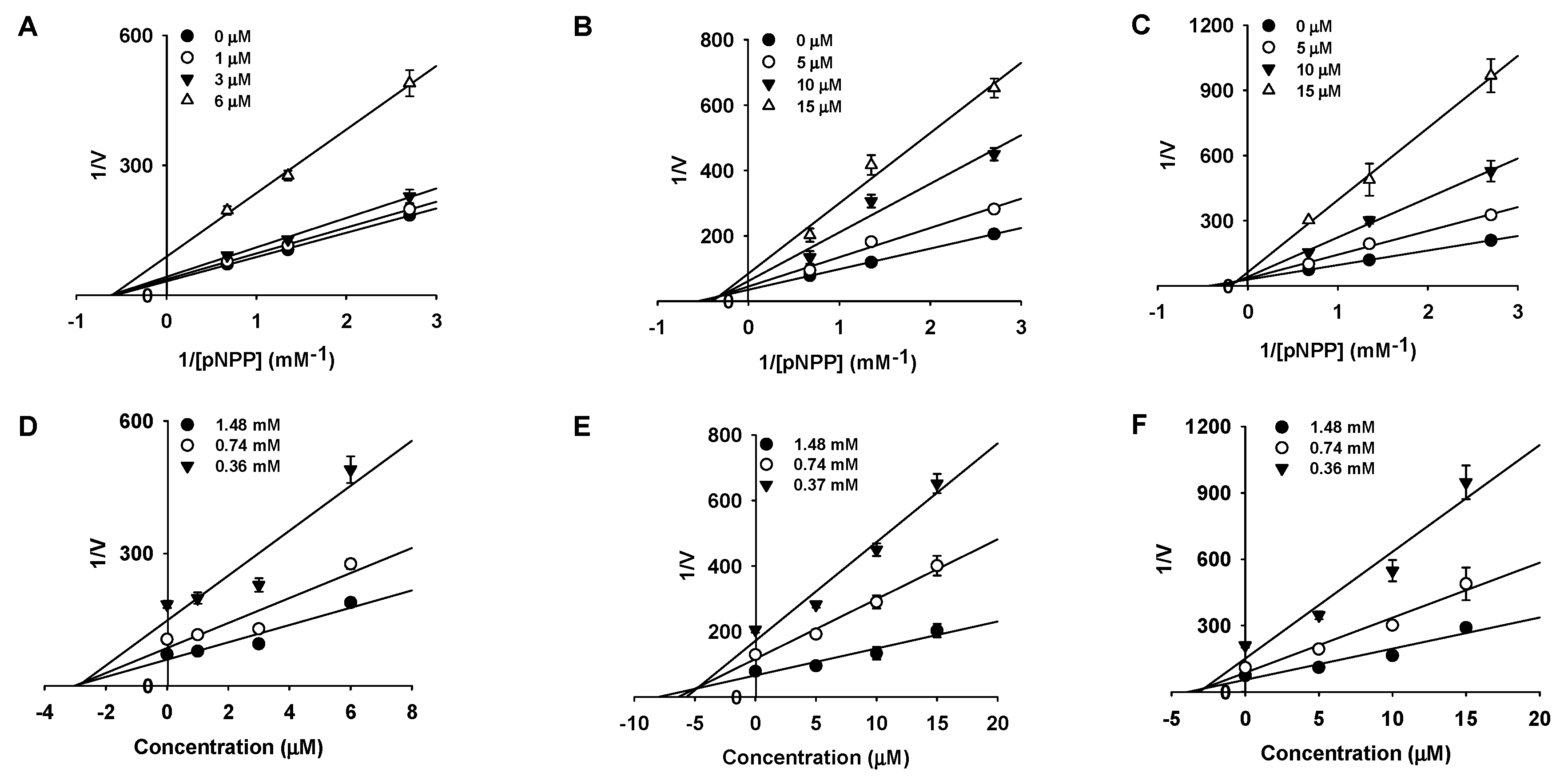
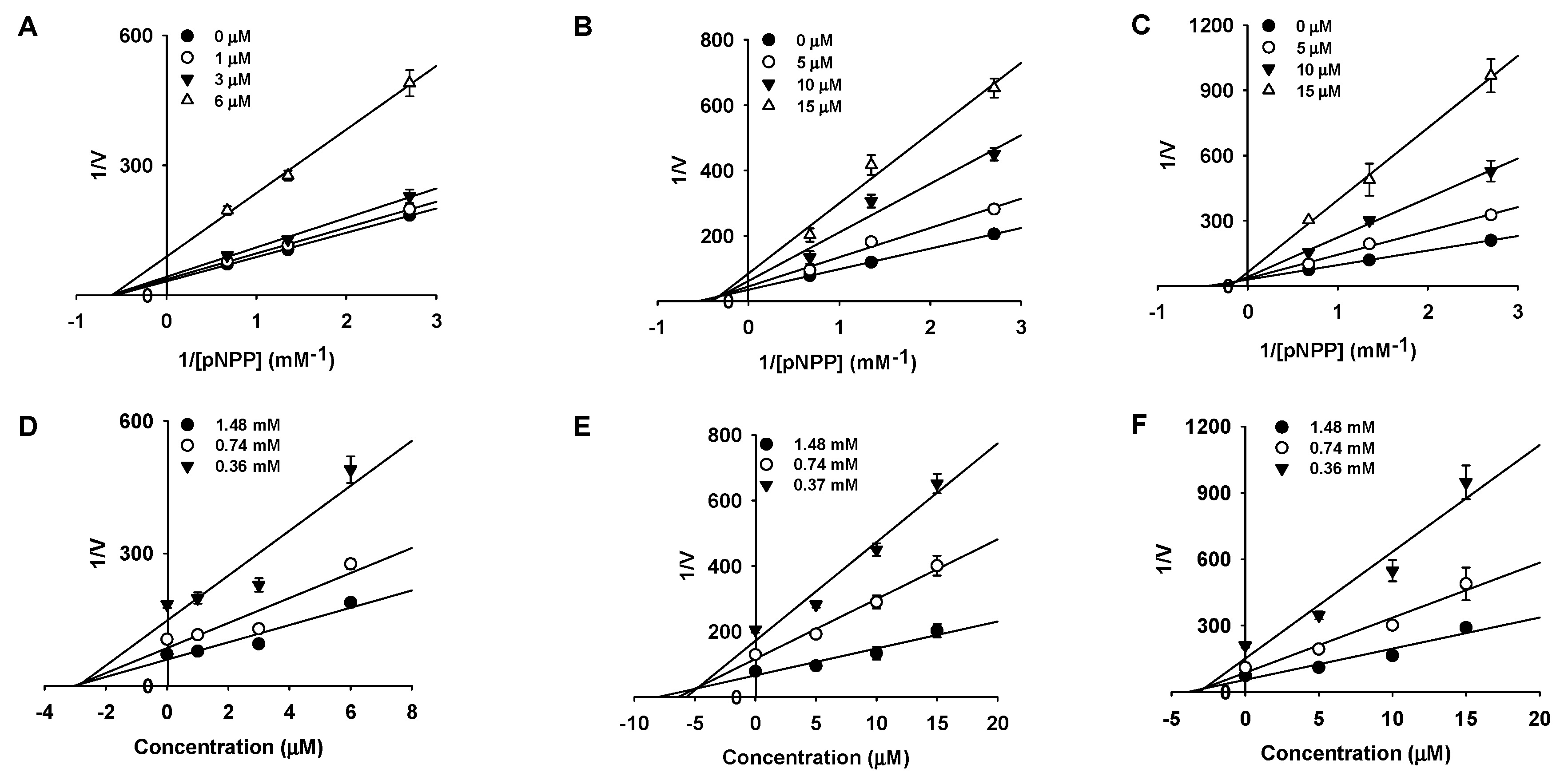
2.2. Kinetic Studies
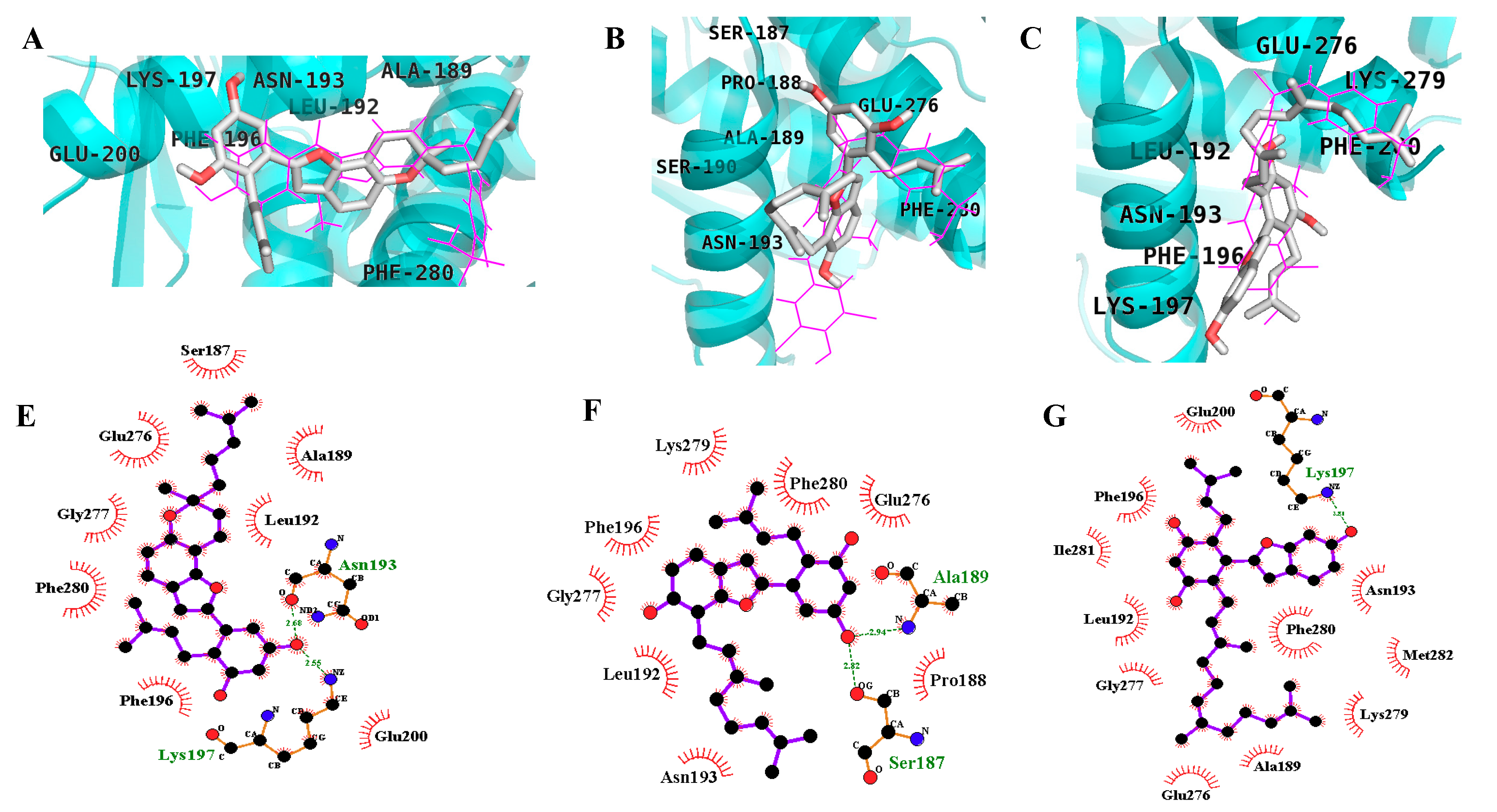
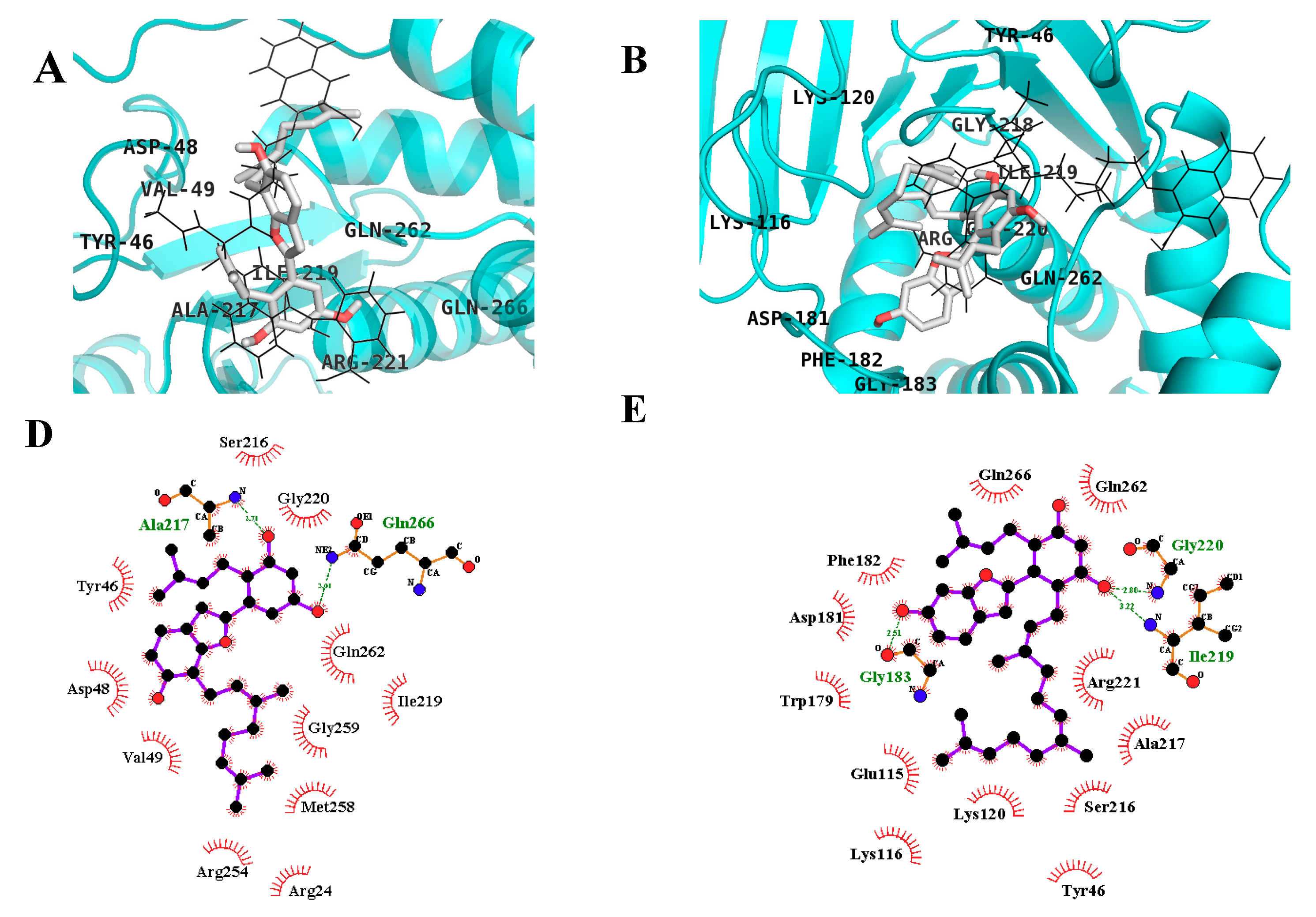
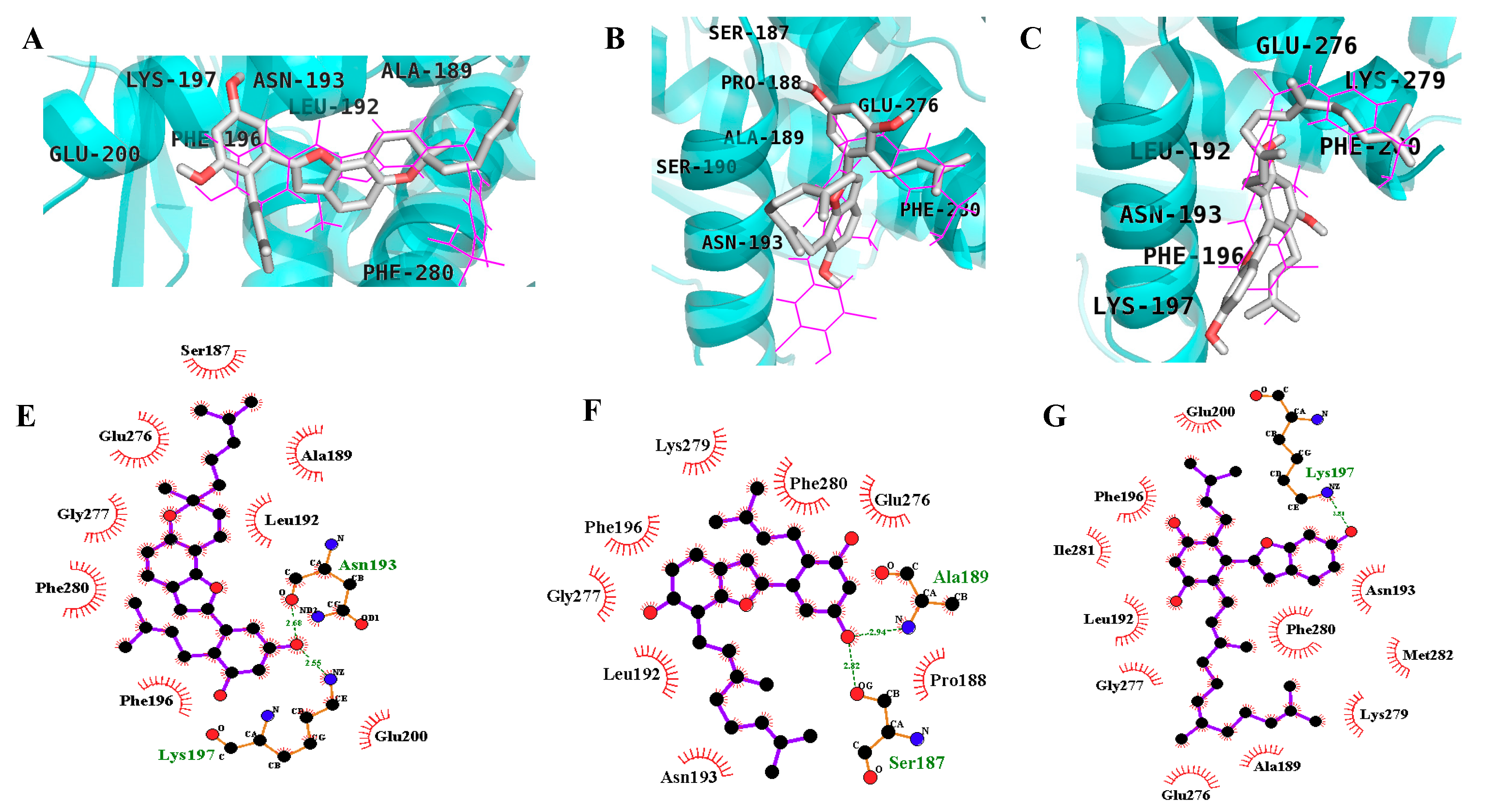
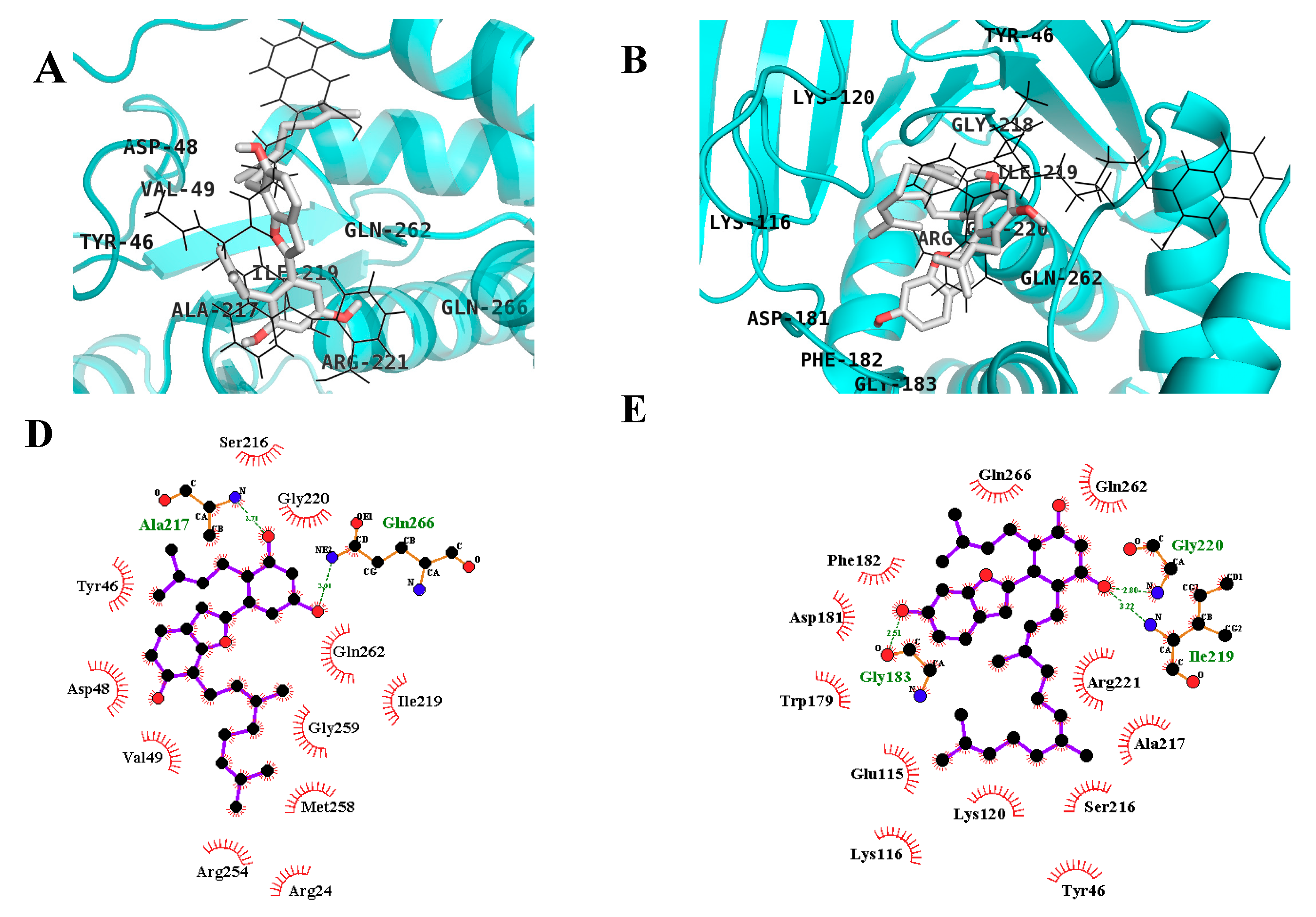
2.3. In Silico Docking Studies
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Plant Materials
4.3. Extraction and Isolation
4.4. PTP1B Inhibitory Assays
4.5. Enzyme Kinetic Analysis
4.6. In Silico Docking Analysis
4.7. Statistics
Author Contributions
Funding
Conflicts of Interest
References
- Johnson, T.O.; Ermolieff, J.; Jirousek, M.R. Protein tyrosine phosphatase 1B inhibitors for diabetes. Nat. Rev. Drug Discovery 2002, 1, 696. [Google Scholar] [CrossRef] [PubMed]
- Elchebly, M.; Payette, P.; Michaliszyn, E.; Cromlish, W.; Collins, S.; Loy, A.L.; Normandin, D.; Cheng, A.; Himms-Hagen, J.; Chan, C.C. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science 1999, 283, 1544–1548. [Google Scholar] [CrossRef] [PubMed]
- Klaman, L.D.; Boss, O.; Peroni, O.D.; Kim, J.K.; Martino, J.L.; Zabolotny, J.M.; Moghal, N.; Lubkin, M.; Kim, Y.B.; Sharpe, A.H. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol. Cell. Biol. 2000, 20, 5479–5489. [Google Scholar] [CrossRef] [PubMed]
