
Inhibition of CpLIP2 Lipase Hydrolytic Activity by Four Flavonols (Galangin, Kaempferol, Quercetin, Myricetin) Compared to Orlistat and Their Binding Mechanisms Studied by Quenching of Fluorescence

 , and
, and
Abstract
:1. Introduction
2. Results
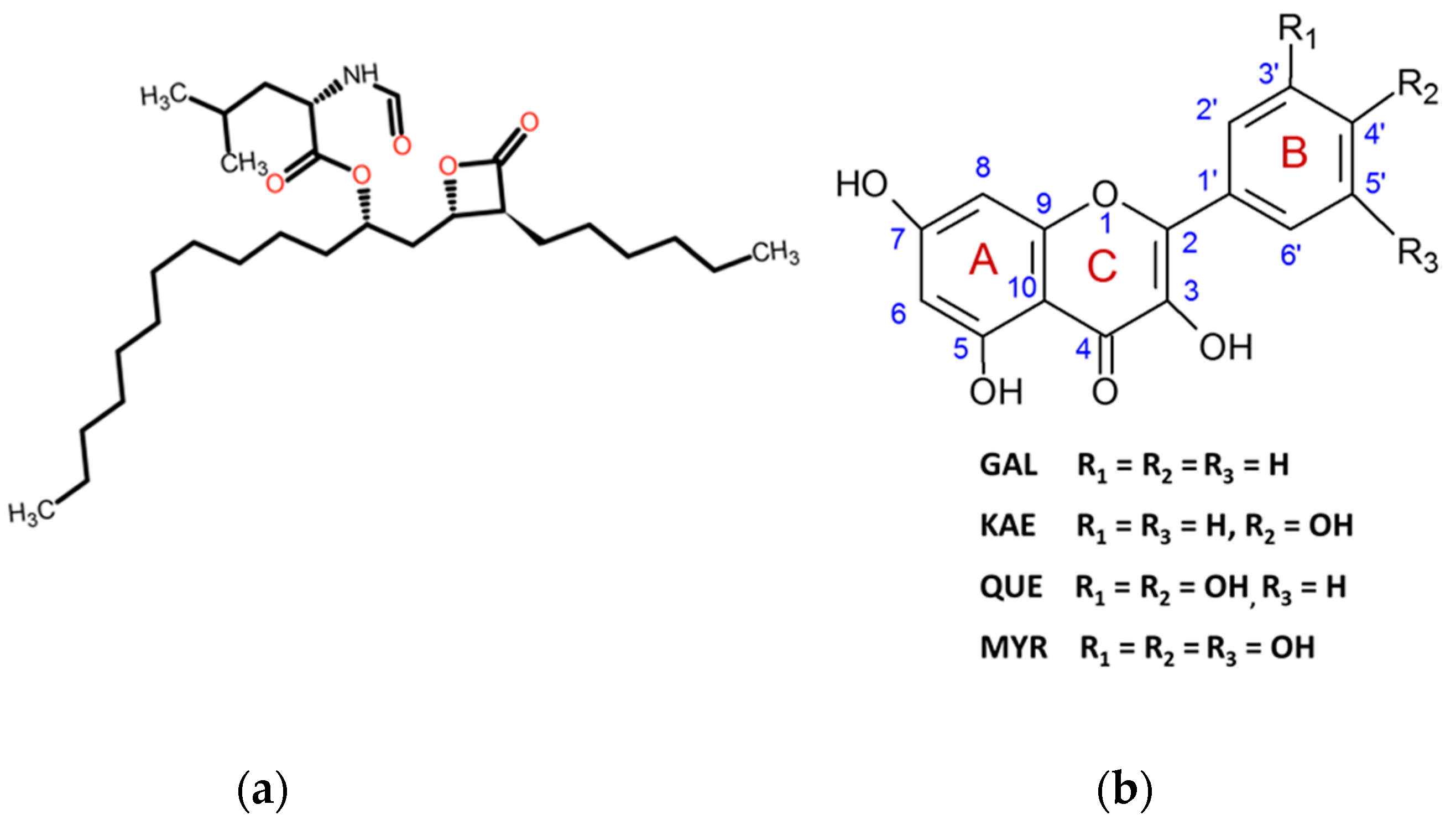
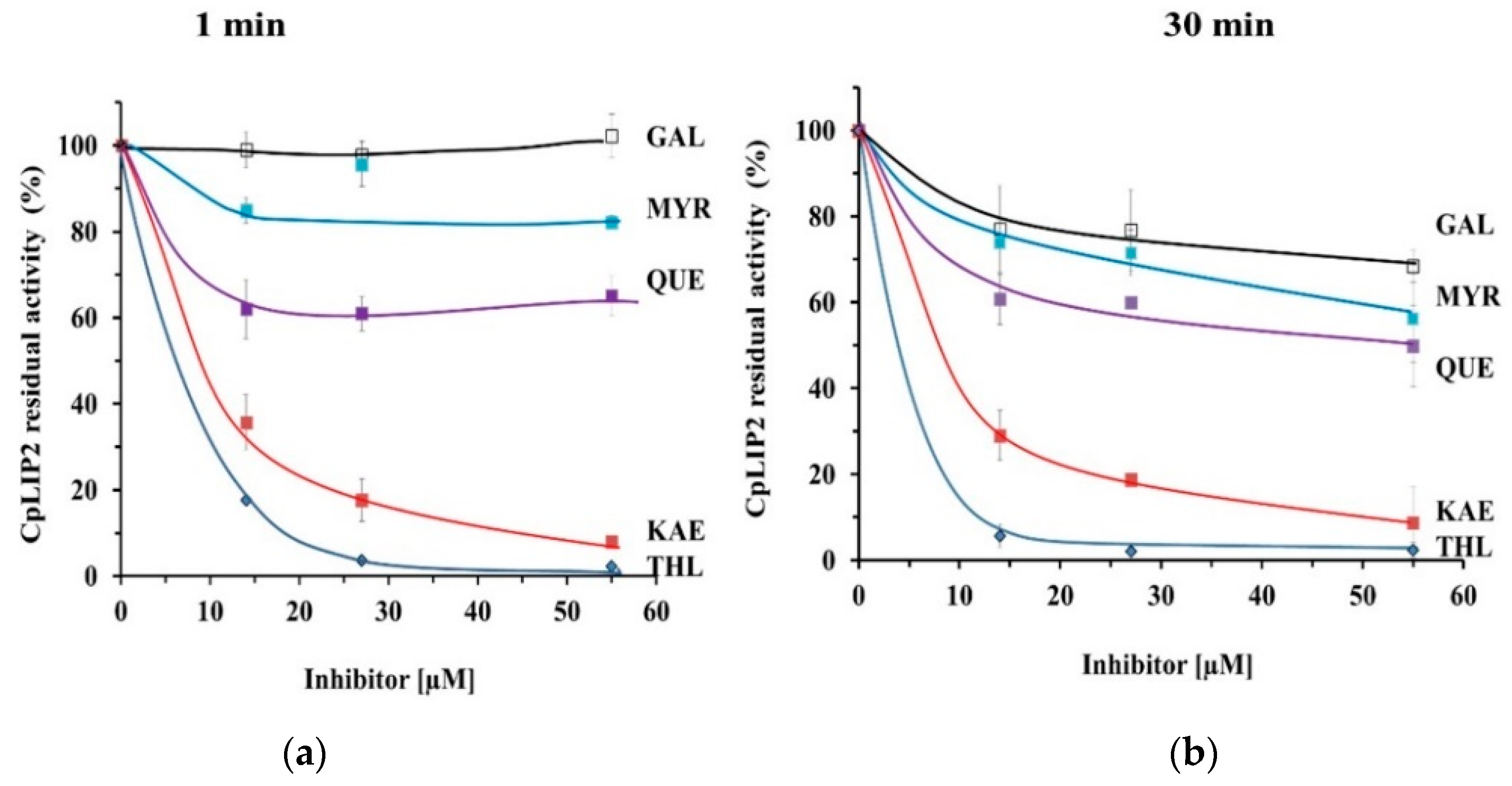
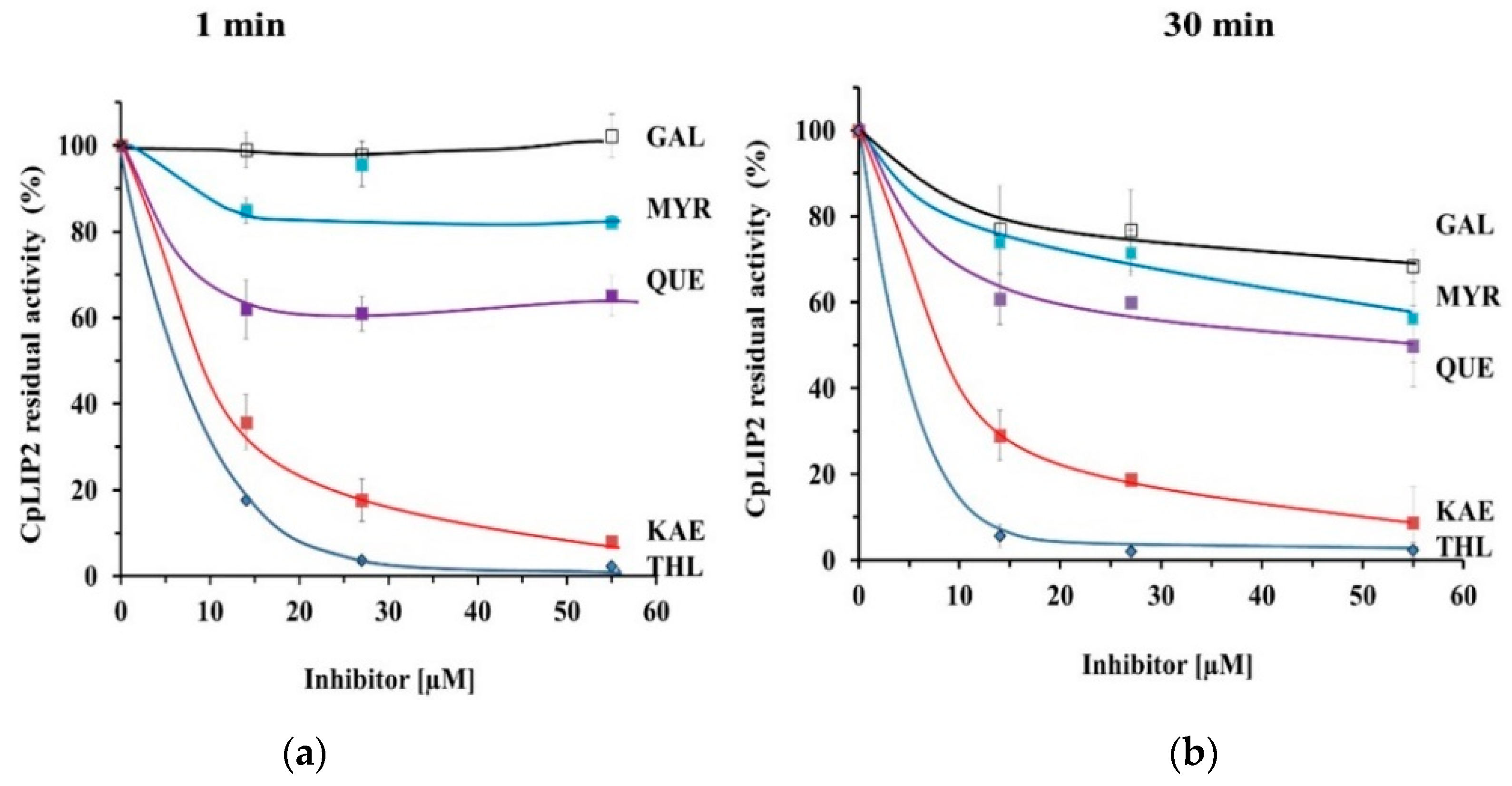
2.1. THL Inhibits Rapidly and Strongly the Hydrolytic Activity of CpLIP2
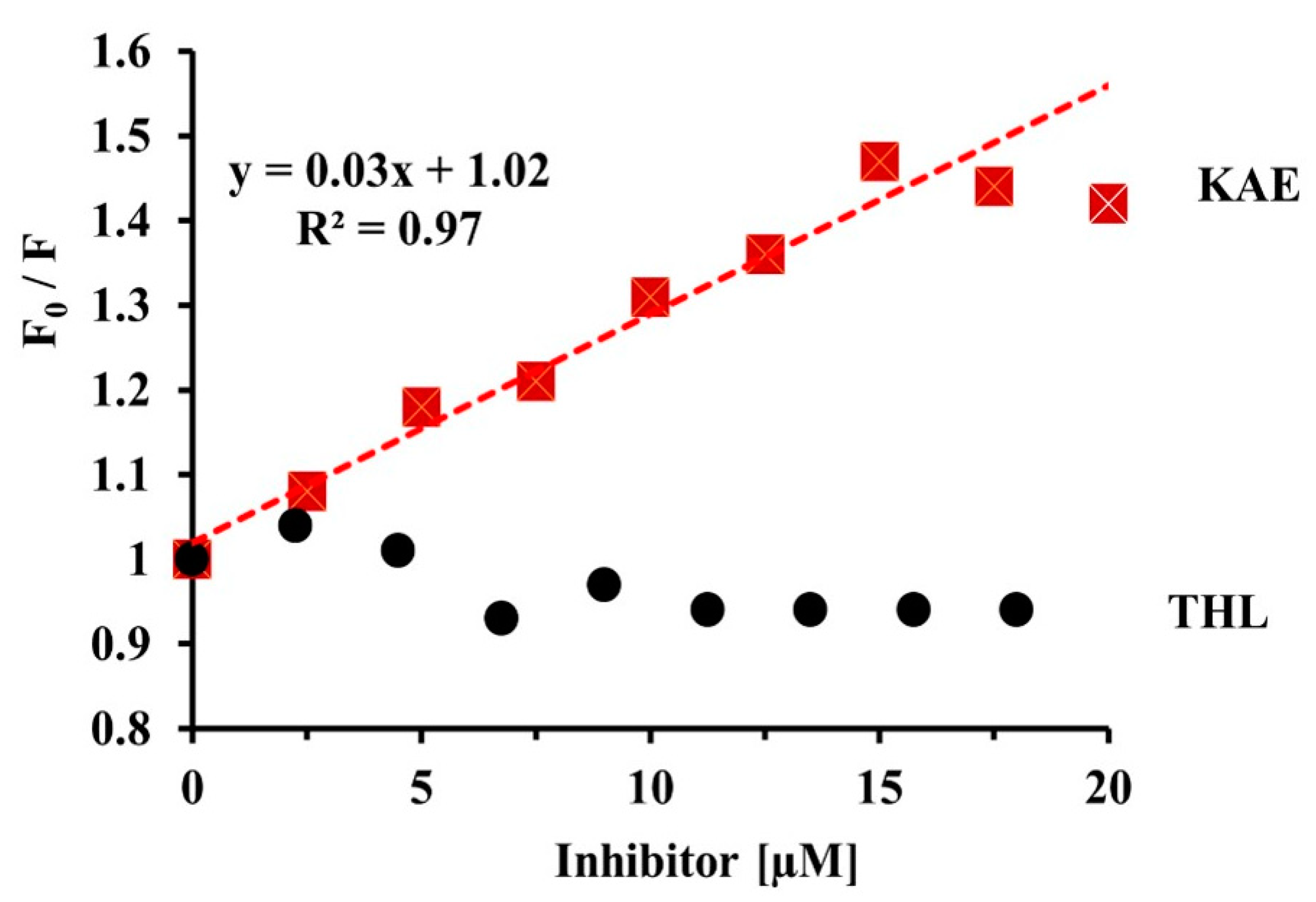
2.2. KAE Is the Strongest Inhibitor among the Flavonols Tested
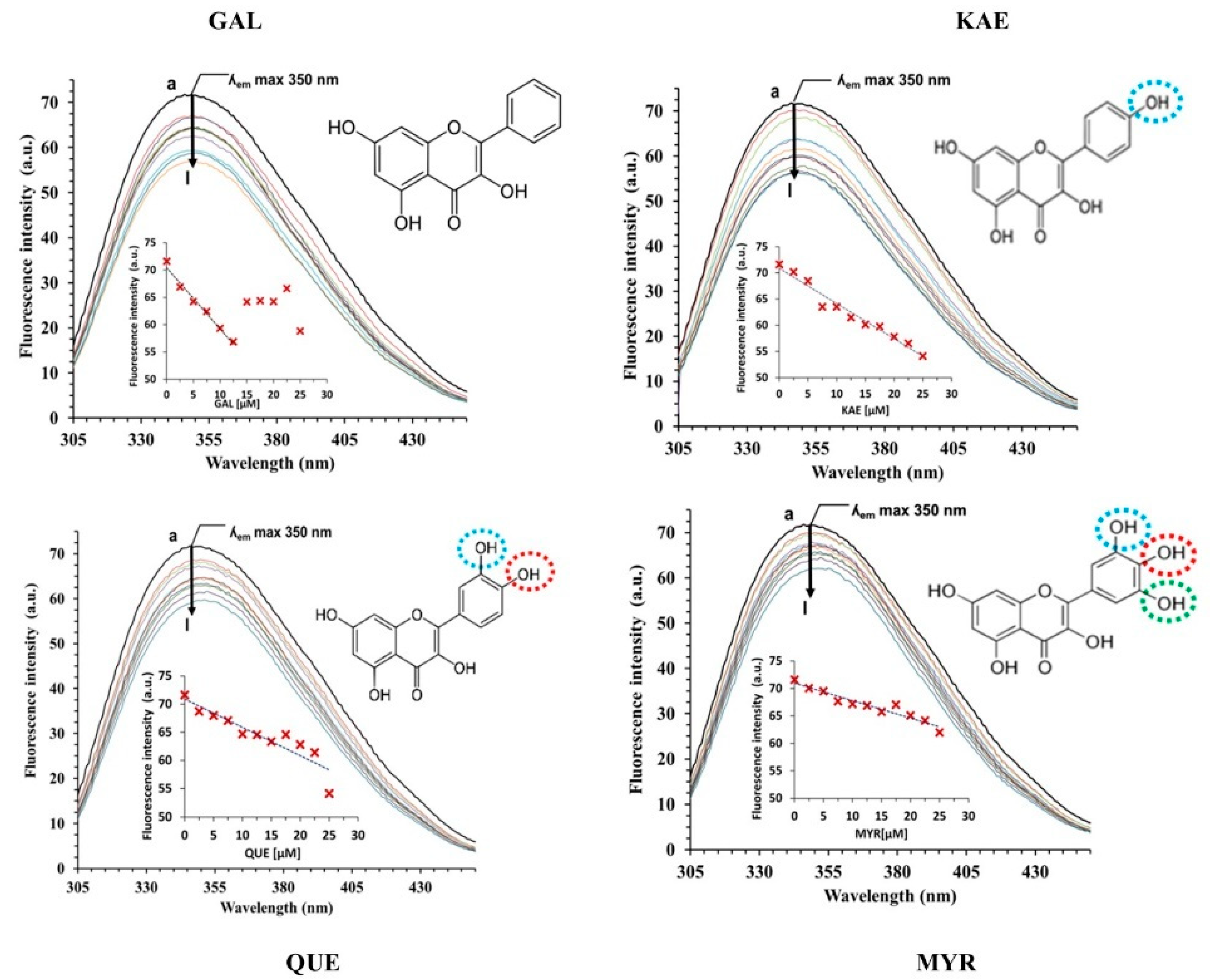
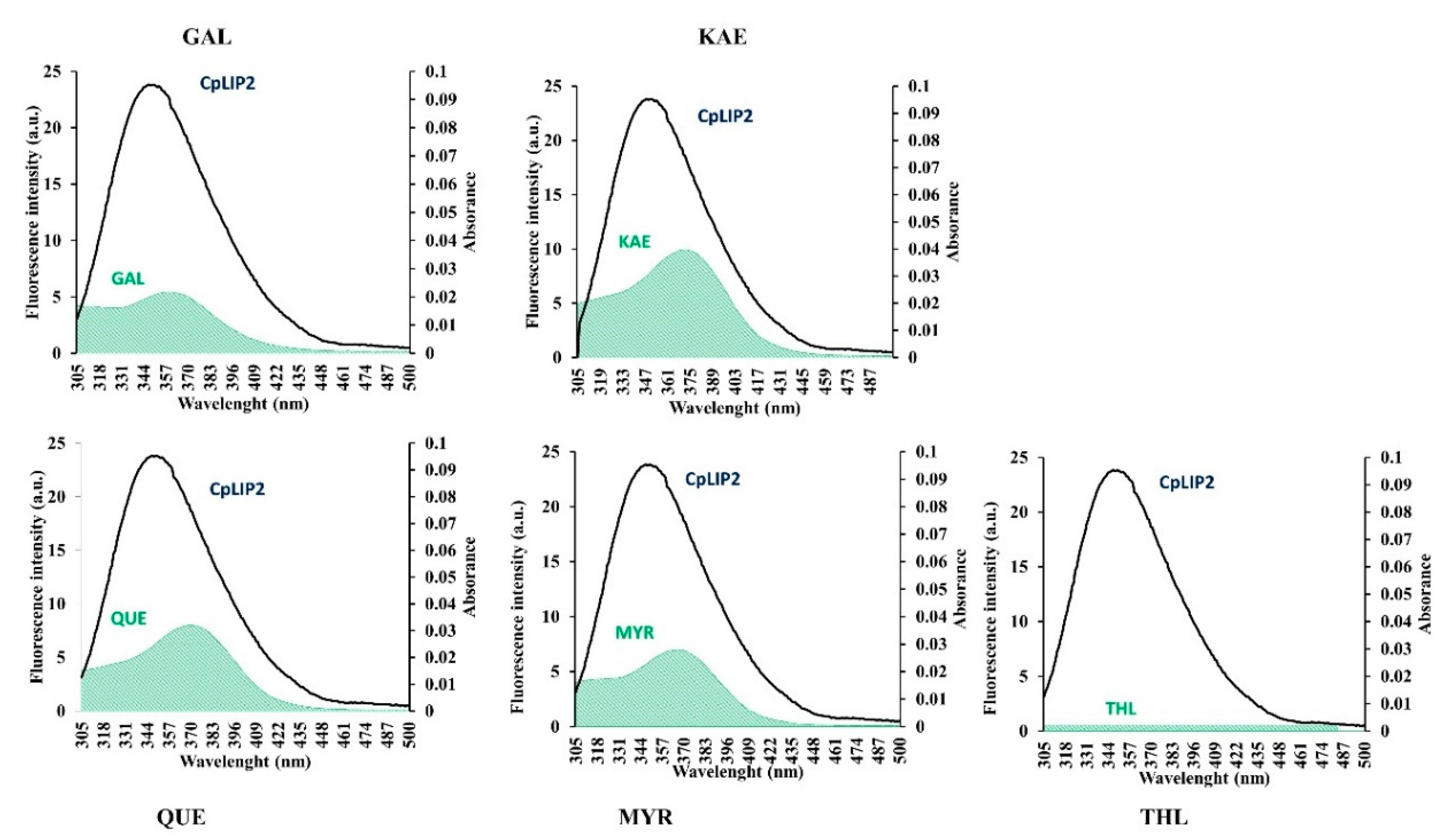
2.3. Interactions between CpLIP2 Lipase and 4 Flavonols by Means of Fluorescence Spectroscopy
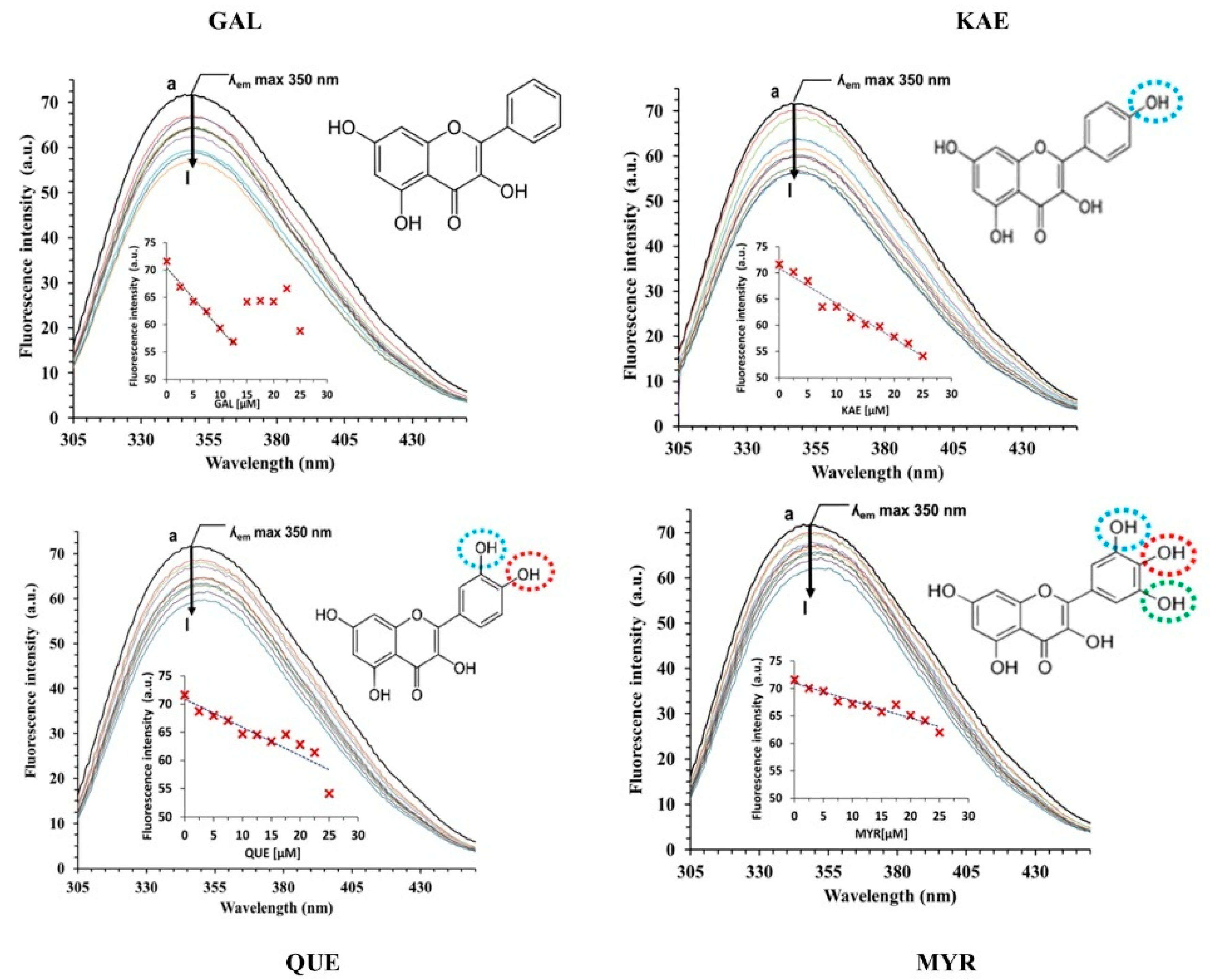
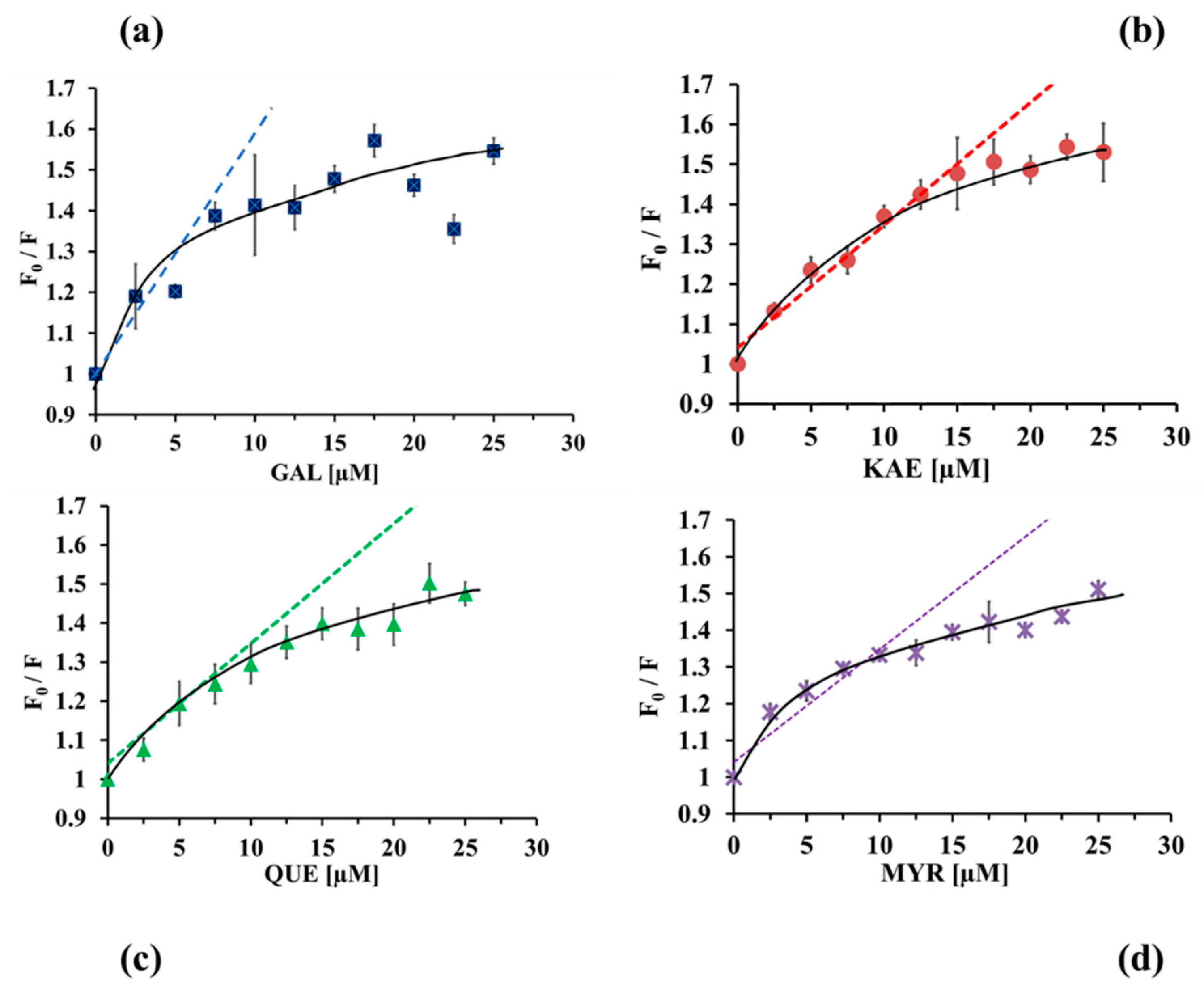
2.3.1. Analysis of CpLIP2 Fluorescence Quenching by Flavonols
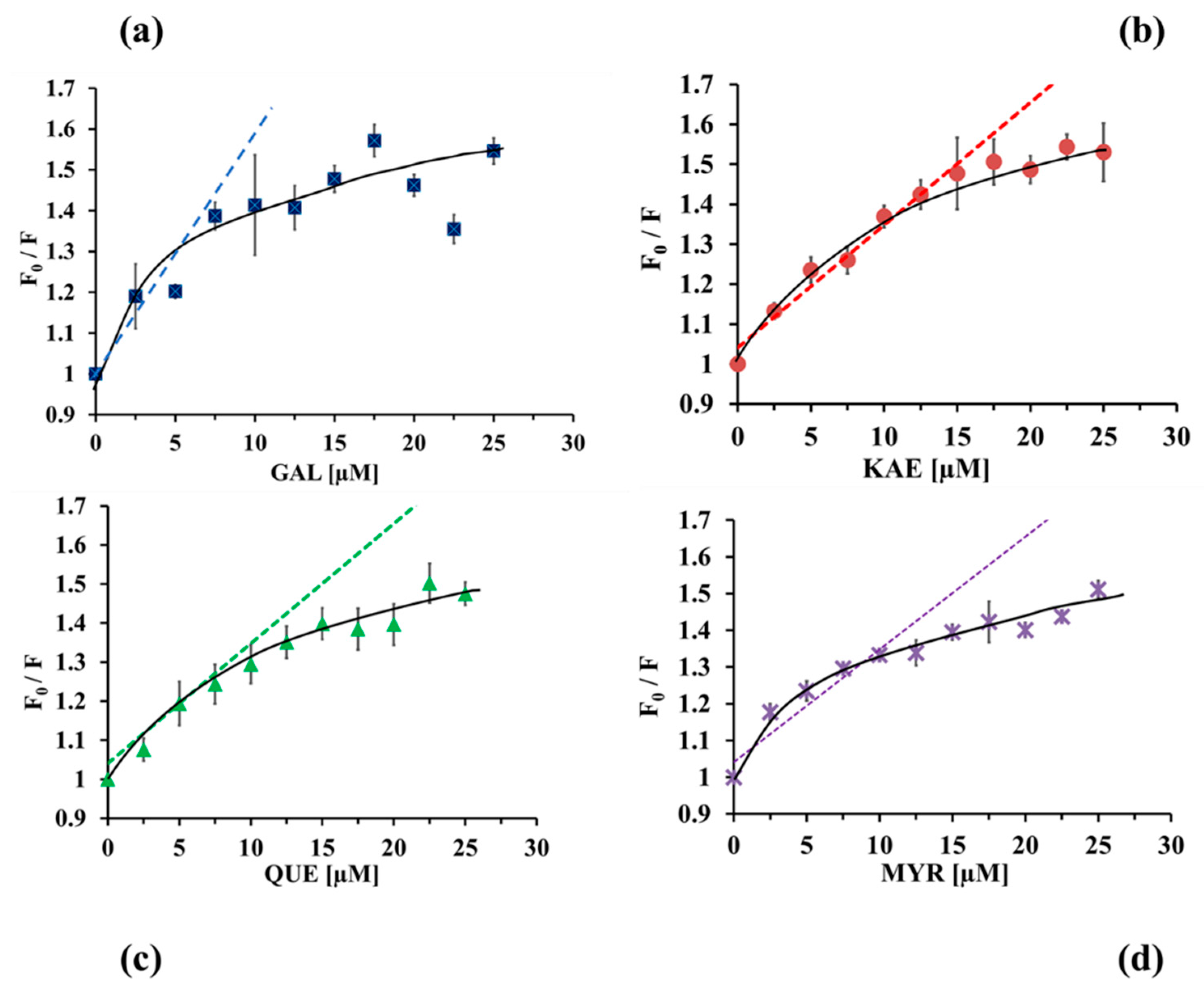
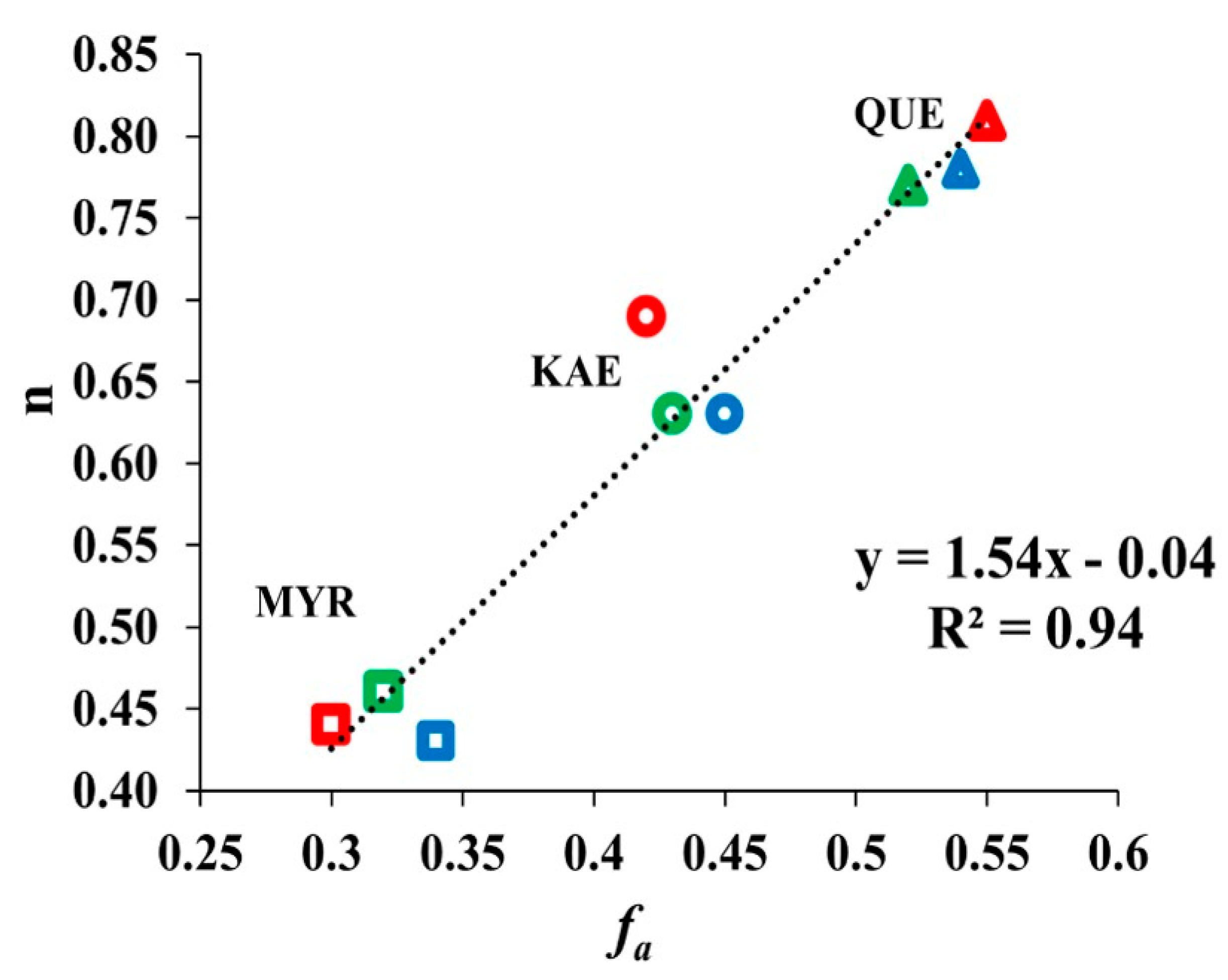
2.3.2. Binding Constants and Number of Binding Sites
2.3.3. Determination of Thermodynamic Parameters and the Nature of the Binding Forces
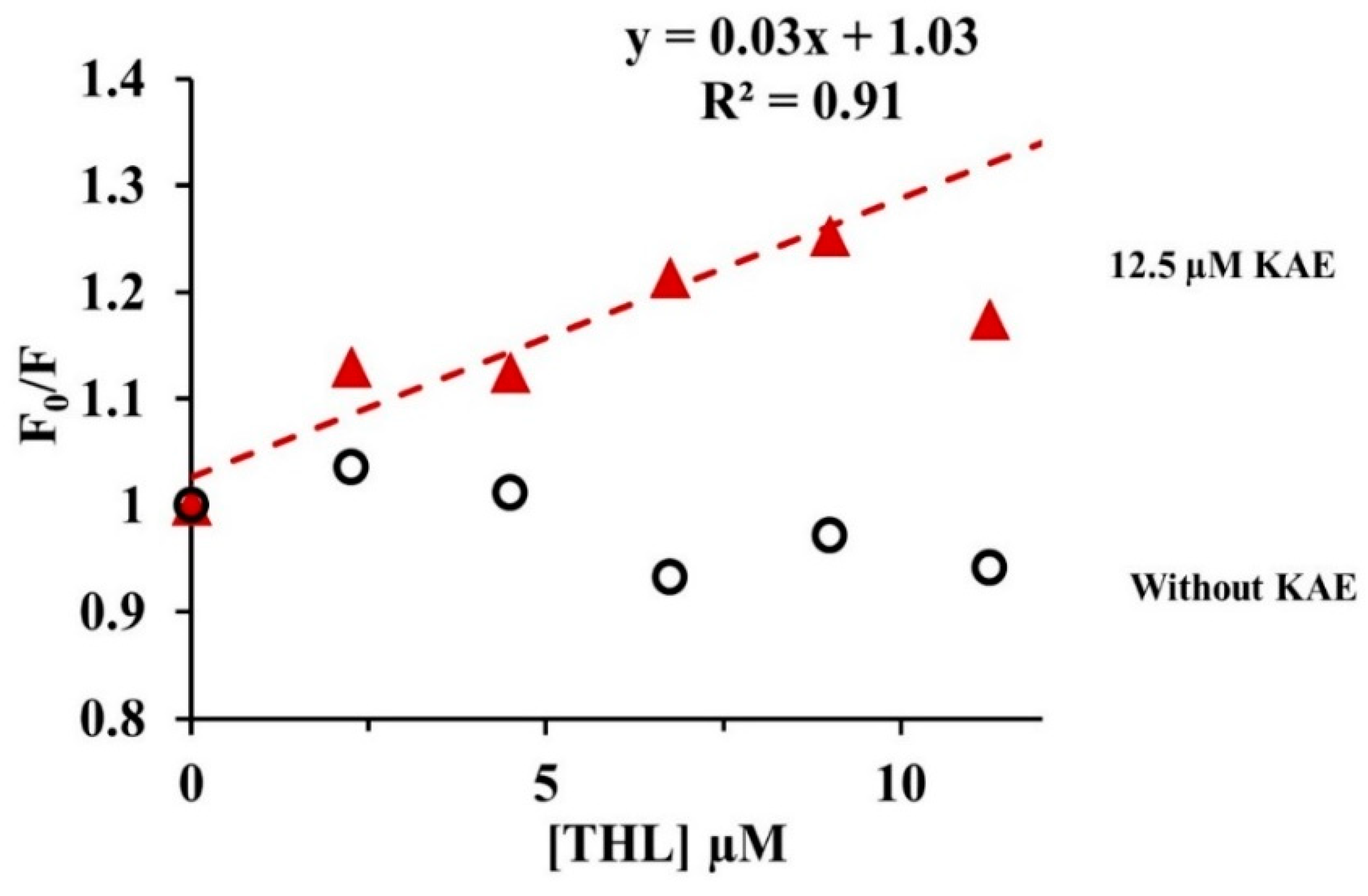
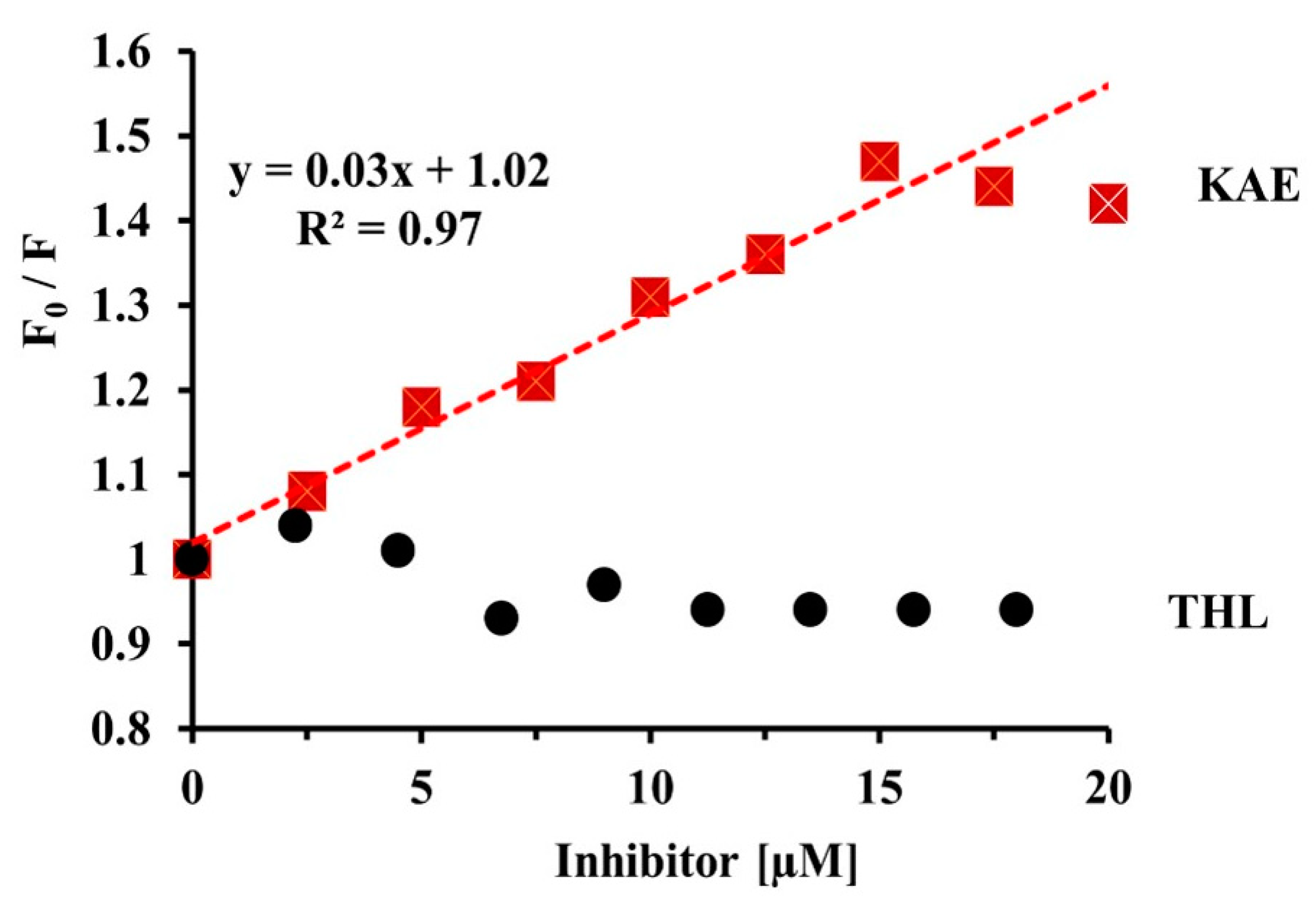
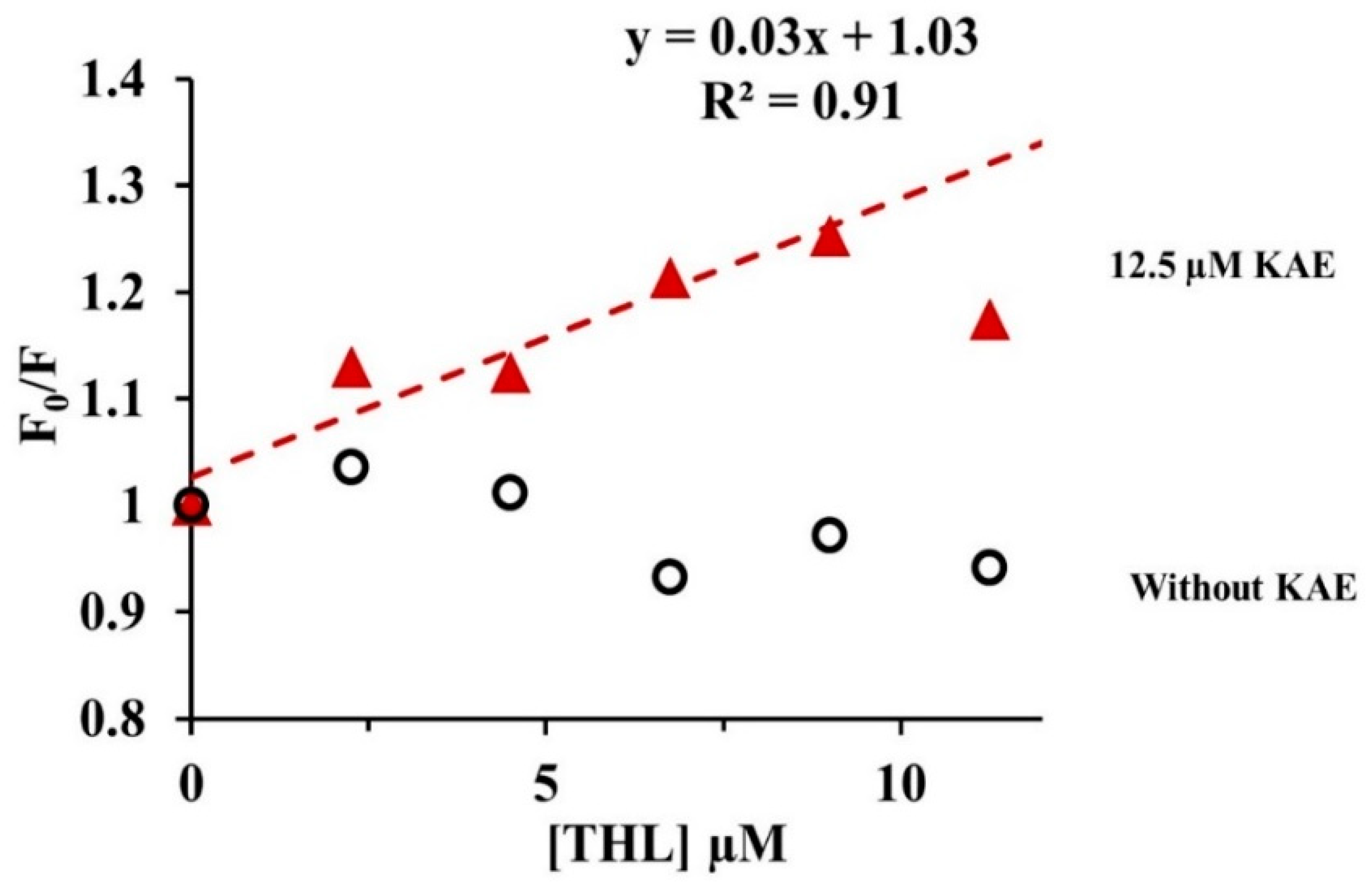
2.3.4. Quenching of CpLIP2 Lipase Fluorescence by KAE Mixed with THL
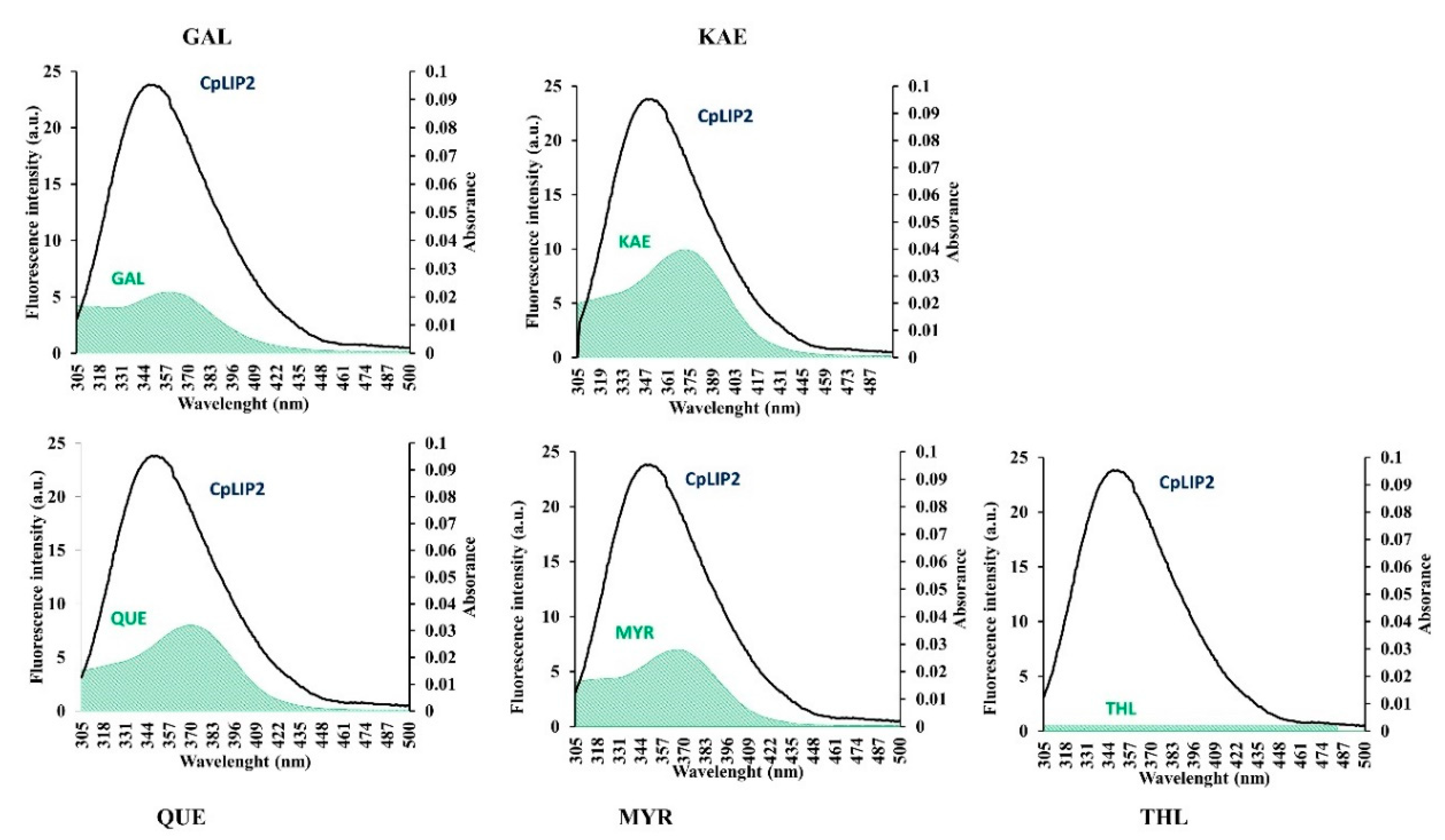
2.4. Energy Transfer between CpLIP2 and Flavonols
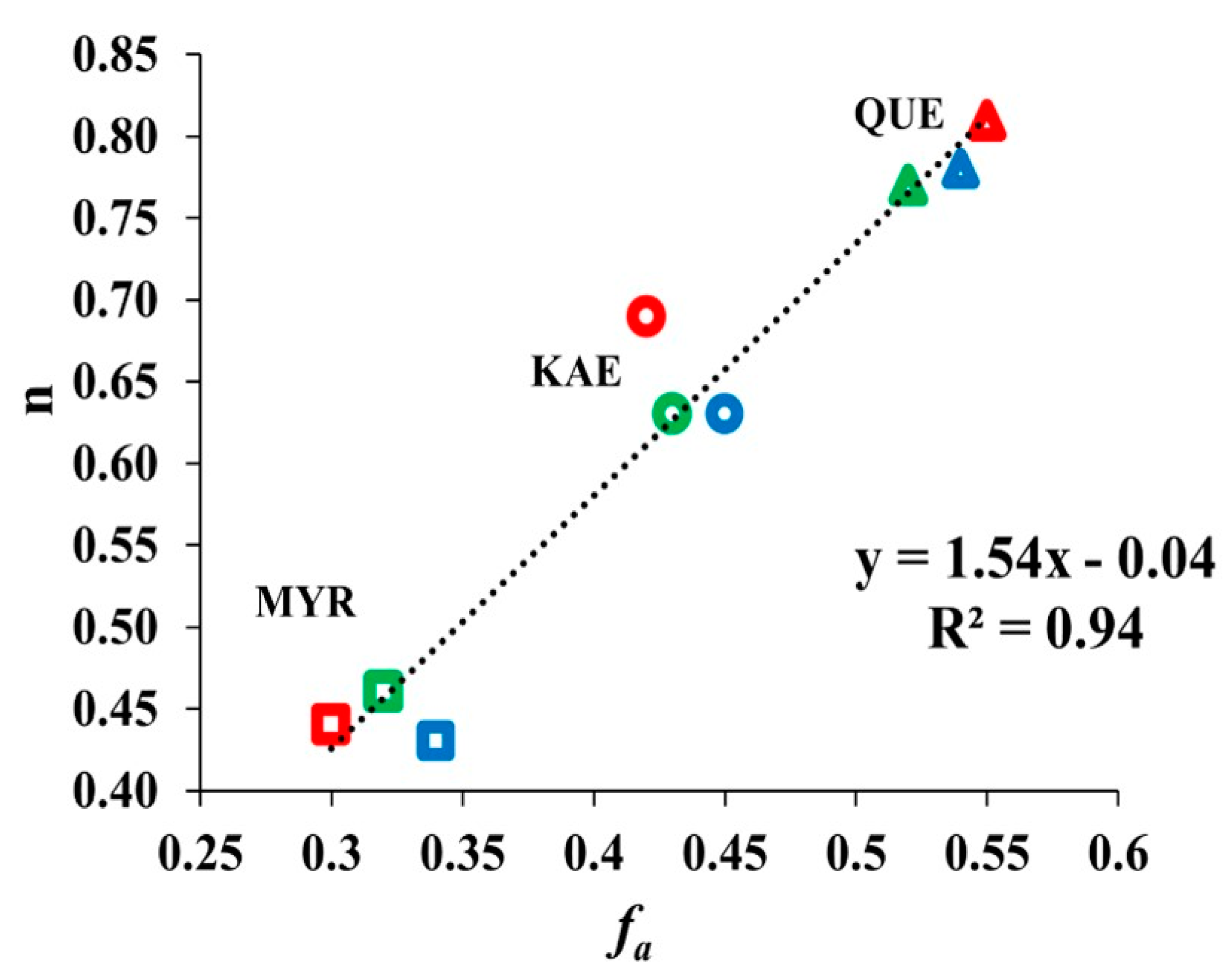
2.5. Relationship between Binding Sites Number and Factors of W Accessibility to Flavonols
2.6. Accessible Surface Area (ASA) to Solvent of the Seven W Residues of CpLIP2
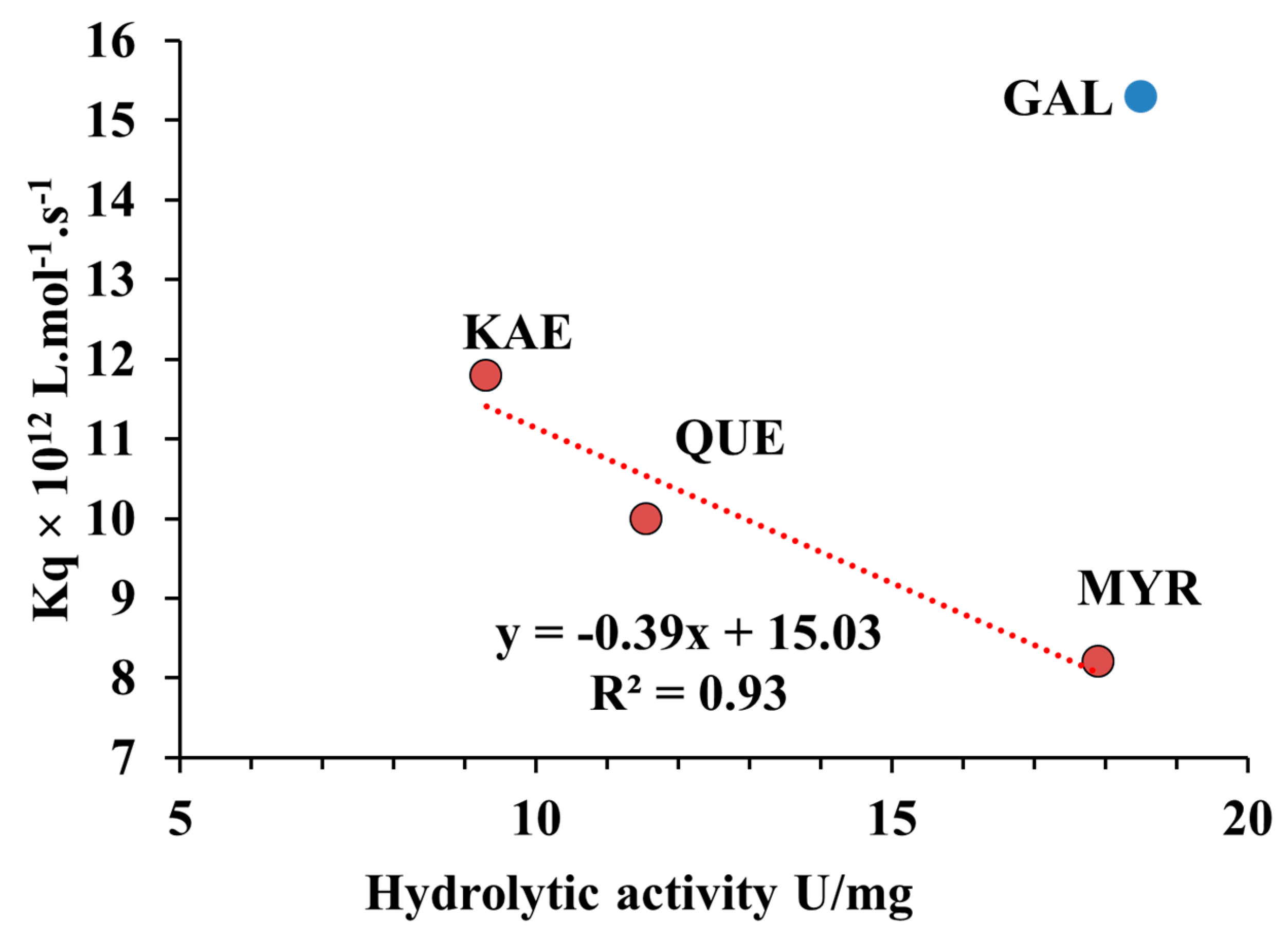
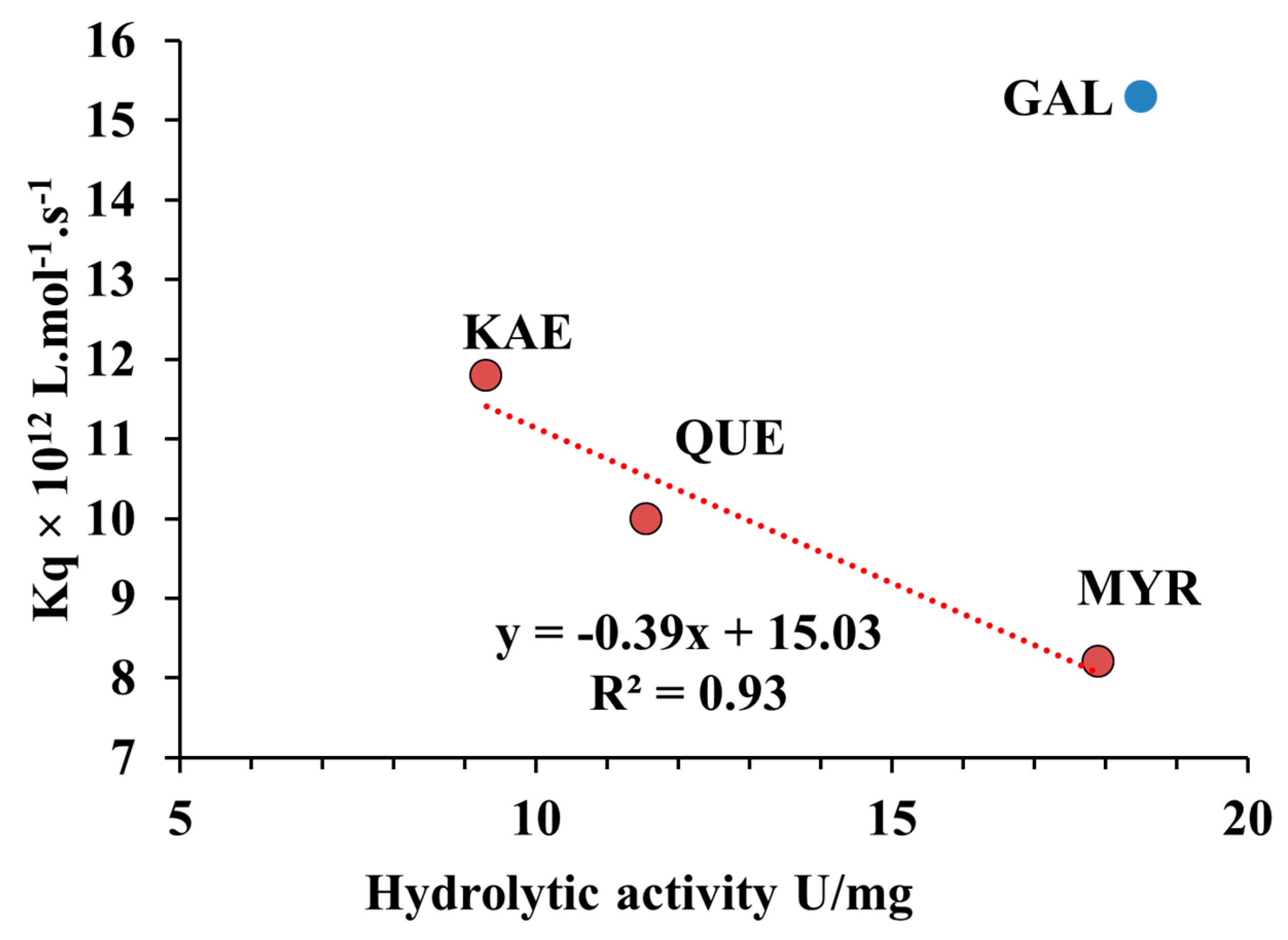
2.7. Relationship between Quenching Constant (Kq) and Hydrolytic Activity of CpLIP2 Lipase when Inhibited by Hydroxylated Flavonols on Their B-Rings
3. Discussion
3.1. THL and KAE Are Potent Inhibitors of CpLIP2 Lipase, with Different Complexation Mechanisms
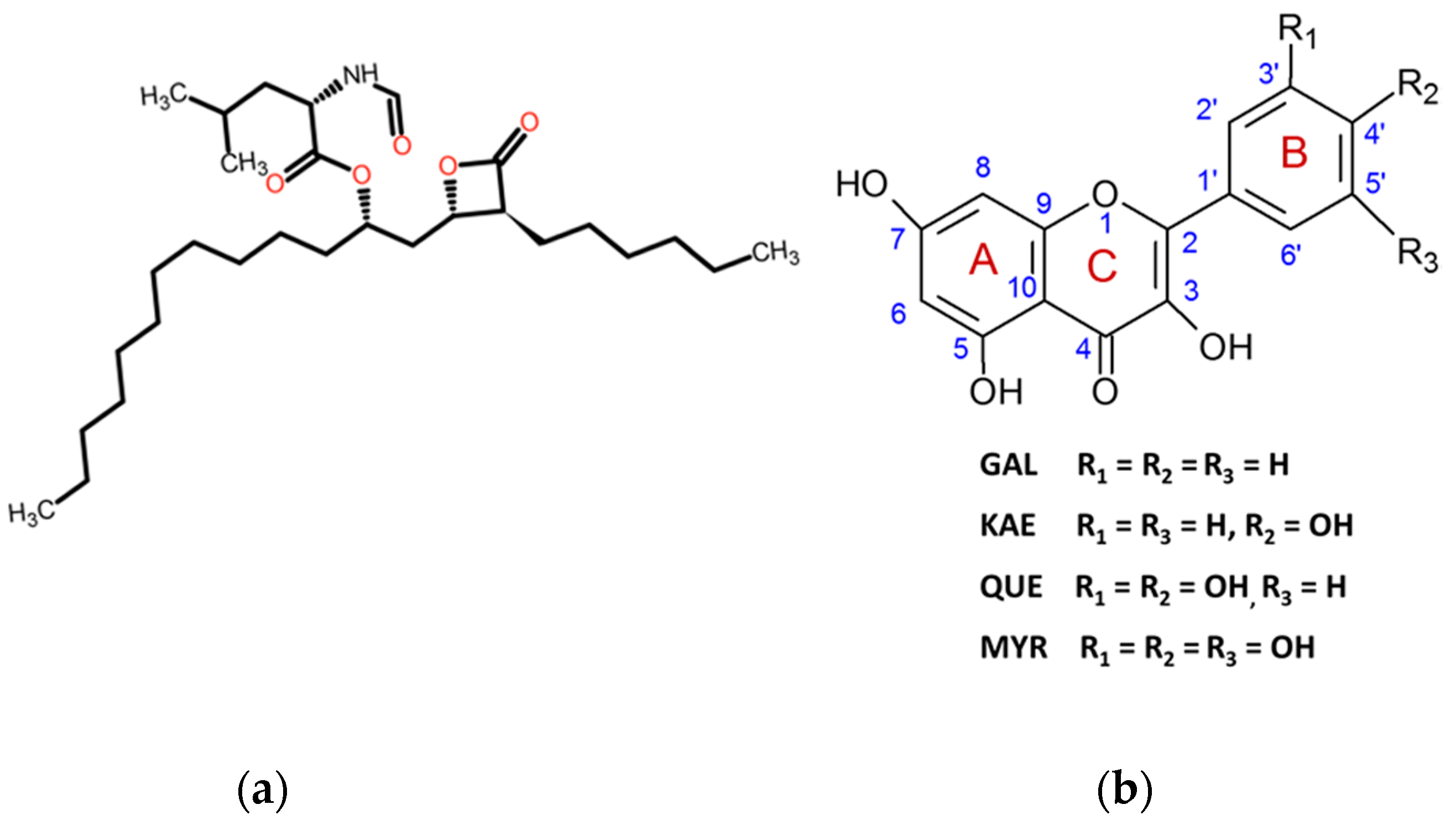
3.2. Hydroxylation Degree of Flavonols Is Essential in CpLIP2 Lipase Inhibitory Mechanisms
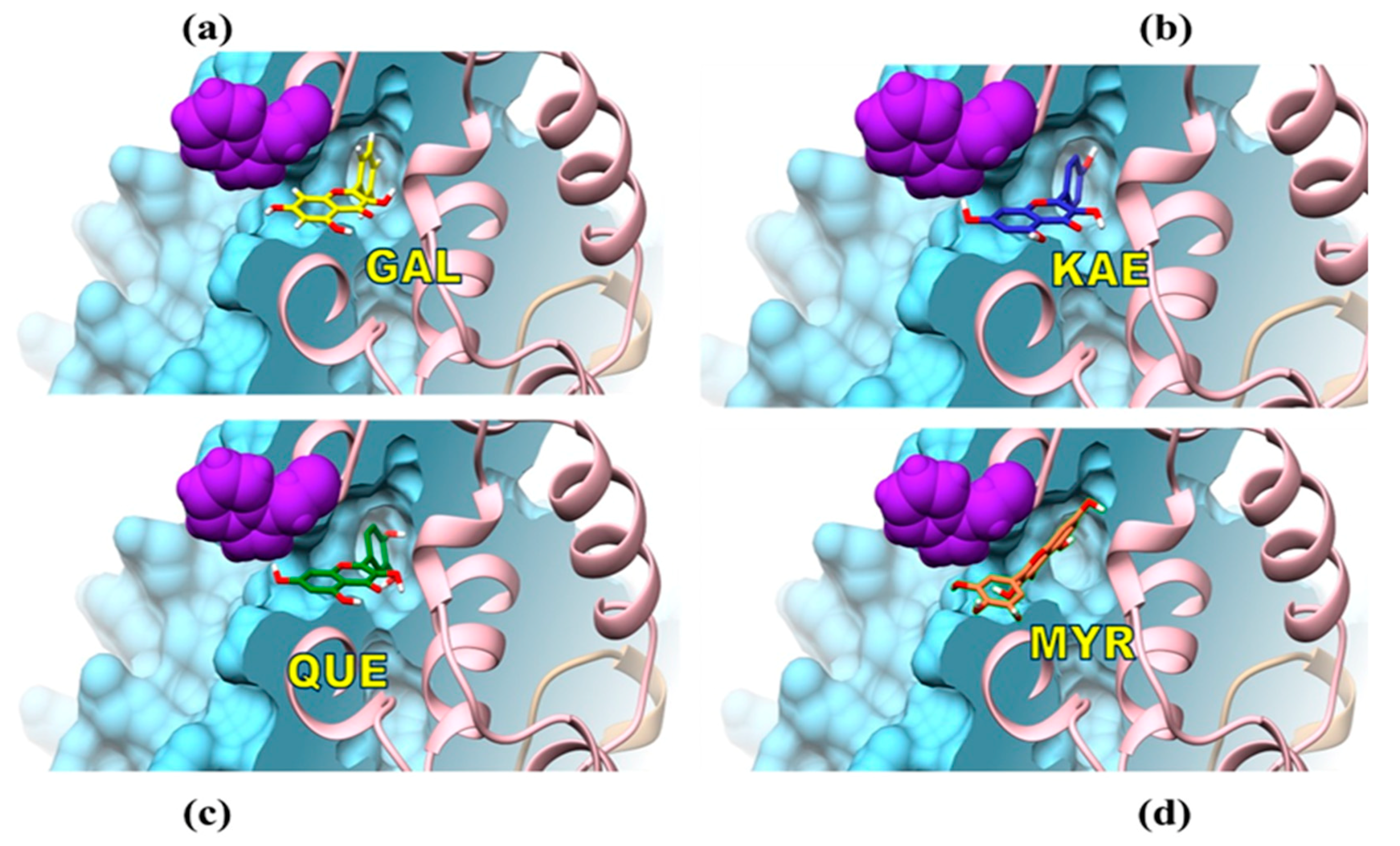
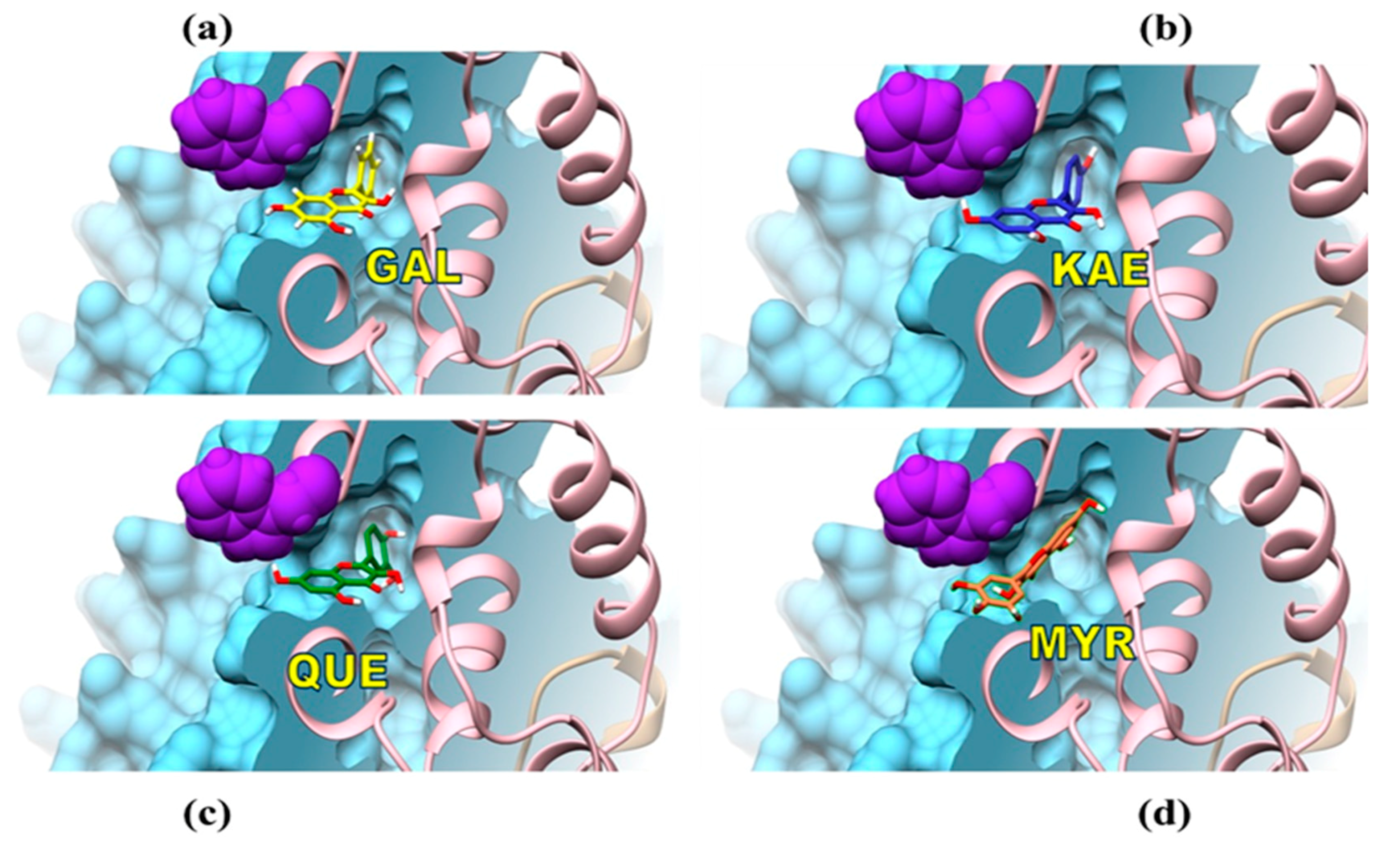
3.3. Mechanisms of Molecular Interactions between CpLIP2 Lipase and Flavonols Investigated by Means of Fluorescence and Docking
3.4. Relationship between Binding Sites Number and Factors of W Accessibility to Flavonols
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Production of Recombinant CpLIP2 Lipase
4.3. Enzyme Activity Assay
4.4. Lipase Inhibition Assay and the Determination of CpLIP2 Inhibitory Rate
4.5. Fluorescence Spectroscopy Measurement
4.5.1. Apparatus
4.5.2. Sample Preparation and Well Constituent
4.5.3. Determination of Excitation and Emission Wavelengths
4.5.4. Effect of Ionic Strength and Temperature on Fluorescence Intensity
4.6. Fluorescence Quenching
4.6.1. Flavonols–CpLIP2 Lipase Interaction
4.6.2. THL–CpLIP2 Lipase Interaction
4.6.3. Quenching of CpLIP2 Fluorescence by KAE in the Presence of THL
