
Thiol-ene "Click" Synthesis and Pharmacological Evaluation of C-Glycoside sp2-Iminosugar Glycolipids

 ,
,  , ,
, ,
Abstract
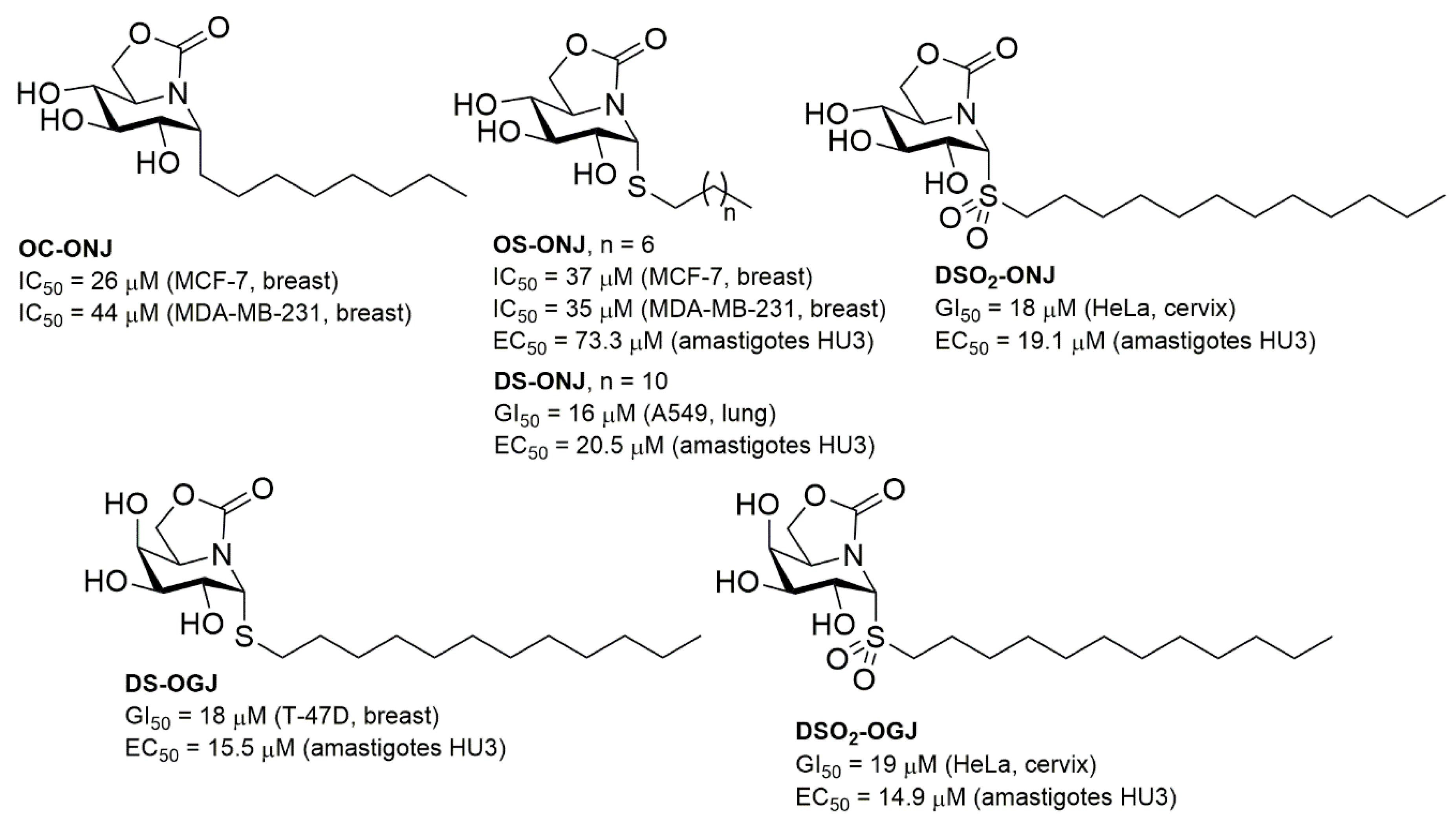
1. Introduction
2. Results and Discussion
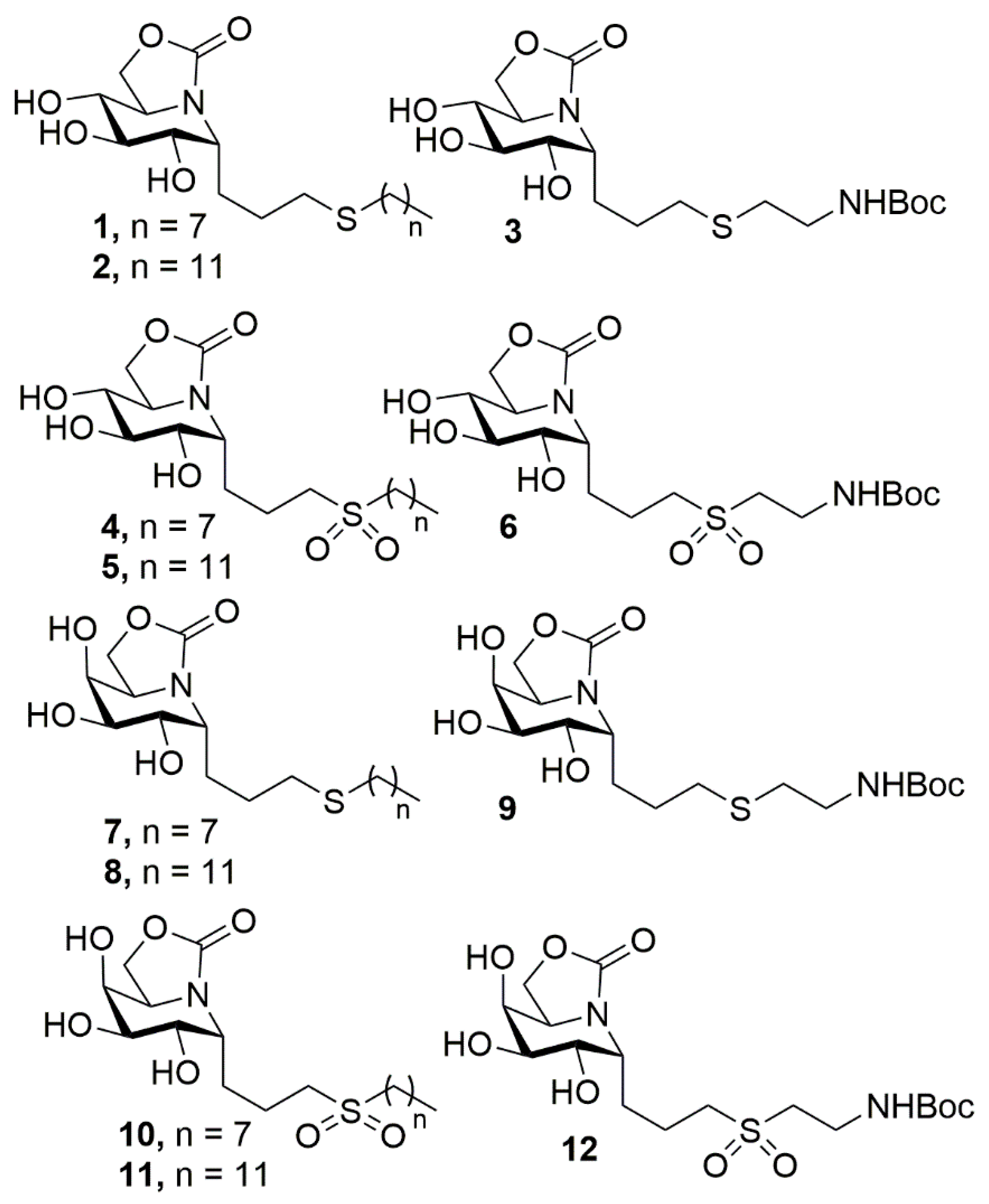
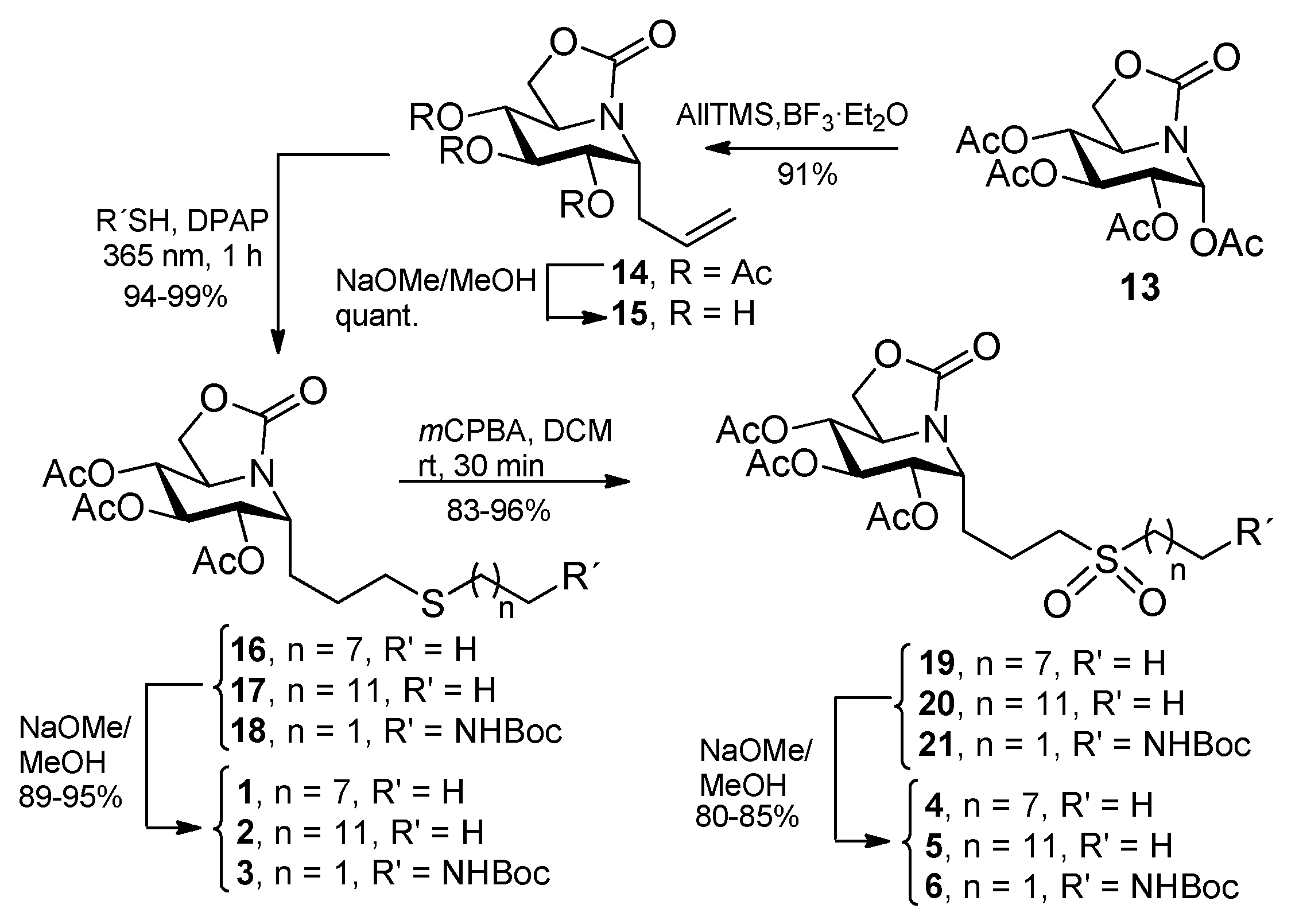
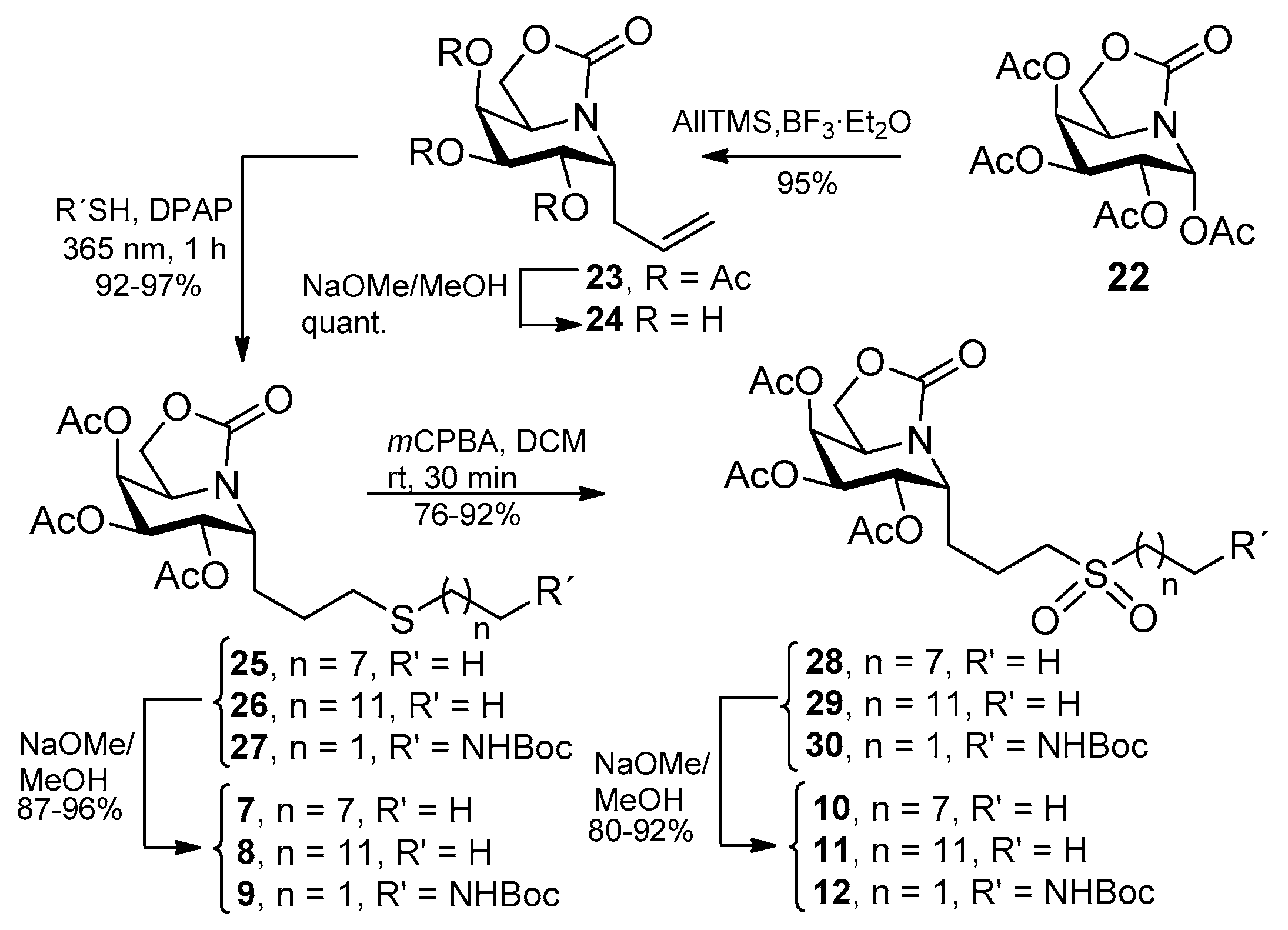
2.1. Synthesis
2.2. Inhibitory Properties against Commercial Enzymes
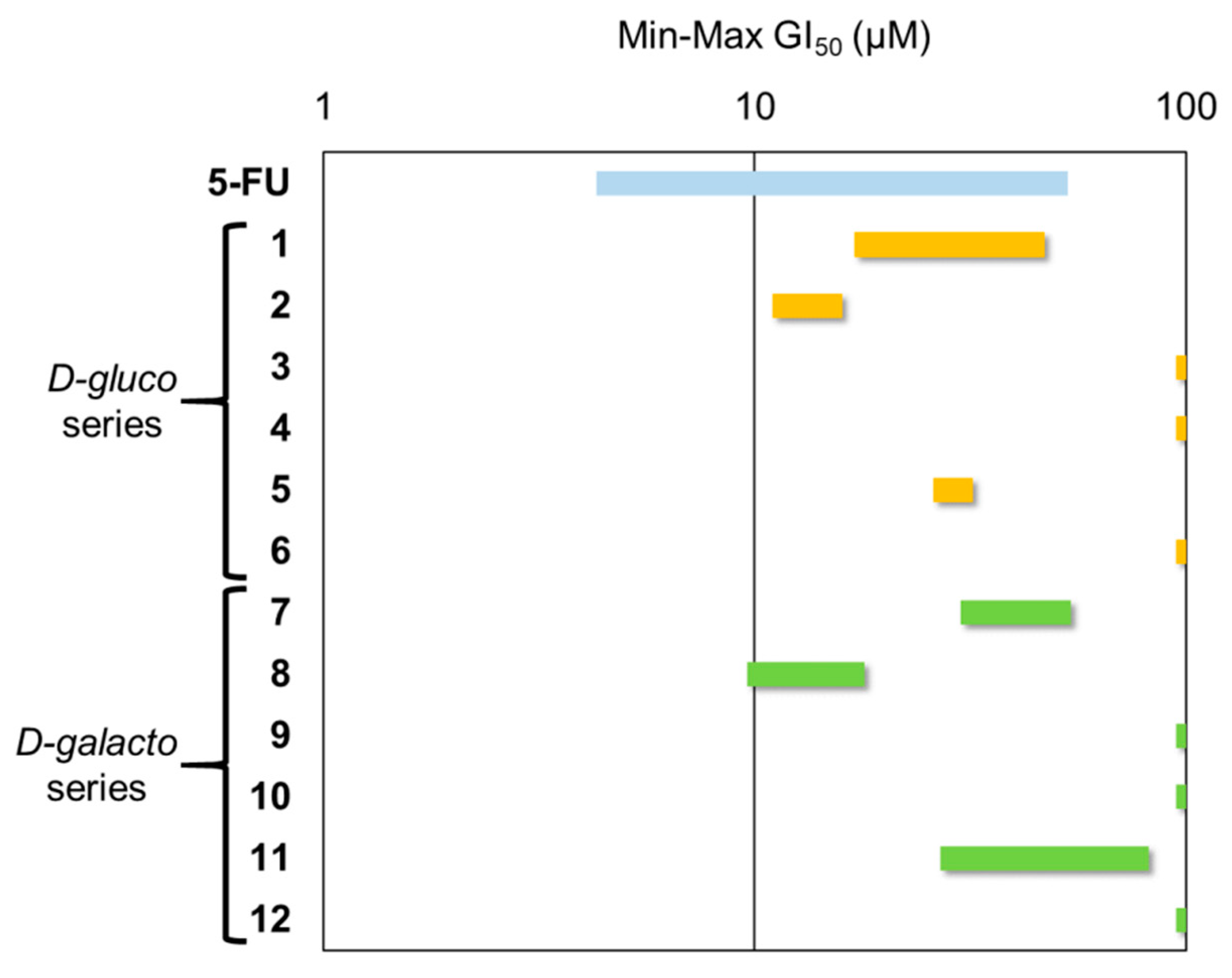
2.3. Antiproliferative Activity
2.4. Antileishmanial Activity and Cellular Toxicity
3. Materials and Methods
3.1. General Methods
3.2. Statistical Analysis
3.3. General Procedure for the Synthesis of the Allylated ONJ and OGJ Precursors 14 and 23
3.4. General Procedure for the Synthesis of ONJ (1–3) and OGJ Derivatives (7–9) by Photoinduced Thiol-Ene Reaction
3.5. General Procedure for the Synthesis of Sulfone Derivatives of ONJ 4–6 and OGJ 10–12
3.6. General Procedure for Antiproliferative Assays
Chemosensitive Testing
3.7. General Procedure for Antileishmanial Assays
3.7.1. Leishmania Culture Conditions
3.7.2. Cell lines Culture and Determination of Cellular Toxicity
3.7.3. Susceptibility Analysis in Intracellular Leishmania Amastigotes.
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- García-Moreno, M.I.; Ortiz Mellet, C.; García Fernández, J.M. Polyhydroxylated N-(thio)carbamoylpiperidines: Nojirimycin-type glycomimetics with controlled anomeric configuration. Tetrahedron: Asymmetry 1999, 10, 4271–4275. [Google Scholar] [CrossRef]
- Aguilar-Moncayo, M.; Díaz-Pérez, P.; García-Moreno, M.I.; Ortiz Mellet, C.; García Fernández, J.M. Synthesis and biological evaluation of guanidine-type iminosugars. J. Org. Chem. 2008, 73, 1995–1998. [Google Scholar] [CrossRef] [PubMed]
- Sevšek, A.; Sastre Toraño, J.; van Ufford, L.Q.; Moret, E.E.; Pieters, R.J.; Martin, N.I. Orthoester functionalized N-guanidino derivatives of 1,5-dideoxy-1,5-imino-d-xylitol as pH-responsive inhibitors of β-glucocerebrosidase. Med. Chem. Commun. 2017, 8, 2050–2054. [Google Scholar] [CrossRef] [PubMed]
- Sevšek, A.; Šrot, L.; Rihter, J.; Celan, M.; van Ufford, L.Q.; Moret, E.E.; Martin, N.I.; Pieters, R.J. N-Guanidino Derivatives of 1,5-Dideoxy-1,5-imino-d-xylitol are Potent, Selective, and Stable Inhibitors of β-Glucocerebrosidase. ChemMedChem 2017, 12, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Mena Barragán, T.; García Moreno, M.I.; Nanba, E.; Higaki, K.; Lisa Concia, A.; Clapés, P.; García Fernández, J.M.; Ortiz Mellet, C. Inhibitor versus chaperone behaviour of fagomine, DAB and LAB sp2-iminosugar conjugates against glycosidases: A structure-activity relationship study in Gaucher fibroblasts. Eur. J. Med. Chem. 2016, 121, 880–891. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, S.; Hirokami, Y.; Kinami, K.; Hoshino, M.; Minehira, D.; Daiki Miyamoto, D.; Nash, R.J.; Fleet, G.W.J.; Adachi, I.; Toyooka, N.; et al. Strategy for designing selective α-L-rhamnosidase inhibitors: Synthesis and biological evaluation of L-DMDP cyclic isothioureas. Bioorg. Med. Chem. 2017, 25, 107–115. [Google Scholar] [CrossRef] [PubMed]