- Shinde, R.N.; Kumar, G.S.; Eqbal, S.; Sobhia, M.E. Screening and identification of potential PTP1B allosteric inhibitors using in silico and in vitro approaches. PLoS ONE 2018, 13, e0199020. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.C.; Chen, W.; Liu, L.X.; Li, Y.; Yang, L.J.; Deng, X.Y.; Zhang, H.B.; Yang, X.D. Synthesis and cytotoxic activity of novel hybrid compounds between 2-alkylbenzofuran and imidazole. Med. Chem. Res. 2014, 23, 1599–1611. [Google Scholar] [CrossRef]
- Rao, G.K.; Venugopala, K.; Sanjay, P. Novel schiff bases of 4-hydroxy 6-carboxhydrazino benzofuran analogs: synthesis and pharmacological study. J. Pharmacol. Toxicol. 2007, 2, 481–488. [Google Scholar]
- Koca, M.; Servi, S.; Kirilmis, C.; Ahmedzade, M.; Kazaz, C.; Özbek, B.; Ötük, G. Synthesis and antimicrobial activity of some novel derivatives of benzofuran: part 1. Synthesis and antimicrobial activity of (benzofuran-2-yl)(3-phenyl-3-methylcyclobutyl) ketoxime derivatives. Eur. J. Med. Chem. 2005, 40, 1351–1358. [Google Scholar] [CrossRef]
- Demirayak, Ş.; Uçucu, Ü.; Benkli, K.; Gündoğdu-Karaburun, N.; Karaburun, A.Ç.; Akar, D.; Karabacak, M.; Kiraz, N. Synthesis and antifungal activities of some aryl (benzofuran-2-yl) ketoximes. Il Farmaco 2002, 57, 609–612. [Google Scholar] [CrossRef]
- Ragab, F.A.F.; El-Sayed, N.A.M.; Eissa, A.A.H.M.; El Kerdawy, A.M. Synthesis and anticonvulsant activity of certain substituted furochromone, benzofuran and flavone derivatives. Chem. Pharm. Bull. 2010, 58, 1148–1156. [Google Scholar] [CrossRef]
