
Synthesis and In Vitro Evaluation of 8-Pyridinyl-Substituted Benzo[e]imidazo[2,1-c][1,2,4]triazines as Phosphodiesterase 2A Inhibitors

 and
and
Abstract
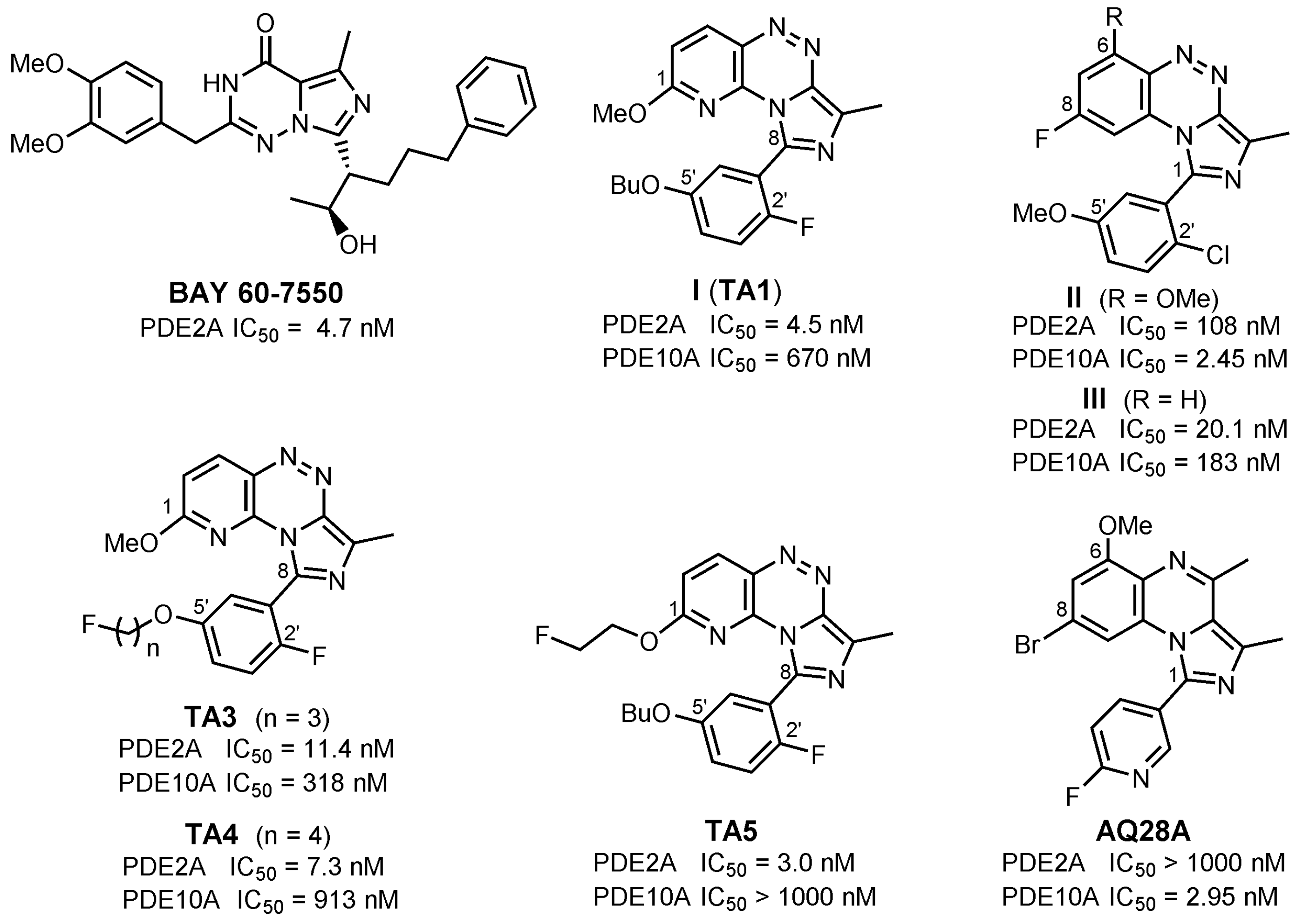
1. Introduction
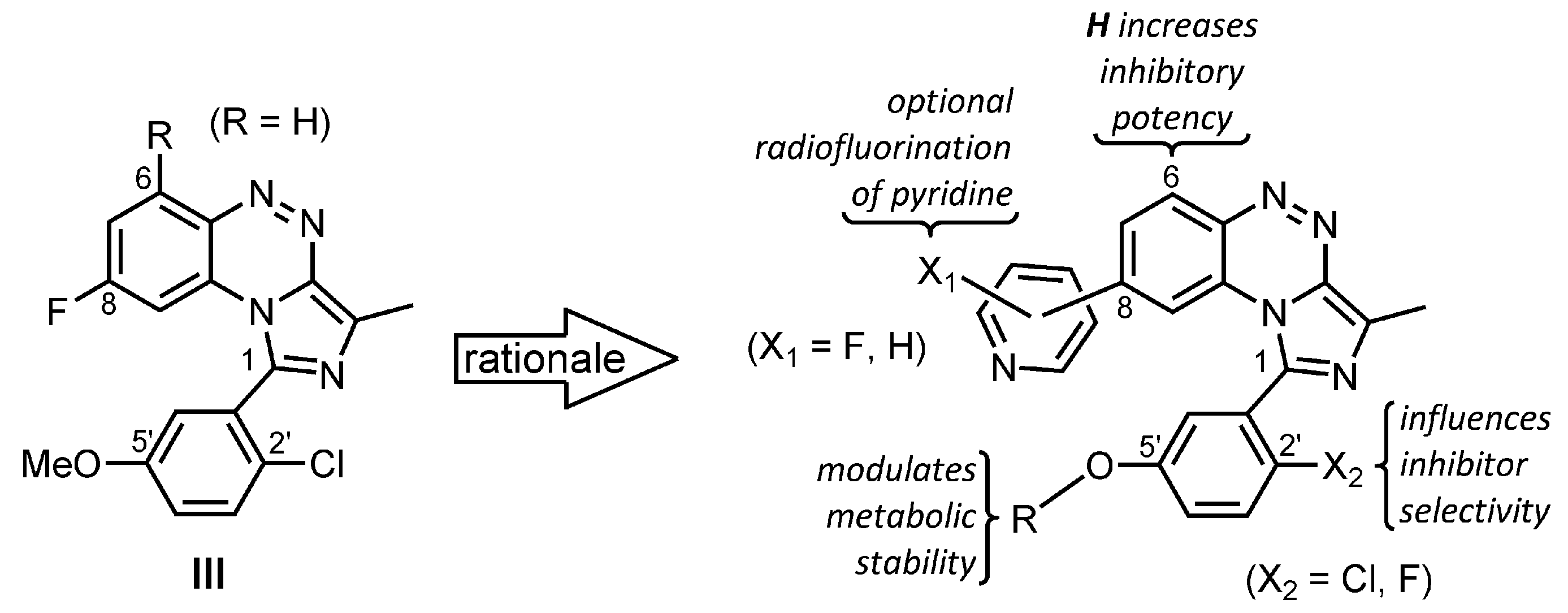
2. Results and Discussion
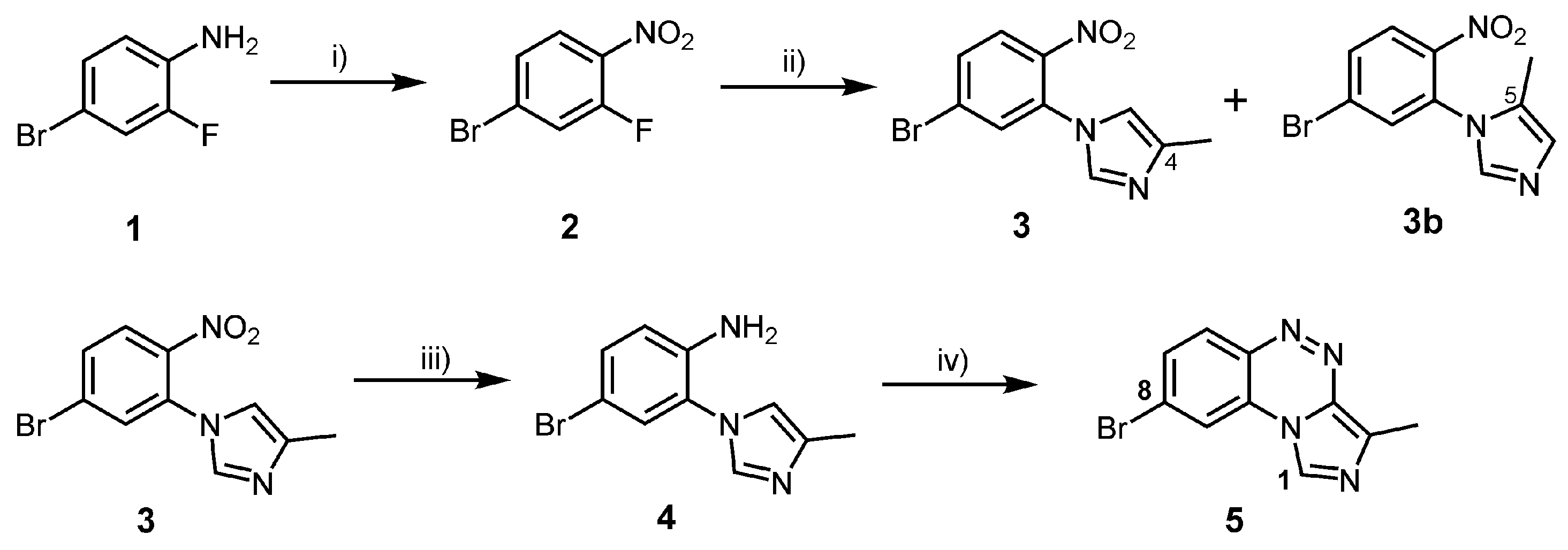
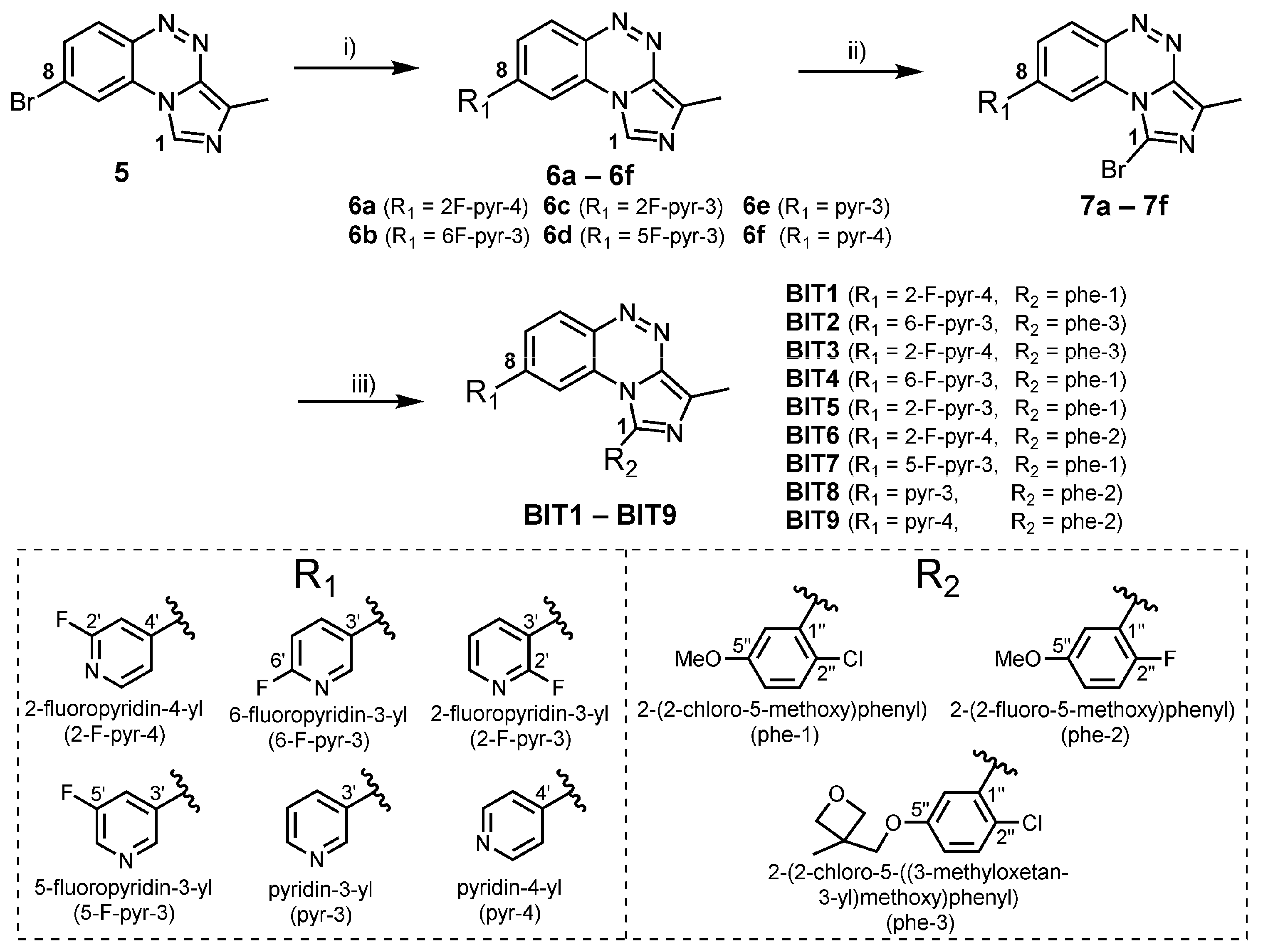
2.1. Chemistry
2.2. In Vitro Evaluation: Structure-Activity Relationships of BIT Derivatives
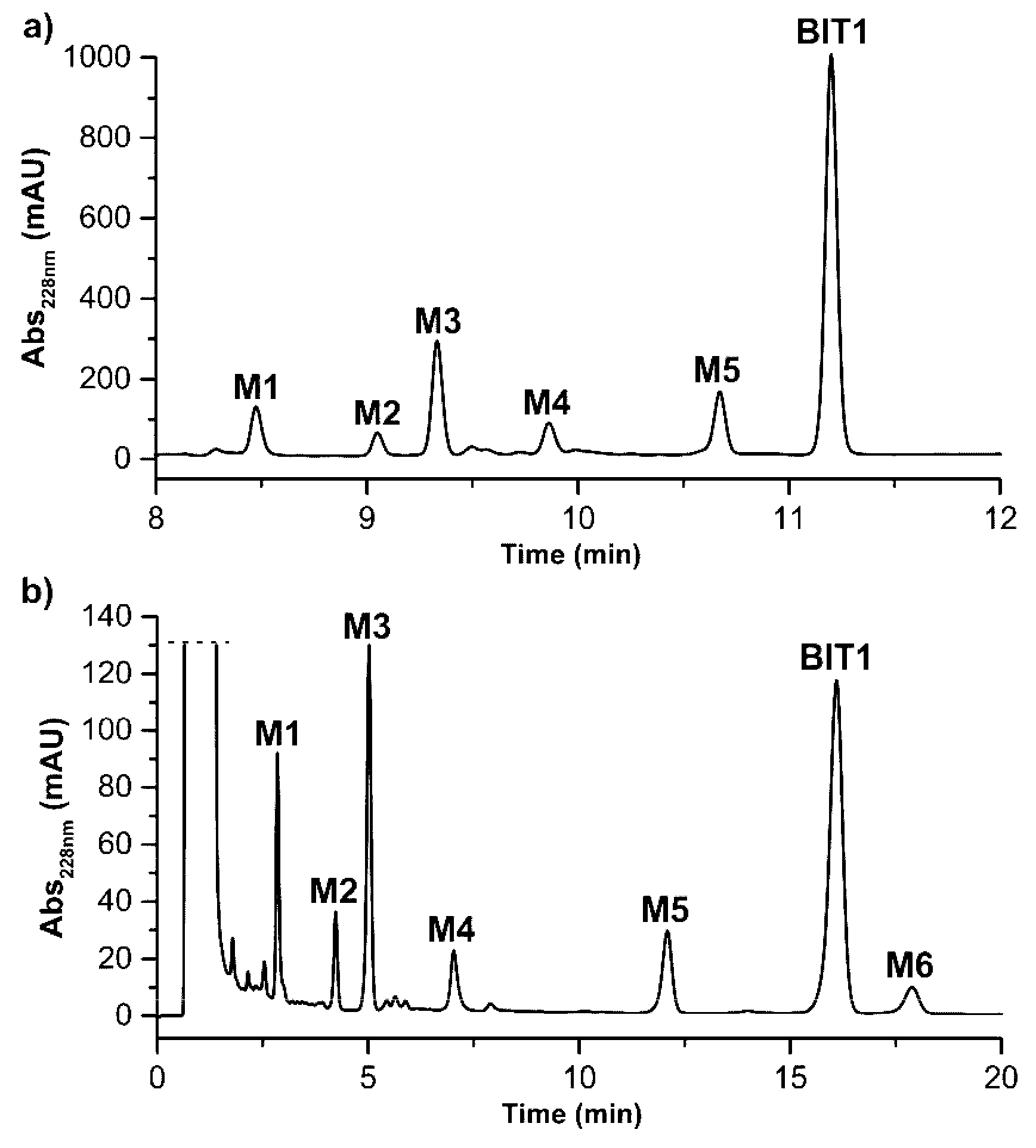
2.3. Incubations with Mouse Liver Microsomes
3. Materials and Methods
3.1. General Information
3.2. Syntheses
3.2.1. General Procedure A for the Suzuki Coupling of Bromo Derivatives 5, 7a, 7b, 7c, 7d, 7e, and 7f
3.2.2. General Procedure B for Bromination of Compounds 6a, 6b, 6c, 6d, 6e, and 6f
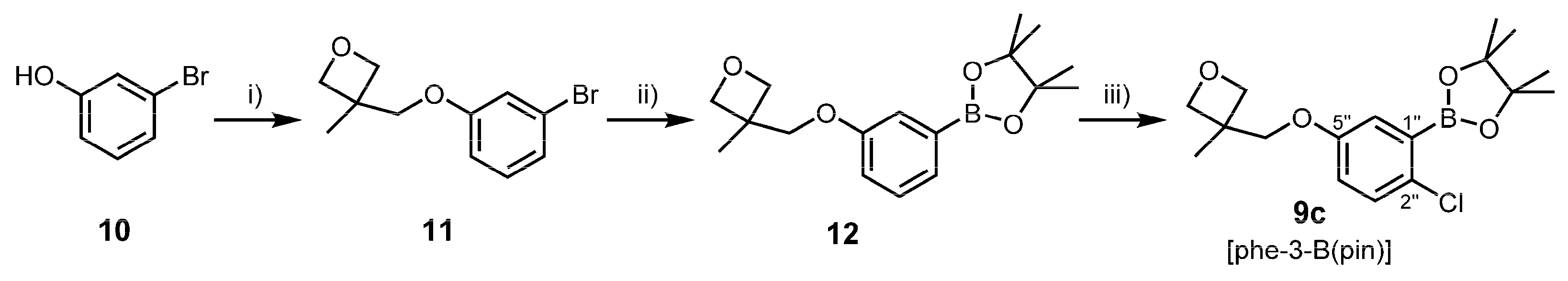
3.2.3. Synthesis of Oxetanyl Building Block 9c
3.2.4. Synthesis of Compounds BIT1–BIT9 via Suzuki Coupling According to General Procedure A
3.3. Biology
3.3.1. In Vitro Evaluation of BIT Derivatives Towards PDEs
3.3.2. Incubations with Mouse Liver Microsomes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Helal, C.J.; Arnold, E.P.; Boyden, T.L.; Chang, C.; Chappie, T.A.; Fennell, K.F.; Forman, M.D.; Hajos, M.; Harms, J.F.; Hoffman, W.E.; et al. Application of Structure-Based Design and Parallel Chemistry to Identify a Potent, Selective, and Brain Penetrant Phosphodiesterase 2A Inhibitor. J. Med. Chem. 2017, 60, 5673–5698. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, A.M.; Piazza, G.A.; Tinsley, H.N. The role of cyclic nucleotide signaling pathways in cancer: Targets for prevention and treatment. Cancers 2014, 6, 436–458. [Google Scholar] [CrossRef] [PubMed]
- Maurice, D.H.; Ke, H.; Ahmad, F.; Wang, Y.; Chung, J.; Manganiello, V.C. Advances in targeting cyclic nucleotide phosphodiesterases. Nat. Rev. Drug Discov. 2014, 13, 290–314. [Google Scholar] [CrossRef] [PubMed]