- García-Moreno, M.I.; Rodríguez-Lucena, D.; Ortiz Mellet, C.; García Fernández, J.M. Pseudoamide-type pyrrolidine and pyrrolizidine glycomimetics and their inhibitory activities against glycosidases. J. Org. Chem. 2004, 69, 3578–3581. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-L.; Shimada, Y.; Adachi, I.; Kato, A.; Jia, Y.-M.; Fleet, G.W.J.; Xiao, M.; Yu, C.-Y. Fluorinated and Conformationally Fixed Derivatives of L-Homo DMDP: Synthesis and Glycosidase Inhibition. J. Org. Chem. 2015, 80, 5151–5158. [Google Scholar] [CrossRef]
- Díaz-Pérez, P.; García-Moreno, M.I.; Ortiz Mellet, C.; García Fernández, J.M. Synthesis of (1S,2S,3R,8S,8aR)-1,2,3,8-tetrahydroxy-6-oxa-5-thioindolizidine: A stable reducing swainsonine analog with controlled anomeric configuration. Synlett 2003, 3, 341–344. [Google Scholar] [CrossRef]
- Benltifa, M.; García-Moreno, M.I.; Ortiz Mellet, C.; García Fernández, J.M.; Wadouachi, A. Synthesis and evaluation of sulfamide-type indolizidines as glycosidase inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 2805–2808. [Google Scholar] [CrossRef]
- Aguilar-Moncayo, M.; García-Moreno, M.I.; Ortiz Mellet, C.; García Fernández, J.M. Synthesis of thiohydantoine-castanospermine glycomimetics as glycosidase inhibitors. J. Org. Chem. 2009, 74, 3595–3598. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.; Sánchez-Fernández, E.M.; Ortiz Mellet, C.; Tatibouët, A.; Rauter, A.P.; Rollin, P. N-Thiocarbonyl iminosugars: Synthesis and evaluation of castanospermine analogues bearing oxazole-2(3H)-thione moieties. Eur. J. Org. Chem. 2013, 7941–7951. [Google Scholar] [CrossRef]
- Sánchez-Fernández, E.M.; Álvarez, E.; Ortiz Mellet, C.; García Fernández, J.M. Synthesis of Multibranched Australine Derivatives from Reducing Castanospermine Analogues through the Amadori Rearrangement of gem-Diamine Intermediates: Selective Inhibitors of β-Glucosidase. J. Org. Chem. 2014, 79, 11722–11728. [Google Scholar] [CrossRef] [PubMed]
- Sevšek, A.; Čelan, M.; Erjavec, B.; van Ufford, L.Q.; Sastre Toraño, J.; Moret, E.E.; Pieters, R.J.; Martin, N.I. Bicyclic isoureas derived from 1-deoxynojirimycinare potent inhibitors of β-glucocerebrosidase. Org. Biomol. Chem. 2016, 14, 8670–8673. [Google Scholar] [CrossRef] [PubMed]
- García-Moreno, M.I.; Benito-Hernández, J.M.; Ortiz Mellet, C.; García Fernández, J.M. Synthesis and evaluation of calystegine B2 analogues as glycosidase inhibitors. J. Org. Chem. 2001, 66, 7604–7614. [Google Scholar] [CrossRef] [PubMed]
- García Fernández, J.M.; Ortiz Mellet, C.; Benito-Hernández, J.M.; Fuentes-Mota, J. Synthesis of calystegine B2 analogs by tandem tautomerization-intramolecular glycosylation of thioureidosugars. Synlett 1998, 3, 316–318. [Google Scholar] [CrossRef]
- Aguilar-Moncayo, M.; Gloster, T.M.; García-Moreno, M.I.; Ortiz Mellet, C.; Davies, G.J.; Llebaria-Soldevilla, A.; Casas-Brugulat, J.; Egido-Gabás, M.; García Fernández, J.M. Molecular basis for β-glucosidase inhibition by ring-modified calystegine analogues. ChemBioChem 2008, 9, 2612–2618. [Google Scholar] [CrossRef] [PubMed]