- Tan, Y.X.; Yang, Y.; Zhang, T.; Chen, R.Y.; Yu, D.Q. Bioactive 2-arylbenzofuran derivatives from Morus wittiorum. Fitoterapia 2010, 81, 742–746. [Google Scholar] [CrossRef]
- Kapche, D.W.; Lekane, N.M.; Kulabas, S.S.; Ipek, H.; Tok, T.T.; Ngadjui, B.T.; Demirtas, I.; Tumer, T.B. Aryl benzofuran derivatives from the stem bark of Calpocalyx dinklagei attenuate inflammation. Phytochemistry 2017, 141, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Byun, J.H.; Kim, H.; Kim, Y.; Mook-Jung, I.; Kim, D.J.; Lee, W.K.; Yoo, K.H. Aminostyrylbenzofuran derivatives as potent inhibitors for Aβ fibril formation. Bioorg. Med. Chem. Lett. 2008, 18, 5591–5593. [Google Scholar] [CrossRef] [PubMed]
- Yamatake, Y.; Shibata, M.; Nagai, M. Pharmacological studies on root bark of mulberry tree (Morus alba L.). Jpn. J. Pharmacol. 1976, 26, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Park, S.J.; Lee, E.-J.; Kim, B.K.; Huh, H.; Lee, B.J. Purification and characterization of Moran 20K from Morus alba. Arch. Pharmacal Res. 1999, 22, 9–12. [Google Scholar] [CrossRef]
- Noumi, E.; Dibakto, T. Medicinal plants used for peptic ulcer in the Bangangte region, western Cameroon. Fitoterapia 2000, 71, 406–412. [Google Scholar] [CrossRef]