- Conti, M.; Beavo, J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: Essential components in cyclic nucleotide signaling. Annu. Rev. Biochem. 2007, 76, 481–511. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.H.; Blount, M.A.; Corbin, J.D. Mammalian cyclic nucleotide phosphodiesterases: Molecular mechanisms and physiological functions. Physiol. Rev. 2011, 91, 651–690. [Google Scholar] [CrossRef] [PubMed]
- Keravis, T.; Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) isozymes as targets of the intracellular signalling network: Benefits of PDE inhibitors in various diseases and perspectives for future therapeutic developments. Br. J. Pharmacol. 2012, 165, 1288–1305. [Google Scholar] [CrossRef]
- Lakics, V.; Karran, E.H.; Boess, F.G. Quantitative comparison of phosphodiesterase mRNA distribution in human brain and peripheral tissues. Neuropharmacology 2010, 59, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Gomez, L.; Massari, M.E.; Vickers, T.; Freestone, G.; Vernier, W.; Ly, K.; Xu, R.; McCarrick, M.; Marrone, T.; Metz, M.; et al. Design and Synthesis of Novel and Selective Phosphodiesterase 2 (PDE2a) Inhibitors for the Treatment of Memory Disorders. J. Med. Chem 2017, 60, 2037–2051. [Google Scholar] [CrossRef] [PubMed]
- Gomez, L.; Breitenbucher, J.G. PDE2 inhibition: Potential for the treatment of cognitive disorders. Bioorg. Med. Chem. Lett 2013, 23, 6522–6527. [Google Scholar] [CrossRef]
- Redrobe, J.P.; Rasmussen, L.K.; Christoffersen, C.T.; Bundgaard, C.; Jørgensen, M. Characterisation of Lu AF33241: A novel, brain-penetrant, dual inhibitor of phosphodiesterase (PDE) 2A and PDE10A. Eur. J. Pharmacol. 2015, 761, 79–85. [Google Scholar] [CrossRef]