- Sánchez Fernández, E.M.; García Fernández, J.M.; Ortiz Mellet, C. Glycomimetic-based pharmacological chaperones for lysosomal storage disorders: Lessons from Gaucher, GM1-gangliosidosis and Fabry diseases. Chem. Commun. 2016, 52, 5497–5515. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; Boland, B.; van der Spoel, A.C. The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J. Cell. Biol. 2012, 199, 723–734. [Google Scholar] [CrossRef] [PubMed]
- García Fernández, J.M.; Ortiz Mellet, C. Novel therapies for orphan diseases. ACS Med. Chem. Lett. 2019, 10, 1020–1023. [Google Scholar] [CrossRef] [PubMed]
- Luan, Z.; Higaki, K.; Aguilar-Moncayo, M.; Ninomiya, H.; Ohno, K.; García-Moreno, M.I.; Ortiz Mellet, C.; García Fernández, J.M.; Suzuki, Y. Chaperone Activity of Bicyclic Nojirimycin Analogues for Gaucher Mutations in Comparison with N-(n-nonyl)-Deoxynojirimycin. ChemBioChem 2009, 10, 2780–2792. [Google Scholar] [CrossRef] [PubMed]
- Luan, Z.; Higaki, K.; Aguilar-Moncayo, M.; Li, L.; Ninomiya, H.; Nanba, E.; Ohno, K.; García-Moreno, M.I.; Ortiz Mellet, C.; García Fernández, J.M.; et al. A Fluorescent sp2-Iminosugar with Pharmacological Chaperone Activity for Gaucher Disease: Synthesis and Intracellular Distribution Studies. ChemBioChem 2010, 11, 2453–2464. [Google Scholar] [CrossRef] [PubMed]
- Alfonso, P.; Andreu, V.; Pino-Ángeles, A.; Moya-García, A.A.; García-Moreno, M.I.; Rodríguez-Rey, J.C.; Sánchez-Jiménez, F.; Pocoví, M.; Ortiz Mellet, C.; García Fernández, J.M.; et al. Bicyclic derivatives of L-idonojirimycin as pharmacological chaperones for neuronopathic forms of Gaucher disease. ChemBioChem 2013, 14, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Tiscornia, G.; Lorenzo Vivas, E.; Matalonga, L.; Berniakovich, I.; Barragán Monasterio, M.; Eguizábal Argaiz, C.; Gort, L.; González, F.; Ortiz Mellet, C.; García Fernández, J.M.; et al. Neuronopathic Gaucher’s disease: Induced pluripotent stem cells for disease modelling and testing chaperone activity of small compounds. Hum. Mol. Genet. 2013, 22, 633–645. [Google Scholar] [CrossRef] [PubMed]
- De la Mata, M.; Cotán, D.; Oropesa-Ávila, M.; Garrido-Maraver, J.; Cordero, M.D.; Villanueva Paz, M.; Delgado Pavón, A.; Alcocer-Gómez, E.; de Lavera, I.; Ybot-González, P.; et al. Pharmacological Chaperones and Coenzyme Q10 Treatment Improves Mutant β-Glucocerebrosidase Activity and Mitochondrial Function in Neuronopathic Forms of Gaucher Disease. Sci. Rep. 2015, 5, 10903. [Google Scholar] [CrossRef] [PubMed]
- Mena-Barragán, T.; García-Moreno, M.I.; Sevšek, A.; Okazaki, T.; Nanba, E.; Higaki, K.; Martin, N.I.; Pieters, R.J.; García Fernández, J.M.; Ortiz Mellet, C. Probing the Inhibitor versus Chaperone Properties of sp2-Iminosugars towards Human β-Glucocerebrosidase: A Picomolar Chaperone for Gaucher Disease. Molecules 2018, 23, 927. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Mena-Barragán, T.; Higaki, K.; Johnson, J.; Drury, J.; Lieberman, R.; Nakasone, N.; Ninomiya, H.; Tsukimura, T.; Sakuraba, H.; et al. Molecular basis of 1-deoxygalactonojirimycin arylthiourea binding to human α-galactosidase: Pharmacological chaperoning efficacy on Fabry disease mutants. ACS. Chem. Biol. 2014, 9, 1460–1469. [Google Scholar] [CrossRef] [PubMed]