- Paudel, P.; Yu, T.; Seong, S.; Kuk, E.; Jung, H.; Choi, J. Protein tyrosine phosphatase 1B inhibition and glucose uptake potentials of mulberrofuran G, albanol B, and kuwanon G from root bark of Morus alba L. in insulin-resistant HepG2 cells: An in vitro and in silico study. Int. J. Mol. Sci. 2018, 19, 1542. [Google Scholar] [CrossRef] [PubMed]
- Seong, S.H.; Ha, M.T.; Min, B.S.; Jung, H.A.; Choi, J.S. Moracin derivatives from Morus radix as dual BACE1 and cholinesterase inhibitors with antioxidant and anti-glycation capacities. Life Sci. 2018, 210, 20–28. [Google Scholar] [CrossRef]
- Paudel, P.; Seong, S.H.; Zhou, Y.; Ha, M.T.; Min, B.S.; Jung, H.A.; Choi, J.S. Arylbenzofurans from the root bark of Morus alba as triple inhibitors of cholinesterase, β-site amyloid precursor protein cleaving enzyme 1, and glycogen synthase kinase-3β: Relevance to alzheimer’s disease. ACS Omega 2019, 4, 6283–6294. [Google Scholar] [CrossRef]
- Wiesmann, C.; Barr, K.J.; Kung, J.; Zhu, J.; Erlanson, D.A.; Shen, W.; Fahr, B.J.; Zhong, M.; Taylor, L.; Randal, M. Allosteric inhibition of protein tyrosine phosphatase 1B. Nat. Struct. Mol. Biol. 2004, 11, 730. [Google Scholar] [CrossRef]
- Roglic, G. WHO Global report on diabetes: A summary. Int. J. Noncommun. Dis. 2016, 1, 3. [Google Scholar] [CrossRef]