- Mikami, S.; Sasaki, S.; Asano, Y.; Ujikawa, O.; Fukumoto, S.; Nakashima, K.; Oki, H.; Kamiguchi, N.; Imada, H.; Iwashita, H.; et al. Discovery of an Orally Bioavailable, Brain-Penetrating, in Vivo Active Phosphodiesterase 2A Inhibitor Lead Series for the Treatment of Cognitive Disorders. J. Med. Chem. 2017, 60, 7658–7676. [Google Scholar] [CrossRef] [PubMed]
- Kehler, J. Targeting Phosphodiesterases in the CNS. In Comprehensive Medicinal Chemistry, 3rd ed.; Chackalamannil, S., Rotela, D., Ward, S.E., Eds.; Elsevier: Oxford, UK, 2017; Volume 7, pp. 1–24. [Google Scholar]
- Stephenson, D.T.; Coskran, T.M.; Kelly, M.P.; Kleiman, R.J.; Morton, D.; O’Neill, S.M.; Schmidt, C.J.; Weinberg, R.J.; Menniti, F.S. The distribution of phosphodiesterase 2A in the rat brain. Neuroscience 2012, 226, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, D.T.; Coskran, T.M.; Wilhelms, M.B.; Adamowicz, W.O.; O’Donnell, M.M.; Muravnick, K.B.; Menniti, F.S.; Kleiman, R.J.; Morton, D. Immunohistochemical localization of phosphodiesterase 2A in multiple mammalian species. J. Histochem. Cytochem 2009, 57, 933–949. [Google Scholar] [CrossRef] [PubMed]
- Schroder, S.; Wenzel, B.; Deuther-Conrad, W.; Scheunemann, M.; Brust, P. Novel Radioligands for Cyclic Nucleotide Phosphodiesterase Imaging with Positron Emission Tomography: An Update on Developments Since 2012. Molecules 2016, 21, 650. [Google Scholar] [CrossRef] [PubMed]
- Buijnsters, P.; de Angelis, M.; Langlois, X.; Rombouts, F.J.R.; Sanderson, W.; Tresadern, G.; Ritchie, A.; Trabanco, A.A.; VanHoof, G.; van Roosbroeck, Y.; et al. Structure-Based Design of a Potent, Selective, and Brain Penetrating PDE2 Inhibitor with Demonstrated Target Engagement. ACS Med. Chem. Lett. 2014, 5, 1049–1053. [Google Scholar] [CrossRef]