- Mena-Barragán, T.; Narita, A.; Matias, D.; Tiscornia, G.; Nanba, E.; Ohno, K.; Suzuki, Y.; Higaki, K.; García Fernández, J.M.; Ortiz Mellet, C. pH-Responsive Pharmacological Chaperones for Rescuing Mutant Glycosidases. Angew. Chem. Int. Ed. 2015, 54, 11696–11700. [Google Scholar] [CrossRef] [PubMed]
- Takai, T.; Higaki, K.; Aguilar-Moncayo, M.; Mena-Barragán, T.; Hirano, Y.; Yura, K.; Yu, L.; Ninomiya, H.; García-Moreno, M.I.; Sakakibara, Y.; et al. A Bicyclic 1 Deoxygalactonojirimycin Derivative as Novel Pharmacological Chaperone for GM1 Gangliosidosis. Mol. Ther. 2013, 21, 526–532. [Google Scholar] [CrossRef]
- Suzuki, H.; Ohto, U.; Higaki, K.; Mena-Barragán, T.; Aguilar-Moncayo, M.; Ortiz Mellet, C.; Nanba, E.; García Fernández, J.M.; Suzuki, Y.; Shimizu, T. Structural basis of pharmacological chaperoning for human β-galactosidase. J. Biol. Chem. 2014, 289, 14560–14568. [Google Scholar] [CrossRef]
- De la Fuente, A.; Rísquez Cuadro, R.; Verdaguer, X.; García Fernández, J.M.; Nanba, K.; Higaki, K.; Ortiz Mellet, C.; Riera, A. Efficient Stereoselective Synthesis of 2-Acetamido-1,2-dideoxyallonojirimycin (DAJNAc) and sp2-Iminosugar Conjugates: Novel Hexosaminidase Inhibitors with Discrimination Capabilities between the Mature and Precursor Forms of the Enzyme. Eur. J. Med. Chem. 2016, 121, 926–938. [Google Scholar] [CrossRef] [PubMed]
- Rísquez-Cuadro, R.; Matsumoto, R.; Ortega-Caballero, F.; Nanba, E.; Katsumi, H.; García Fernández, J.M.; Ortiz Mellet, C. Pharmacological Chaperones for the Treatment of α-Mannosidosis. J. Med. Chem. 2019, 62, 5832–5843. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Pérez, V.; García-Moreno, M.I.; Ortiz Mellet, C.; Fuentes-Mota, J.; Díaz-Arribas, J.C.; Cañada, J.; García Fernández, J.M. Generalized anomeric effect in action: synthesis and evaluation of stable reducing indolizidine glycomimetics as glycosidase inhibitors. J. Org. Chem. 2000, 65, 136–143. [Google Scholar] [CrossRef] [PubMed]
- García-Moreno, M.I.; Díaz-Pérez, P.; Ortiz Mellet, C.; García Fernández, J.M. Castanospermine-trehazoline hybrids: a new family of glycomimetics with tuneable glycosidase inhibitory properties. Chem. Commun. 2002, 8, 848–849. [Google Scholar] [CrossRef]
- Sánchez-Fernández, E.M.; Rísquez-Cuadro, R.; Aguilar-Moncayo, M.; García-Moreno, M.I.; Ortiz Mellet, C.; García Fernández, J.M. Generalized anomeric effect in gem-diamines: Stereoselective synthesis of α-N-linked disaccharide mimics. Org. Lett. 2009, 11, 3306–3309. [Google Scholar] [CrossRef]