4.7. Molecular Docking Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Macheix, J.J.; Fleuriet, A.; Jay-Allemand, C. Les composés phénoliques des végétaux, Un exemple de métabolites secondaires d’importance économique; Presses Polytechniques et universitaires romandes: Lausanne, Switzerland, 2005; ISBN 2880746256. [Google Scholar]
- Bidel, L.P.R.; Meyer, S.; Goulas, Y.; Cadot, Y.; Cerovic, Z.G. Responses of epidermal phenolic compounds to light acclimation: In vivo qualitative and quantitative assessment using chlorophyll fluorescence excitation spectra in leaves of three woody species. J. Photochem. Photobiol. B. 2007, 88, 163–179. [Google Scholar] [CrossRef]
- Bidel, L.P.R.; Chomicki, G.; Bonini, F.; Mondolot, L.; Soulé, J.; Coumans, M.; La Fisca, P.; Baissac, Y.; Petit, V.; Loiseau, A.; et al. Dynamics of flavonol accumulation in leaf tissues under different UV-B regimes in Centella asiatica (Apiaceae). Planta 2015, 242, 545–559. [Google Scholar] [CrossRef] [Green Version]
- Bidel, L.P.R.; Coumans, M.; Baissac, Y.; Doumas, P.; Jay-Allemand, C. Biological Activity of Phenolics in Plant Cells. In Recent Advances in Polyphenol Research; Santos-Buelga, C., Escribano-Bailon, M.T., Lattenzio, V., Eds.; In Wiley-Blackwell publishing: Oxford, UK, 2010; Volume 2, pp. 163–205. ISBN 9781405193993. [Google Scholar]
- Jay-Allemand, C.; Tattini, M.; Gould, K.S. New evidences of the functional roles of secondary metabolites in plant-environment interactions. Environ. Exp. Bot. 2015, 119, 1–3. [Google Scholar] [CrossRef]
- Mierziak, J.; Kostyn, K.; Kulma, A. Flavonoids as Important Molecules of Plant Interactions with the Environment. Molecules 2014, 19, 16240–16265. [Google Scholar] [CrossRef]
- Mathesius, U. Flavonoid Functions in Plants and Their Interactions with Other Organisms. Plants 2018, 7, 30. [Google Scholar] [CrossRef]
- Harborne, J.B.; Williams, C.A. Advances in flavonoid research since 1992. Phytochemistry 2000, 55, 481–504. [Google Scholar] [CrossRef]
- Agrawal, A.D. Pharmacological Activities of Flavonoids: A Review. Int. J. Pharm. Sci. Nanotechnol. 2011, 4, 1394–1398. [Google Scholar]
- Friedman, M. Overview of antibacterial, antitoxin, antiviral, and antifungal activities of tea flavonoids and teas. Mol. Nutr. Food Res. 2007, 51, 116–134. [Google Scholar] [CrossRef]
- Felice, M.R.; Giuffre, L.; El Aamri, L.; Hafidi, M.; Criseo, G.; Romeo, O.; Scordino, F. Looking for new antifungal drugs from flavonoids: Impact of the genetic diversity of Candida albicans on the in-vitro response. Curr. Med. Chem. 2017, 25, 1–14. [Google Scholar] [CrossRef]
- Weidenbörner, M.; Jha, H.C. Antifugal activiy of flavonoids in reslation to degree of hydrolxylation, methoxylation and glycosidation. In Acta Horticulturae; International Society for Horticultural Science (ISHS): Leuven, Belgium, 1994; pp. 702–709. [Google Scholar]
- Seleem, D.; Pardi, V.; Murata, R.M. Review of flavonoids: A diverse group of natural compounds with anti-Candida albicans activity in vitro. Arch. Oral Biol. 2017, 76, 76–83. [Google Scholar] [CrossRef]