- Blokland, A.; Menniti, F.S.; Prickaerts, J. PDE inhibition and cognition enhancement. Expert. Opin. Ther. Pat. 2012, 22, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Helal, C.J.; Arnold, E.; Boyden, T.; Chang, C.; Chappie, T.A.; Fisher, E.; Hajos, M.; Harms, J.F.; Hoffman, W.E.; Humphrey, J.M.; et al. Identification of a Potent, Highly Selective, and Brain Penetrant Phosphodiesterase 2A Inhibitor Clinical Candidate. J. Med. Chem. 2018, 61, 1001–1018. [Google Scholar] [CrossRef]
- Malamas, M.S.; Ni, Y.; Erdei, J.J.; Egerland, U.; Langen, B. The Substituted Imidazo [1,5-a]Quinoxalines as Inhibitors of Phosphodiesterase 10. WO 2010/138833 A1, 2 December 2010. [Google Scholar]
- Am Ende, C.W.; Kormos, B.L.; Humphrey, J.M. The State of the Art in Selective PDE2A Inhibitor Design. In Phosphodiesterases and their inhibitors; Liras, S., Bell, A.S., Eds.; Wiley-VCH: Weinheim an der Bergstrasse, Germany, 2014; Volume 61, pp. 83–104. [Google Scholar]
- Schroder, S.; Wenzel, B.; Deuther-Conrad, W.; Teodoro, R.; Egerland, U.; Kranz, M.; Scheunemann, M.; Hofgen, N.; Steinbach, J.; Brust, P. Synthesis, 18F-Radiolabelling and Biological Characterization of Novel Fluoroalkylated Triazine Derivatives for in Vivo Imaging of Phosphodiesterase 2A in Brain via Positron Emission Tomography. Molecules 2015, 20, 9591–9615. [Google Scholar] [CrossRef]
- Schröder, S.; Wenzel, B.; Deuther-Conrad, W.; Teodoro, R.; Kranz, M.; Scheunemann, M.; Egerland, U.; Höfgen, N.; Briel, D.; Steinbach, J.; et al. Investigation of an 18F-labelled Imidazopyridotriazine for Molecular Imaging of Cyclic Nucleotide Phosphodiesterase 2A. Molecules 2018, 23, 556. [Google Scholar] [CrossRef]
- Wagner, S.; Scheunemann, M.; Dipper, K.; Egerland, U.; Hoefgen, N.; Steinbach, J.; Brust, P. Development of highly potent phosphodiesterase 10A (PDE10A) inhibitors: Synthesis and in vitro evaluation of 1,8-dipyridinyl- and 1-pyridinyl-substituted imidazo[1,5-a]quinoxalines. Eur. J. Med. Chem. 2016, 107, 97–108. [Google Scholar] [CrossRef]