- Sánchez-Fernández, E.M.; Rísquez-Cuadro, R.; Chasseraud, M.; Ahidouch, A.; Ortiz Mellet, C.; Ouadid-Ahidouch, H.; García Fernández, J.M. Synthesis of N-, S-, and C-Glycoside castanospermine analogues with selective neutral α-glucosidase inhibitory activity as antitumor agents. Chem. Commun. 2010, 46, 5328–5330. [Google Scholar] [CrossRef] [PubMed]
- Sánchez Fernández, E.M.; Rísquez-Cuadro, R.; Ortiz Mellet, C.; García Fernández, J.M.; Nieto, P.M.; Angulo, J. sp2-Iminosugar O-, S- and N-glycosides as conformational mimics of α-linked disaccharides: Implications for glycosidase inhibition. Chem. Eur. J. 2012, 18, 8527–8539. [Google Scholar] [CrossRef]
- Rísquez-Cuadro, R.; García Fernández, J.M.; Nierengarten, J.-F.; Ortiz Mellet, C. Fullerene-sp2-iminosugarballs as multimodal ligands for lectins and glycosidases: A mechanistic hypothesis for the inhibitory multivalent. Chem. Eur. J. 2013, 19, 16791–16803. [Google Scholar] [CrossRef]
- García Fernández, J.M.; Nierengarten, J.-F.; Ortiz Mellet, C. Multivalency as an action principle in multimodal lectin recognition and glycosidase inhibition: A paradigm shift driven by carbon-based glyconanomaterials. J. Mater. Chem. B 2017, 5, 6428–6436. [Google Scholar] [CrossRef]
- García-Moreno, M.I.; Ortega-Caballero, F.; Rísquez-Cuadro, R.; Ortiz Mellet, C.; García Fernández, J.M. The Impact of Heteromultivalency in Lectin Recognition and Glycosidase Inhibition: An Integrated Mechanistic Study. Chem. Eur. J. 2017, 23, 6295–6304. [Google Scholar] [CrossRef]
- Abellán Flos, M.; García-Moreno, M.I.; Ortiz Mellet, C.; García Fernández, J.M.; Nierengarten, J.-F.; Vincent, S.P. Potent glycosidase inhibition with heterovalent fullerenes: Unveiling the binding modes triggering multivalent inhibition. Chem. Eur. J. 2016, 22, 11450–11460. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Fernández, E.M.; Navo, C.; Martínez-Saez, N.; Gonçalves-Pereira, R.; Somovilla, V.J.; Avenoza, A.; Busto, J.H.; Bernardes, G.J.L.; Jiménez-Osés, G.; Corzana, F.; et al. Tn Antigen Mimics based on sp2-Iminosugars with Affinity for an anti-MUC1 Antibody. Org. Lett. 2016, 18, 3890–3893. [Google Scholar] [CrossRef] [PubMed]
- Alcalde-Estévez, E.; Arroba, A.I.; Sánchez-Fernández, E.M.; Ortiz Mellet, C.; García Fernández, J.M.; Masgrau, L.; Valverde, A.M. The sp2-iminosugar glycolipid 1-dodecylsulfonyl-5N,6O-oxomethylidenenojirimycin (DSO2-ONJ) as selective anti-inflammatory agent by modulation of hemeoxygenase-1 in Bv.2 microglial cells and retinal explants. Food. Chem. Toxicol. 2018, 454–466. [Google Scholar] [CrossRef]
- Fontelle, N.; Yamamoto, A.; Arda, A.; Jiménez-Barbero, J.; Kato, A.; Désiré, J.; Blériot, Y. 2-Acetamido-2-deoxy-L-iminosugar C-Alkyl and C-Aryl Glycosides: Synthesis and Glycosidase Inhibition. Eur. J. Org. Chem. 2018, 5477–5488. [Google Scholar] [CrossRef]
- Compain, P.; Chagnault, V.; Martin, O.R.; Auberger, N.; Onfroy, B.; Fontelle, N.; Foucart, Q.; Blériot, Y. Lewis acid-catalysed nucleophilic opening of a bicyclic hemiaminal followed by ring contraction: Access to functionalized L-idonojirimycin derivatives. Carbohydr. Res. 2019, 472, 65–71. [Google Scholar] [CrossRef]