- De Oliveira, F.A.; de Oliveira, H.; Sousa, J.; Meireles, D.; de Azevedo, M.G.; Filho, J.; Lima, E.O. In vitro anti-Candida activity and mechanism of action of the flavonoid isolated from Praxelis clematidea against Candida albicans species. J. Appl. Pharm. Sci. 2016, 6, 066–069. [Google Scholar] [CrossRef] [Green Version]
- Salazar-Aranda, R.; Granados-Guzmán, G.; Pérez-Meseguer, J.; González, G.M.; De Torres, N.W. Activity of polyphenolic compounds against candida glabrata. Molecules 2015, 20, 17903–17912. [Google Scholar] [CrossRef] [PubMed]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Lättig, J.; Böhl, M.; Fischer, P.; Tischer, S.; Tietböhl, C.; Menschikowski, M.; Gutzeit, H.O.; Metz, P.; Pisabarro, M.T. Mechanism of inhibition of human secretory phospholipase A2 by flavonoids: Rationale for lead design. J. Comput. Aided. Mol. Des. 2007, 21, 473–483. [Google Scholar] [CrossRef]
- Cao, H.; Chen, X. Structures required of flavonoids for inhibiting digestive enzymes. Anticancer. Agents Med. Chem. 2012, 12, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Agullo, G.; Gamet-Payrastre, L.; Manenti, S.; Viala, C.; Rémésy, C.; Chap, H.; Payrastre, B. Relationship between flavonoid structure and inhibition of phosphatidylinositol 3-kinase: A comparison with tyrosine kinase and protein kinase C inhibition. Biochem. Pharmacol. 1997, 53, 1649–1657. [Google Scholar] [CrossRef]
- Shiomi, K.; Kuriyama, I.; Yoshida, H.; Mizushina, Y. Inhibitory effects of myricetin on mammalian DNA polymerase, topoisomerase and human cancer cell proliferation. Food Chem. 2013, 139, 910–918. [Google Scholar] [CrossRef]
- Ruiz, C.; Falcocchio, S.; Xoxi, E.; Villo, L.; Nicolosi, G.; Pastor, F.I.J.; Diaz, P.; Saso, L. Inhibition of Candida rugosa lipase by saponins, flavonoids and alkaloids. J. Mol. Catal. B Enzym. 2006, 40, 138–143. [Google Scholar] [CrossRef]
- Yordanov, M.; Dimitrova, P.; Patkar, S.; Saso, L.; Ivanovska, N. Inhibition of Candida albicans extracellular enzyme activity by selected natural substances and their application in Candida infection. Can. J. Microbiol. 2008, 54, 435–440. [Google Scholar] [CrossRef]
- Benarous, K.; Bombarda, I.; Iriepa, I.; Moraleda, I.; Gaetan, H.; Linani, A.; Tahri, D.; Sebaa, M.; Yousfi, M. Harmaline and hispidin from Peganum harmala and Inonotus hispidus with binding affinity to Candida rugosa lipase: In silico and in vitro studies. Bioorg. Chem. 2015, 62, 1–7. [Google Scholar] [CrossRef]
- Aouf, C.; Durand, E.; Lecomte, J.; Figueroa-Espinoza, M.-C.; Dubreucq, E.; Fulcrand, H.; Villeneuve, P. The use of lipases as biocatalysts for the epoxidation of fatty acids and phenolic compounds. Green Chem. 2014, 16, 1740–1754. [Google Scholar] [CrossRef]
- Gaillardin, C. Lipases as Pathogenicity Factors of Fungi BT. In Handbook of Hydrocarbon and Lipid Microbiology; Timmis, K.N., Ed.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2010; pp. 3259–3268. ISBN 978-3-540-77587-4. [Google Scholar]
- Brunel, L.; Neugnot, V.; Landucci, L.; Boze, H.; Moulin, G.; Bigey, F.; Dubreucq, E. High-level expression of Candida parapsilosis lipase/acyltransferase in Pichia pastoris. J. Biotechnol. 2004, 111, 41–50. [Google Scholar] [CrossRef]