- Wagner, S.; Teodoro, R.; Deuther-Conrad, W.; Kranz, M.; Scheunemann, M.; Fischer, S.; Wenzel, B.; Egerland, U.; Hoefgen, N.; Steinbach, J.; et al. Radiosynthesis and biological evaluation of the new PDE10A radioligand [18F]AQ28A. J. Label. Compd. Radiopharm. 2017, 60, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Stange, H.; Langen, B.; Egerland, U.; Hoefgen, N.; Priebs, M.; Malamas, M.S.; Erdei, J.; Ni, Y. Triazine Derivatives as Inhibitors of Phosphodiesterases. Patent US 2010/0120762 A1, 13 May 2010. [Google Scholar]
- Stange, H.; Langen, B.; Egerland, U.; Hoefgen, N.; Priebs, M.; Malamas, M.S.; Erdei, J.J.; Ni, Y. Imidazo[5,1-C][1,2,4]Benzotriazine Derivatives As Inhibitors of. Patent US 2010/0120763 A1, 13 May 2010. [Google Scholar]
- Malamas, M.S.; Stange, H.; Schindler, R.; Lankau, H.-J.; Grunwald, C.; Langen, B.; Egerland, U.; Hage, T.; Ni, Y.; Erdei, J.; et al. Novel triazines as potent and selective phosphodiesterase 10A inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 5876–5884. [Google Scholar] [CrossRef] [PubMed]
- Trabanco, A.A.; Buijnsters, P.; Rombouts, F.J.R. Towards selective phosphodiesterase 2A (PDE2A) inhibitors: A patent review (2010 - present). Expert. Opin. Ther. Pat. 2016, 26, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wenzel, B.; Dukic-Stefanovic, S.; Teodoro, R.; Ludwig, F.-A.; Deuther-Conrad, W.; Schröder, S.; Chezal, J.-M.; Moreau, E.; Brust, P.; et al. Development of a new radiofluorinated quinoline analog for PET imaging of phosphodiesterase 5 (PDE5) in brain. Pharmaceuticals 2016, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Gujral, S.S.; Khatri, S.; Riyal, P. Suzuki Cross Coupling Reaction- A Review. Indo Glob. J. Pharm. Sci. 2012, 2, 351–367. [Google Scholar]