- Taylor, S.D.; Hill, B. Recent advances in protein tyrosine phosphatase 1B inhibitors. Expert Opin. Investig. Drugs 2004, 13, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Fantus, I.G. Inhibition of the protein tyrosine phosphatase PTP1B: Potential therapy for obesity, insulin resistance and type-2 diabetes mellitus. Best Pract. Res. Clin. Endocrinol. Metab. 2007, 21, 621–640. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.J.; Jiang, B.; Wu, N.; Wang, S.Y.; Shi, D.Y. Natural and semisynthetic protein tyrosine phosphatase 1B (PTP1B) inhibitors as anti-diabetic agents. RSC Adv. 2015, 5, 48822–48834. [Google Scholar] [CrossRef]
- Zhang, Y.L.; Luo, J.G.; Wan, C.X.; Zhou, Z.B.; Kong, L.Y. Geranylated 2-arylbenzofurans from Morus alba var. tatarica and their α-glucosidase and protein tyrosine phosphatase 1B inhibitory activities. Fitoterapia 2014, 92, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Szczepankiewicz, B.G.; Liu, G.; Hajduk, P.J.; Abad-Zapatero, C.; Pei, Z.; Xin, Z.; Lubben, T.H.; Trevillyan, J.M.; Stashko, M.A.; Ballaron, S.J. Discovery of a potent, selective protein tyrosine phosphatase 1B inhibitor using a linked-fragment strategy. J. Am. Chem. Soc. 2003, 125, 4087–4096. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.Q.; Fukai, T.; Chang, W.J.; Yang, P.Q.; Wang, F.P.; Nomura, T. Phenolic constituents of the root bark of Chinese Morus australis. Nat. Med. 2001, 55, 143–146. [Google Scholar]