- Neugnot, V.; Moulin, G.; Dubreucq, E.; Bigey, F. The lipase/acyltransferase from Candida parapsilosis: Molecular cloning and characterization of purified recombinant enzymes. Eur. J. Biochem. 2002, 269, 1734–1745. [Google Scholar] [CrossRef]
- Neang, P.M.; Subileau, M.; Perrier, V.; Dubreucq, E. Peculiar features of four enzymes of the CaLA superfamily in aqueous media: Differences in substrate specificities and abilities to catalyze alcoholysis. J. Mol. Catal. B Enzym. 2013, 94, 36–46. [Google Scholar] [CrossRef]
- Briand, D.; Dubreucq, E.; Galzy, P. Factors affecting the acyltransfer activity of the lipase fromCandida parapsilosis in aqueous media. J. Am. Oil Chem. Soc. 1995, 72, 1367–1373. [Google Scholar] [CrossRef]
- Toth, R.; Toth, A.; Vagvolgyi, C.; Gacser, A. Candida parapsilosis Secreted Lipase as an Important Virulence Factor. Curr. Protein Pept. Sci. 2017, 18, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Gácser, A.; Trofa, D.; Schäfer, W.; Nosanchuk, J.D. Targeted gene deletion in Candida parapsilosis demonstrates the role of secreted lipase in virulence. J. Clin. Invest. 2007, 117, 3049–3058. [Google Scholar] [CrossRef] [Green Version]
- Gacser, A. Adhesins in Candida parapsilosis: Understudied players in virulence. Virulence 2016, 7, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Carriere, F.; Renou, C.; Ransac, S.; Lopez, V.; De Caro, J.; Ferrato, F.; De Caro, A.; Fleury, A.; Sanwald-Ducray, P.; Lengsfeld, H.; et al. Inhibition of gastrointestinal lipolysis by Orlistat during digestion of test meals in healthy volunteers. Am. J. Physiol. Liver Physiol. 2001, 281, G16–G28. [Google Scholar] [CrossRef]
- Hadváry, P.; Sidler, W.; Meister, W.; Vetter, W.; Wolfer, H. The lipase inhibitor tetrahydrolipstatin binds covalently to the putative active site serine of pancreatic lipase. J. Biol. Chem. 1991, 266, 2021–2027. [Google Scholar]
- Lüthi-Peng, Q.; Märki, H.P.; Hadváry, P. Identification of the active-site serine in human pancreatic lipase by chemical modification with tetrahydrolipstatin. FEBS Lett. 1992, 299, 111–115. [Google Scholar] [CrossRef] [Green Version]
- Tiss, A.; Lengsfeld, H.; Carrière, F.; Verger, R. Inhibition of human pancreatic lipase by tetrahydrolipstatin: Further kinetic studies showing its reversibility. J. Mol. Catal. B Enzym. 2009, 58, 41–47. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer US: New York, NY, USA, 2006; ISBN1 978-0-387-31278-1. (Print); ISBN2 978-0-387-46312-4. (Online). [Google Scholar]
- Ghisaidoobe, A.; Chung, S. Intrinsic Tryptophan Fluorescence in the Detection and Analysis of Proteins: A Focus on Förster Resonance Energy Transfer Techniques. Int. J. Mol. Sci. 2014, 15, 22518–22538. [Google Scholar] [CrossRef]
- Goncalves, R.; Mateus, N.; de Freitas, V. Study of the interaction of pancreatic lipase with procyanidins by optical and enzymatic methods. J. Agric. Food Chem. 2010, 58, 11901–11906. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chen, X.; Xiao, J.; Zhao, J.; Jiao, F.; Jiang, X. Effect of hydrogenation on ring C of flavonols on their affinity for bovine serum albumin. J. Solution Chem. 2010, 39, 533–542. [Google Scholar] [CrossRef]