- Dollé, F. [18F]Fluoropyridines: From Conventional Radiotracers to the Labeling of Macromolecules Such as Proteins and Oligonucleotides. In PET Chemistry The Driving Force in Molecular Imaging; Schubinger, P.A., Lehmann, L., Friebe, M., Eds.; Springer: Berlin, Germany, 2006; Volume 62, pp. 113–157. [Google Scholar]
- Goldstein, S.W.; Bill, A.; Dhuguru, J.; Ghoneim, O. Nucleophilic Aromatic Substitution—Addition and Identification of an Amine. J. Chem. Educ. 2017, 94, 1388–1390. [Google Scholar] [CrossRef]
- Artamkina, G.A.; Egorov, M.P.; Beletskaya, I.P. Some aspects of anionic sigma complexes. Chem. Rev. 1982, 82, 427–459. [Google Scholar] [CrossRef]
- Gamble, A.B.; Garner, J.; Gordon, C.P.; O’Conner, S.M.J.; Keller, P.A. Aryl Nitro Reduction with Iron Powder or Stannous Chloride under Ultrasonic Irradiation. Synth. Commun. 2007, 37, 2777–2786. [Google Scholar] [CrossRef]
- Wuitschik, G.; Rogers-Evans, M.; Muller, K.; Fischer, H.; Wagner, B.; Schuler, F.; Polonchuk, L.; Carreira, E.M. Oxetanes as promising modules in drug discovery. Angew. Chem. Int. Ed. 2006, 45, 7736–7739. [Google Scholar] [CrossRef]
- Bull, J.A.; Croft, R.A.; Davis, O.A.; Doran, R.; Morgan, K.F. Oxetanes: Recent Advances in Synthesis, Reactivity, and Medicinal Chemistry. Chem. Rev. 2016, 116, 12150–12233. [Google Scholar] [CrossRef]
- Talele, T.T. Natural-Products-Inspired Use of the gem-Dimethyl Group in Medicinal Chemistry. J. Med. Chem. 2018, 61, 2166–2210. [Google Scholar] [CrossRef] [PubMed]
- Kamei, T. Metal-free halogenation of arylboronate with N-halosuccinimide. Tetrahedron Lett. 2014, 55, 4245–4247. [Google Scholar] [CrossRef]
- Zhu, J.; Yang, Q.; Dai, D.; Huang, Q. X-ray crystal structure of phosphodiesterase 2 in complex with a highly selective, nanomolar inhibitor reveals a binding-induced pocket important for selectivity. J. Am. Chem. Soc. 2013, 135, 11708–11711. [Google Scholar] [CrossRef] [PubMed]