- Nomura, T.; Fukai, T.; Shimada, T.; Chen, I.S. Components of root bark of Morus australis. Planta Med. 1983, 49, 90–94. [Google Scholar] [CrossRef]
- Dat, N.T.; Jin, X.; Lee, K.; Hong, Y.S.; Kim, Y.H.; Lee, J.J. Hypoxia-inducible factor-1 inhibitory benzofurans and chalcone-derived diels− alder adducts from Morus species. J. Nat. Prod. 2008, 72, 39–43. [Google Scholar] [CrossRef]
- Ha, M.T.; Tran, M.H.; Ah, K.J.; Jo, K.-J.; Kim, J.; Kim, W.D.; Cheon, W.J.; Woo, M.H.; Ryu, S.H.; Min, B.S. Potential pancreatic lipase inhibitory activity of phenolic constituents from the root bark of Morus alba L. Bioorg. Med. Chem. Lett. 2016, 26, 2788–2794. [Google Scholar] [CrossRef]
- Jung, H.A.; Paudel, P.; Seong, S.H.; Min, B.S.; Choi, J.S. Structure-related protein tyrosine phosphatase 1B inhibition by naringenin derivatives. Bioorg. Med. Chem. Lett. 2017, 27, 2274–2280. [Google Scholar] [CrossRef]
- Lineweaver, H.; Burk, D. The determination of enzyme dissociation constants. J. Am. Chem. Soc. 1934, 56, 658–666. [Google Scholar] [CrossRef]
- Dixon, M. The determination of enzyme inhibitor constants. Biochem. J. 1953, 55, 170. [Google Scholar] [CrossRef] [PubMed]