- Sun, L.; Gidley, M.J.; Warren, F.J. The mechanism of interactions between tea polyphenols and porcine pancreatic alpha-amylase: Analysis by inhibition kinetics, fluorescence quenching, differential scanning calorimetry and isothermal titration calorimetry. Mol. Nutr. Food Res. 2017, 61, 1700324. [Google Scholar] [CrossRef]
- Du, X.; Li, Y.; Xia, Y.L.; Ai, S.M.; Liang, J.; Sang, P.; Ji, X.L.; Liu, S.Q. Insights into Protein-Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.D.; Subramanian, S. Thermodynamics of protein association reactions: Forces contributing to stability. Biochemistry 1981, 20, 3096–3102. [Google Scholar] [CrossRef]
- Song, H.; Chen, C.; Zhao, S.; Ge, F.; Liu, D.; Shi, D.; Zhang, T. Interaction of gallic acid with trypsin analyzed by spectroscopy. J. Food Drug Anal. 2015, 23, 234–242. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Lin, G.; Xie, Y.; Ma, P.; Li, G.; Meng, Q.; Wu, T. Preformulation studies of myricetin: A natural antioxidant flavonoid. Pharmazie 2014, 69, 19–26. [Google Scholar] [CrossRef]
- Jovanovic, S.V.; Steenken, S.; Tosic, M.; Marjanovic, B.; Simic, M.G. Flavonoids as Antioxidants. J. Am. Chem. Soc. 1994, 116, 4846–4851. [Google Scholar] [CrossRef]
- Benarous, K.; Linani, A. Selection of orlistat as a potential inhibitor for lipase from Candida species. Bioinformation 2011, 7, 973–2063. [Google Scholar] [CrossRef]
- Kamoun, J.; Rahier, R.; Sellami, M.; Koubaa, I.; Mansuelle, P.; Lebrun, R.; Berlioz-Barbier, A.; Fiore, M.; Alvarez, K.; Abousalham, A.; et al. Identification of a new natural gastric lipase inhibitor from star anise. Food Funct. 2019, 10, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Kawabata, J. Importance of the B ring and its substitution on the alpha-glucosidase inhibitory activity of baicalein, 5,6,7-trihydroxyflavone. Biosci. Biotechnol. Biochem. 2004, 68, 1858–1864. [Google Scholar] [CrossRef] [PubMed]
- Moreira, M.R.; Kanashiro, A.; Kabeya, L.M.; Polizello, A.C.M.; Azzolini, A.E.C.S.; Curti, C.; Oliveira, C.A.; -do Amaral, A.T.; Lucisano-Valim, Y.M. Neutrophil effector functions triggered by Fc-gamma and/or complement receptors are dependent on B-ring hydroxylation pattern and physicochemical properties of flavonols. Life Sci. 2007, 81, 317–326. [Google Scholar] [CrossRef]
- Kah, M.; Brown, C.D. LogD: Lipophilicity for ionisable compounds. Chemosphere 2008, 72, 1401–1408. [Google Scholar] [CrossRef] [PubMed]
- Martin, H.J.; Kornmann, F.; Fuhrmann, G.F. The inhibitory effects of flavonoids and antiestrogens on the Glut1 glucose transporter in human erythrocytes. Chem. Biol. Interact. 2003, 146, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Matei, I.; Tablet, C.; Ionescu, S.; Hillebrand, M. Close-lying pKa values of kaempferol determined by second-derivative synchronous fluorescence. Rev. Roum. Chim. 2014, 59, 401–405. [Google Scholar]
- Shimura, S.; Itoh, Y.; Yamashita, A.; Kitano, A.; Hatano, T.; Yoshida, T.; Okuda, T. Inhibitory Effects of Flavonoidson Lipase. J. Japanese Soc. Food Sci. Technol. 1994, 41, 847–850. [Google Scholar] [CrossRef]