- Gomez, L.; Xu, R.; Sinko, W.; Selfridge, B.; Vernier, W.; Ly, K.; Truong, R.; Metz, M.; Marrone, T.; Sebring, K.; et al. Mathematical and Structural Characterization of Strong Nonadditive Structure-Activity Relationship Caused by Protein Conformational Changes. J. Med. Chem. 2018, 61, 7754–7766. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.-J. Hydrogen bonds with fluorine. Studies in solution, in gas phase and by computations, conflicting conclusions from crystallographic analyses. Chem. Sci. 2012, 3, 1381–1394. [Google Scholar] [CrossRef]
- Taylor, R. The hydrogen bond between N-H or O-H and organic fluorine: favourable yes, competitive no. Acta. Crys. 2017, B73, 474–488. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, F.-A.; Smits, R.; Fischer, S.; Donat, C.K.; Hoepping, A.; Brust, P.; Steinbach, J. LC-MS Supported Studies on the in Vitro Metabolism of both Enantiomers of Flubatine and the in Vivo Metabolism of (+)-[18F]Flubatine-A Positron Emission Tomography Radioligand for Imaging α4β2 Nicotinic Acetylcholine Receptors. Molecules 2016, 21, 1200. [Google Scholar] [CrossRef] [PubMed]
- Testa, B.; Kraemer, S.D. The Biochemistry of Drug Metabolism—An The Biochemistry of Drug Metabolism-Introduction Part 2. Redox Reaction and Their Enzymes. Chem. Biodivers. 2007, 4, 257–405. [Google Scholar] [CrossRef]
- Russell, T.R.; Thompson, W.J.; Schneider, F.; Schneider, F.W.; Appleman, M.M. 3′:5′-Cyclic Adenosine Monophosphate Phosphodiesterase: Negative Cooperativity. Proc. Natl. Acad. Sci. USA 1972, 69, 1791–1795. [Google Scholar] [CrossRef]
Sample Availability: BIT1, BIT2, and BIT6 are avaliable from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Starting Boronic Acid (BA) | Product of First Suzuki Coupling | Yield | Product of Bromination | Yield |
|---|---|---|---|---|---|
| 1 | 2-fluoro-pyridine-4-yl-BA (8a) | 6a | 76% | 7a | 79% |
| 2 | 6-fluoro-pyridine-3-yl-BA (8b) | 6b | 28% | 7b | 63% |