- Bergeron-Brlek, M.; Meanwell, M.; Britton, R. Direct synthesis of imino-C-nucleoside analogues and other biologically active iminosugars. Nat. Commun. 2015, 6, 6903. [Google Scholar] [CrossRef] [PubMed]
- Bergeron-Brlek, M.; Goodwin-Tindall, J.; Cekic, N.; Roth, C.; Zandberg, W.F.; Shan, X.; Varghese, V.; Chan, S.; Davies, G.J.; Vocadlo, D.J.; et al. A Convenient Approach to Stereoisomeric Iminocyclitols: Generation of Potent Brain-Permeable OGA Inhibitors. Angew. Chem. Int. Ed. 2015, 54, 15429–15433. [Google Scholar] [CrossRef]
- Compain, P.; Compain, O.R. Iminosugars: From Synthesis to Therapeutic Applications; Martin, Ed.; Wiley & Sons: Chichester, UK, 2007; pp. 63–86. [Google Scholar]
- Schaeffer, E.; Sánchez-Fernández, E.M.; Gonçalves-Pereira, R.; Flacher, V.; Lamon, D.; Duval, M.; Fauny, J.-D.; García Fernández, J.M.; Mueller, C.G.; Ortiz Mellet, C. sp2-Iminosugar glycolipids as inhibitors of lipopolysaccharide mediated human dendritic cell activation in vitro and of acute inflammation in mice in vivo. Eur. J. Med. Chem. 2019, 169, 111–120. [Google Scholar] [CrossRef]
- Arroba, A.; Alcalde-Estévez, E.; García-Ramírez, M.; Cazzoni, D.; de la Villa, P.; Sánchez-Fernández, E.M.; Ortiz Mellet, C.; García Fernández, J.M.; Hernández, C.; Simo, R.; et al. Modulation of microglia polarization dynamics during diabetic retinopathy in db/db mice. BBA Mol. Basis. Dis. 2016, 1862, 1663–1674. [Google Scholar] [CrossRef]
- Allan, G.; Ouadid-Ahidouch, H.; Sánchez-Fernández, E.M.; Rísquez Cuadro, R.; García Fernández, J.M.; Ortiz Mellet, C.; Ahidouch, A. New Castanospermine glycoside analogues inhibit breast cancer cell proliferation and induce apoptosis without affecting normal cells. PLoS ONE 2013, 8, e76411. [Google Scholar] [CrossRef]
- Allan, G.; Gueder, N.; Telliez, M.-S.; Hague, F.; Sánchez-Fernández, E.M.; García Fernández, J.M.; Ortiz Mellet, C.; Ahidouch, A.; Ouadid-Ahidouch, H. sp2-Iminosugar α-glucosidase inhibitor 1-C-octyl-2-oxa-3-oxocastanospermine specifically affected breast cancer cell migration through Stim1, beta1-integrin, and FAK signaling pathways. J. Cell. Physiol. 2017, 232, 3631–3640. [Google Scholar] [CrossRef]
- Sánchez-Fernández, E.M.; Gómez-Pérez, V.; García-Hernández, R.; García Fernández, J.M.; Plata, G.B.; Padrón, J.M.; Ortiz Mellet, C.; Castanys, S.; Gamarro, F. Antileishmanial activity of sp2-iminosugar derivatives. RSC Adv. 2015, 5, 21812–21822. [Google Scholar] [CrossRef]
- Sinha, A.K.; Equbal, D. Thiol-Ene Reaction: Synthetic Aspects and Mechanistic Studies of an Anti-Markovnikov-Selective Hydrothiolation of Olefins. Asian. J. Org. Chem. 2019, 8, 32–47. [Google Scholar] [CrossRef]
- McSweeney, L.; Denes, F.; Scanlan, E.M. Thiyl-Radical Reactions in Carbohydrate Chemistry: From Thiosugars to Glycoconjugate Synthesis. Eur. J. Org. Chem. 2016, 2080–2095. [Google Scholar] [CrossRef]
- Dondoni, A.; Marra, A. Recent applications of thiol–ene coupling as a click process for glycoconjugation. Chem. Soc. Rev. 2012, 41, 573–586. [Google Scholar] [CrossRef]