- Tadera, K.; Minami, Y.; Takamatsu, K.; Matsuoka, T. Inhibition of alpha-glucosidase and alpha-amylase by flavonoids. J. Nutr. Sci. Vitaminol. 2006, 52, 149–153. [Google Scholar] [CrossRef]
- Kumar, S.; Alagawadi, K.R. Anti-obesity effects of galangin, a pancreatic lipase inhibitor in cafeteria diet fed female rats. Pharm. Biol. 2013, 51, 607–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyama, M.; Rodrigues, S.D.; Toyama, D.O.; Soares, V.C.G.; Cotrim, C.; Ximenes, R.; Santos, M.L. Phospholipases A2 Protein Structure and Natural Products Interactions in Development of New Pharmaceuticals. In Protein Structure; IntechOpen: London, UK, 2012; ISBN 978-953-51-0555-8. [Google Scholar] [Green Version]
- Lindahl, M.; Tagesson, C. Flavonoids as phospholipase A2 inhibitors: Importance of their structure for selective inhibition of group II phospholipase A2. Inflammation 1997, 21, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, M.; Tagesson, C. Selective inhibition of group II phospholipase A2 by quercetin. Inflammation 1993, 17, 573–582. [Google Scholar] [CrossRef]
- Guerrero, L.; Castillo, J.; Quinones, M.; Garcia-vallve, S.; Arola, L.; Pujadas, G.; Muguerza, B. Inhibition of Angiotensin-Converting Enzyme Activity by Flavonoids: Structure-Activity Relationship Studies. PLoS ONE 2012, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Q.; Yang, P.; Gao, F.; Zhang, Z.W.; Wu, B. Probing the interaction between 3 flavonoids and pancreatic lipase by methods of fluorescence spectroscopy and enzymatic kinetics. Eur. Food Res. Technol. 2011, 233, 63–69. [Google Scholar] [CrossRef]
- Martinez-Gonzalez, A.I.; Alvarez-Parrilla, E.; Díaz-Sánchez, Á.G.; de la Rosa, L.A.; Núñez-Gastélum, J.A.; Vazquez-Flores, A.A.; Gonzalez-Aguilar, G.A. In vitro inhibition of pancreatic lipase by polyphenols: A kinetic, Fluorescence spectroscopy and molecular docking study. Food Technol. Biotechnol. 2017, 55, 519–530. [Google Scholar] [CrossRef]
- Daniel Boese, A.; Codorniu-Hernández, E. Cross-talk between amino acid residues and flavonoid derivatives: Insights into their chemical recognition. Phys. Chem. Chem. Phys. 2012, 14, 15682–15692. [Google Scholar] [CrossRef]
- Kessel, A.; Ben-Tal, N. Introduction to Proteins: Structure, Function, and Motion, 2nd ed.; Chapman & Hall/CRC Mathematical and Computational Biology; CRC Press: Boca Raton, FL, USA, 2018; ISBN 9781498747172. [Google Scholar]
- Zacharias, N.; Dougherty, D.A. Cation-π interactions in ligand recognition and catalysis. Trends Pharmacol. Sci. 2002, 23, 281–287. [Google Scholar] [CrossRef]
- Dougherty, D.A. The cation-π interaction. Acc. Chem. Res. 2013, 46, 885–893. [Google Scholar] [CrossRef]