- Cornish-Bowden, A. A simple graphical method for determining the inhibition constants of mixed, uncompetitive and non-competitive inhibitors. Biochem. J. 1974, 137, 143–144. [Google Scholar] [CrossRef] [PubMed]
- Goodsell, D.S.; Morris, G.M.; Olson, A.J. Automated docking of flexible ligands: applications of AutoDock. J. Mol. Recognit. 1996, 9, 1–5. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 (μM) a | Inhibition Type b | Ki Value (μM) c |
|---|---|---|---|
| SA | 31.85 ± 2.98 | ‒ | ‒ |
| MD2 | 3.11 ± 0.10 | Noncompetitive | 2.63 |
| MD | 11.61 ± 0.19 | Mixed | 4.79 |
| MB | 12.00 ± 0.75 | Mixed | 2.84 |
| MH | 53.47 ± 12.5 | ‒ | ‒ |
| Ursolic acid d | 7.47 ± 1.24 | ‒ | ‒ |
| Compound | Binding Energy (kcal/mol) | No. of H-Bonds | H-Bond Interacting Residues | Hydrophobic Interacting Residues |
|---|---|---|---|---|
| Compound C a | −10.18 | 11 | Ser216, Arg221, Ala217, Ile219, Gly220, Arg24, Arg254, Asp48 | Tyr46, Cys215, Lys120, Thr263, Cln266, Val149, Met258, Gln262, Asp29, Arg24, Ser28 |
| Compound A b | −10.98 | 2 | Asn193, Glu276 | Ala189, Leu192, Phe196, Gly277, Lys279, Phe280, Ile281, Met282 |
| MD2 | −9.51 | 2 | Lys197, Asn193 | Phe280, Phe196, Leu192, Ala189, Glu200, Ser187, Glu276, Gly277 |
| MD | −6.71 | 2 | Gln266, Ala217 | Asp48, Tyr46, Val49, Met258, Arg254, Gly259, Gln262, Arg24, Ile219, Gly220, Ser216 |
| −7.93 | 2 | Ala189, Ser187 | Asn193, Phe196, Gly277, Leu192, Lys279, Phe280, Glu276, Pro188 | |