- Lázár, L.; Borbás, A.; Somsák, L. Synthesis of thiomaltooligosaccharides by a thio-click approach. Carbohydr. Res. 2018, 470, 8–12. [Google Scholar] [CrossRef] [PubMed]
- József, J.; Juhász, L.; Somsák, L. Thio-click reaction of 2-deoxy-exo-glycals towards new glycomimetics: stereoselective synthesis of C-2-deoxy-D-glycopyranosyl compounds. New J. Chem. 2019, 43, 5670. [Google Scholar] [CrossRef]
- Eszenyi, D.; Kelemen, V.; Balogh, F.; Bege, M.; Csávás, M.; Herczegh, P.; Borbás, A. Promotion of a Reaction by Cooling: Stereoselective 1,2-cis-α-Thioglycoconjugation by Thiol-Ene Coupling at -80 °C. Chem. Eur. J. 2018, 24, 4532–4536. [Google Scholar] [CrossRef]
- Horton, D.; Miyake, T. Stereoselective syntheses of C-(D-glucopyranosyl)alkenes and alkadienes. Carbohydr. Res. 1988, 184, 221–229. [Google Scholar] [CrossRef]
- Dondoni, A.; Massi, A.; Nanni, P.; Roda, A. A New Ligation Strategy for Peptide and Protein Glycosylation: Photoinduced Thiol–Ene Coupling. Chem. Eur. J. 2009, 15, 11444–11449. [Google Scholar] [CrossRef]
- Aguilar-Moncayo, M.; Takai, T.; Higaki, K.; Mena-Barragán, T.; Hirano, Y.; Yura, K.; Li, L.; Yu, Y.; Ninomiya, H.; García-Moreno, M.I.; et al. Tuning glycosidase inhibition through aglycone interactions: pharmacological chaperones for Fabry disease and GM1 gangliosidosis. Chem. Commun. 2012, 48, 6514–6516. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.A.; Cabezas, J.A.; Calvo, P. β-Fucosidase, β-glucosidase and β-galactosidase activities associated in bovine liver. Int. J. Biochem. 1982, 14, 695–698. [Google Scholar] [CrossRef]
- Hossain, F.; Andreana, P.R. Developments in Carbohydrate-Based Cancer Therapeutics. Pharmaceuticals 2019, 12, 84. [Google Scholar] [CrossRef] [PubMed]
- Wrodnigg, T.M.; Steiner, A.J.; Ueberbache, B.J. Natural and Synthetic Iminosugars as Carbohydrate Processing Enzyme Inhibitors for Cancer Therapy. Anti-Cancer Agents Med. Chem. 2008, 8, 77–85. [Google Scholar] [CrossRef]
- García-Hernández, R.; Gómez-Pérez, V.; Castanys, S.; Gamarro, F. Fitness of Leishmania donovani parasites resistant to drug combinations. PLoS. Negl. Trop. Dis. 2015, 9, e0003704. [Google Scholar] [CrossRef]
- Ríos-Marco, P.; Marco, C.; Gálvez, X.; Jiménez-López, J.M.; Carrasco, M.P. Alkylphospholipids: An update on molecular mechanisms and clinical relevance. Biochim. Biphys. Acta-Biomembr. 2017, 1859, 1657–1667. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Pérez, P.; García-Moreno, M.I.; Ortiz Mellet, C.; García Fernández, J.M. Synthesis and comparative glycosidase inhibitory properties of reducing castanospermine analogues. Eur. J. Org. Chem. 2005, 14, 2903–2913. [Google Scholar] [CrossRef]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolff, A.; et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Natl. Cancer Inst. 1991, 83, 757–766. [Google Scholar] [CrossRef]
- El Fadili, K.; Imbeault, M.; Messier, N.; Roy, G.; Gourbal, B.; Bergeron, M.; Tremblay, M.J.; Légaré, D.; Ouellette, D.M. Modulation of gene expression in human macrophages treated with the anti-Leishmania pentavalent antimonial drug sodium stibogluconate. Antimicrob. Agents Chemother. 2008, 52, 526. [Google Scholar] [CrossRef]