- Rabbani, N.; Tabrez, S.; Islam, B.ul.; Rehman, M.T.; Alsenaidy, A.M.; AlAjmi, M.F.; Khan, R.A.; Alsenaidy, M.A.; Khan, M.S. Characterization of colchicine binding with normal and glycated albumin: In vitro and molecular docking analysis. J. Biomol. Struct. Dyn. 2018, 36, 3453–3462. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, Z.; Wang, N.; Han, X.; Yu, W.; Wang, R.; Chang, J. Identification of the binding between three fluoronucleoside analogues and fat mass and obesity-associated protein by isothermal titration calorimetry and spectroscopic techniques. J. Pharm. Biomed. Anal. 2018, 149, 290–295. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Subileau, M.; Jan, A.H.; Nozac’h, H.; Pérez-Gordo, M.; Perrier, V.; Dubreucq, E. The 3D model of the lipase/acyltransferase from Candida parapsilosis, a tool for the elucidation of structural determinants in CAL-A lipase superfamily. Biochim. Biophys. Acta 2015, 1854, 1400–1411. [Google Scholar] [CrossRef]
- Vaysse, L.; Ly, A.; Moulin, G.; Dubreucq, E. Chain-length selectivity of various lipases during hydrolysis, esterification and alcoholysis in biphasic aqueous medium. Enzyme Microb. Technol. 2002, 31, 648–655. [Google Scholar] [CrossRef]
- Gargouri, Y.; Chahinian, H.; Moreau, H.; Ransac, S.; Verger, R. Inactivation of pancreatic and gastric lipases by THL and C12 ’-TNB: A kinetic study with emulsified tributyrin. Biochim. Biophys. Acta 1991, 708, 322–328. [Google Scholar] [CrossRef]
- Wang, S.; Sun, Z.; Dong, S.; Liu, Y.; Liu, Y. Molecular interactions between (-)-epigallocatechin gallate analogs and pancreatic lipase. PLoS ONE 2014, 9, e111143. [Google Scholar] [CrossRef] [PubMed]
- Calabrone, L.; Larocca, M.; Marzocco, S.; Martelli, G. Total Phenols and Flavonoids Content, Antioxidant Capacity and Lipase Inhibition of Root and Leaf Horseradish ( Armoracia rusticana ) Extracts. Food Nutr. Sci. 2015, 64–74. [Google Scholar] [CrossRef]
- Zhou, H.X. Interactions of macromolecules with salt ions: An electrostatic theory for the Hofmeister effect. Proteins Struct. Funct. Genet. 2005, 61, 69–78. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Sanner, M.F.; Olson, A.J.; Spehner, J.C. Reduced surface: An efficient way to compute molecular surfaces. Biopolymers 1996, 38, 305–320. [Google Scholar] [CrossRef]
Sample Availability: Not available. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Equations | ||||||||
|---|---|---|---|---|---|---|---|---|
| Flavonol | T (K) | KSV × 104 L·mol−1 | aR | Kq × 1012 L.mol−1 s−1 | Kd × 10−5 mol·L−1 | bR | fa | KSV (accessible) × 105 L·mol−1 |
| 298 | 2.9 ± 0.1 | 0.91 | 18 | 0.8 ± 0.3 | 0.72 | 0.37 | 3.6 | |
| GAL** | 308 | 2.4 ± 0.9 | 0.92 | 15 | 0.8 ± 0.4 | 0.69 | 0.34 | 3.6 |
| 318 | 2.1 ± 0.8 | 0.91 | 13 | 1.1 ± 0.6 | 0.73 | 0.33 | 2.9 | |
| 328 | 1.5 ± 0.6 | 0.86 | 10 | 1.0 ± 0.6 | 0.80 | 0.25 | 4.2 | |
| KAE* | 298 | 2.1 ± 0.2 | 0.92 | 13 | 1.6 ± 0.3 | 0.97 | 0.45 | 1.4 |
| 308 | 1.9 ± 0.7 | 0.95 | 12 | 1.4 ± 0.5 | 0.97 | 0.43 | 1.7 | |
| 318 | 1.8 ± 0.7 | 0.95 | 11 | 1.6 ± 0.7 | 0.96 | 0.42 | 1.5 | |
| 328 | 1.6 ± 0.6 | 0.95 | 10 | 1.9 ± 0.9 | 0.90 | 0.34 | 1.5 | |
| QUE* | 298 | 1.8 ± 0.1 | 0.94 | 11 | 2.7 ± 0.6 | 0.98 | 0.54 | 0.7 |
| 308 | 1.6 ± 0.6 | 0.93 | 10 | 2.4 ± 1.0 | 0.98 | 0.52 | 0.8 | |
| 318 | 1.6 ± 0.6 | 0.95 | 10 | 2.5 ± 1.0 | 0.98 | 0.55 | 0.7 | |
| 328 | 2.0 ± 0.7 | 0.91 | 13 | 3.1 ± 1.3 | 0.97 | 0.69 | 0.5 | |
| MYR* | 298 | 1.6 ± 0.1 | 0.92 | 10 | 1.0 ± 0.1 | 0.96 | 0.34 | 2.9 |
| 308 | 1.3 ± 0.5 | 0.93 | 8.2 | 1.1 ± 0.4 | 0.96 | 0.32 | 2.8 | |