| MB | −6.44 | 3 | Gly183, Gly220, Ile219 | Phe182, Gln266, Gln262, Asp181, Trp179, Glu115, Lys116, Lys120, Tyr46, Ser216, Ala217, Arg221 |
| −7.13 | 1 | Lys197 | Phe280, Asn193, Met282, Lys279, Ala189, Glu276, Gly277, Leu192, Ile281, Phe196, Glu200 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shrestha, S.; Seong, S.H.; Park, S.G.; Min, B.S.; Jung, H.A.; Choi, J.S. Insight into the PTP1B Inhibitory Activity of Arylbenzofurans: An In Vitro and In Silico Study. Molecules 2019, 24, 2893. https://doi.org/10.3390/molecules24162893
Shrestha S, Seong SH, Park SG, Min BS, Jung HA, Choi JS. Insight into the PTP1B Inhibitory Activity of Arylbenzofurans: An In Vitro and In Silico Study. Molecules. 2019; 24(16):2893. https://doi.org/10.3390/molecules24162893
Chicago/Turabian StyleShrestha, Srijan, Su Hui Seong, Seul Gi Park, Byung Sun Min, Hyun Ah Jung, and Jae Sue Choi. 2019. "Insight into the PTP1B Inhibitory Activity of Arylbenzofurans: An In Vitro and In Silico Study" Molecules 24, no. 16: 2893. https://doi.org/10.3390/molecules24162893
APA StyleShrestha, S., Seong, S. H., Park, S. G., Min, B. S., Jung, H. A., & Choi, J. S. (2019). Insight into the PTP1B Inhibitory Activity of Arylbenzofurans: An In Vitro and In Silico Study. Molecules, 24(16), 2893. https://doi.org/10.3390/molecules24162893