- Gómez-Pérez, V.; Manzano, J.I.; García-Hernández, R.; Castanys, S.; Campos, J.M.; Gamarro, F. 4-Amino bis-pyridinium derivatives as novel antileishmanial agents. Antimicrob. Agents Chemother. 2014, 58, 4103–4112. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1–12 are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound a | α-Glcase (Yeast Maltase) | β-Glcase (Bovine Liver) |
|---|---|---|
| 15 | 79 ± 8 | n.i. |
| 1 | 0.34 ± 0.02 | 400 ± 20 |
| 2 | 0.74 ± 0.03 | 54 ± 5 |
| 3 | 0.28 ± 0.02 | 151 ± 10 |
| 4 | 2.6 ± 0.3 | 172 ± 14 |
| 5 | 2.5 ± 0.2 | 85 ± 18 |
| 6 | 0.75 ± 0.02 | 342 ± 20 |
| 7 | n.i. | n.i. |
| 8 | n.i. | 53 ± 12 |
| 9 | n.i. | n.i. |
| 10 | n.i. | 422 ± 22 |
| 11 | n.i. | 134 ± 10 |
| 12 | n.i. | 770 ± 45 |
| 24 | n.i. | n.i. |
| Compound | Intracellular Amastigotes | THP-1 (SI) | MRC-5 (SI) |
|---|---|---|---|
| 1 | 7.68 ± 0.29 | 130.96 ± 4.29 (17.06) | 32.20 ± 1.03 (4.19) |
| 2 | >20 | 72.24 ± 4.18 (<3.61) | 19.92 ± 2.76 (<1) |
| 3 | 14.96 ± 1.28 | >200 | 133.89 ± 6.02 (8.95) |
| 4 | >20 | >200 | 188.98 ± 15.59 (<9.45) |
| 5 | 11.44 ± 5.08 | 100.43 ± 10.87 (8.78) | 71.49 ± 3.75 (6.25) |
| 6 | >20 | >200 | 155.18 ± 3.10 (<7.76) |
| 7 | 10.58 ± 1.51 | 127.55 ± 5.71 (12.06) | 60.44 ± 2.80 (5.72) |
| 8 | >20 | 68.30 ± 3.04 (<3.42) | 32.08 ± 1.94 (<1.60) |
| 9 | 13.39 ± 1.13 | >200 | 139.92 ± 17.31 (10.44) |
| 10 | 13.81 ± 1.72 | >200 | 184.97 ± 21.25 (13.40) |
| 11 | >20 | >200 | 161.89 ± 20.18 (<8.10) |
| 12 | >20 | >200 | 181.51 ± 14.93 (<9.08) |
| AmB | 0.15 ± 0.01 | 20.07 ± 4.43 (133.80) | 16.93 ± 2.44 (112.87) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Fernández, E.M.; García-Moreno, M.I.; García-Hernández, R.; Padrón, J.M.; García Fernández, J.M.; Gamarro, F.; Ortiz Mellet, C. Thiol-ene "Click" Synthesis and Pharmacological Evaluation of C-Glycoside sp2-Iminosugar Glycolipids. Molecules 2019, 24, 2882. https://doi.org/10.3390/molecules24162882
Sánchez-Fernández EM, García-Moreno MI, García-Hernández R, Padrón JM, García Fernández JM, Gamarro F, Ortiz Mellet C. Thiol-ene "Click" Synthesis and Pharmacological Evaluation of C-Glycoside sp2-Iminosugar Glycolipids. Molecules. 2019; 24(16):2882. https://doi.org/10.3390/molecules24162882
Chicago/Turabian StyleSánchez-Fernández, Elena M., M. Isabel García-Moreno, Raquel García-Hernández, José M. Padrón, José M. García Fernández, Francisco Gamarro, and Carmen Ortiz Mellet. 2019. "Thiol-ene "Click" Synthesis and Pharmacological Evaluation of C-Glycoside sp2-Iminosugar Glycolipids" Molecules 24, no. 16: 2882. https://doi.org/10.3390/molecules24162882
APA StyleSánchez-Fernández, E. M., García-Moreno, M. I., García-Hernández, R., Padrón, J. M., García Fernández, J. M., Gamarro, F., & Ortiz Mellet, C. (2019). Thiol-ene "Click" Synthesis and Pharmacological Evaluation of C-Glycoside sp2-Iminosugar Glycolipids. Molecules, 24(16), 2882. https://doi.org/10.3390/molecules24162882