| 318 | 1.2 ± 0.4 | 0.92 | 7.7 | 1.0 ± 0.4 | 0.95 | 0.30 | 3.2 | |
| 328 | 1.4 ± 0.5 | 0.94 | 9.0 | 1.2 ± 0.7 | 0.94 | 0.33 | 2.5 | |
| Equations | |||||||
|---|---|---|---|---|---|---|---|
| Flavonol | T (K) | Ka (L·mol−1) | n | cR | ΔH (kJ·mol−1) | ΔG (kJ·mol−1) | ΔS (kJ·mol−1·K−1) |
| GAL** (Without OH) | 298 | 323 ± 8 | 0.58 ± 0.18 | 0.85 | 0.07 | −84.6 | 0.28 |
| 308 | 502 ± 10 | 0.63 ± 0.21 | 0.85 | −87.5 | |||
| 318 | 616 ± 10 | 0.66 ± 0.21 | 0.82 | −90.3 | |||
| 328 | 5817 ± 2 | 0.66 ± 0.04 | 0.81 | −93.1 | |||
| KAE* (1 OH) | 298 | 458 ± 2. | 0.63 ± 0.07 | 0.97 | 0.02 | −39.3 | 0.13 |
| 308 | 476 ± 4 | 0.63 ± 0.07 | 0.98 | −40.6 | |||
| 318 | 862 ± 3 | 0.69 ± 0.09 | 0.97 | −41.9 | |||
| 328 | 1290 ± 2 | 0.75 ± 0.07 | 0.94 | −43.3 | |||
| QUE* (2 OH) | 298 | 2034 ± 2 | 0.78 ± 0.06 | 0.95 | 0.02 | −42.3 | 0.14 |
| 308 | 1824 ± 9 | 0.77 ± 0.05 | 0.96 | −43.4 | |||
| 318 | 3041 ± 2 | 0.81 ± 0.07 | 0.95 | −45.2 | |||
| 328 | 4616 ± 3 | 0.85 ± 0.11 | 0.96 | −46.6 | |||
| MYR* (3 OH) | 298 | 45.21 ± 1 | 0.43 ± 0.02 | 0.97 | 0.02 | −26.2 | 0.10 |
| 308 | 53.09 ± 2 | 0.46 ± 0.03 | 0.96 | −27.0 | |||
| 318 | 44.79 ± 1 | 0.44 ± 0.02 | 0.96 | −27.9 | |||
| 328 | 97.14 ± 2 | 0.51 ± 0.09 | 0.96 | −28.8 | |||
| Flavonols | J (nm4·cm−1·M−1) | R0 (Å) | E | r (Å) | r/R0 |
|---|---|---|---|---|---|
| GAL | 0.8 × 1014 | 23.6 | 0.24 | 28.5 | 1.2 |
| KAE | 10 × 1014 | 36.0 | 0.12 | 50.1 | 1.4 |
| QUE | 12 × 1014 | 36.9 | 0.06 | 58.7 | 1.6 |
| MYR | 15 × 1014 | 38.4 | 0.23 | 46.7 | 1.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nasri, R.; Bidel, L.P.R.; Rugani, N.; Perrier, V.; Carrière, F.; Dubreucq, E.; Jay-Allemand, C. Inhibition of CpLIP2 Lipase Hydrolytic Activity by Four Flavonols (Galangin, Kaempferol, Quercetin, Myricetin) Compared to Orlistat and Their Binding Mechanisms Studied by Quenching of Fluorescence. Molecules 2019, 24, 2888. https://doi.org/10.3390/molecules24162888
Nasri R, Bidel LPR, Rugani N, Perrier V, Carrière F, Dubreucq E, Jay-Allemand C. Inhibition of CpLIP2 Lipase Hydrolytic Activity by Four Flavonols (Galangin, Kaempferol, Quercetin, Myricetin) Compared to Orlistat and Their Binding Mechanisms Studied by Quenching of Fluorescence. Molecules. 2019; 24(16):2888. https://doi.org/10.3390/molecules24162888
Chicago/Turabian StyleNasri, Ruba, Luc P R Bidel, Nathalie Rugani, Véronique Perrier, Frédéric Carrière, Eric Dubreucq, and Christian Jay-Allemand. 2019. "Inhibition of CpLIP2 Lipase Hydrolytic Activity by Four Flavonols (Galangin, Kaempferol, Quercetin, Myricetin) Compared to Orlistat and Their Binding Mechanisms Studied by Quenching of Fluorescence" Molecules 24, no. 16: 2888. https://doi.org/10.3390/molecules24162888
APA StyleNasri, R., Bidel, L. P. R., Rugani, N., Perrier, V., Carrière, F., Dubreucq, E., & Jay-Allemand, C. (2019). Inhibition of CpLIP2 Lipase Hydrolytic Activity by Four Flavonols (Galangin, Kaempferol, Quercetin, Myricetin) Compared to Orlistat and Their Binding Mechanisms Studied by Quenching of Fluorescence. Molecules, 24(16), 2888. https://doi.org/10.3390/molecules24162888