| 3 | 2-fluoro-pyridine-3-yl-BA (8c) | 6c | 98% | 7c | 88% |
| 4 | 3-fluoro-pyridine-5-yl-BA (8d) | 6d | 56% | 7d | 98% |
| 5 | pyridine-3-yl-BA (8e) | 6e | 77% | 7e | 87% |
| 6 | pyridine-4-yl-BA (8f) | 6f | 87% | 7f | 92% |
 | |||||
|---|---|---|---|---|---|
| Entry | Bromo Compound | Boronic Acid Derivative | R1/R2 | Product | Yield |
| 1 | 7a | 9a | 2-F-pyr-4/phe-1 | BIT1 | 80% |
| 2 | 7b | 9c | 6-F-pyr-3/phe-3 | BIT2 | 79% |
| 3 | 7a | 9c | 2-F-pyr-4/phe-3 | BIT3 | 68% |
| 4 | 7b | 9a | 6-F-pyr-3/phe-1 | BIT4 | 64% |
| 5 | 7c | 9a | 2-F-pyr-3/phe-1 | BIT5 | 69% |
| 6 | 7a | 9b | 2-F-pyr-4/phe-2 | BIT6 | 72% |
| 7 | 7d | 9a | 5-F-pyr-3/phe-1 | BIT7 | 56% |
| 8 | 7e | 9b | pyr-3/phe-2 | BIT8 | 82% |
| 9 | 7f | 9b | pyr-4/phe-2 | BIT9 | 39% |
| Inhibition of PDE Sub-Types (%) | |||||
|---|---|---|---|---|---|
| 2A3 | 4A1 | 5A1 | 9A1 | 10A1 | |
| BIT1 | 82.9 | 58.6 | 29.3 | NI | 32.4 |
| BIT2 | 8.52 | 64.1 | NI | NI | 2.46 |
| BIT3 | 13.2 | 72.7 | NI | NI | 28.2 |
| BIT4 | 2.63 | NI | NI | NI | 10.8 |
| BIT5 | NI | NI | NI | NI | 22.1 |
| BIT6 | 56.6 | 54.2 | 13.6 | 28.4 | 74.0 |
| BIT7 | 50.6 | 65.7 | 25.1 | 11.5 | 87.0 |
| BIT8 | NI | 13.9 | 36.2 | NI | 55.5 |
| BIT9 | 80.5 | 86.8 | 57.7 | 17.5 | 96.6 |
| TA1 | 93.5 | 24.9 | 3.08 | NI | 41 |
| NI: No Inhibition | |||||
| Compounds | IC50 PDE2A (nM) | IC50 PDE10A (nM) | Selectivity Ratio |
|---|---|---|---|
| BIT1 | 3.33 | 53.23 | 16 |
| BIT6 | 65.06 | 168.4 | 2.5 |
| BIT9 | 17.7 | 20.24 | 1.1 |
| PDE Subtypes | %Inhibition |
|---|---|
| PDE1A3 | 12.6 |
| PDE3A | 20 |
| PDE6AB | 16.8 |
| PDE7A | 16.4 |
| PDE8A1 | 23.3 |
| PDE11A | 14.5 |
| Metabolite | tR (min) Method A Gradient | tR (min) Method B Isocratic | m/z Found | m/z Theoret. | Identification |
|---|---|---|---|---|---|
| BIT1 | 11.28 | 16.10 | 420.7 | 420.1 | parent (M + H)+ |
| M1 | 8.47 | 2.85 | 422.7 | 422.1 | reduction (M + 2H + H)+ |
| M2 | 9.05 | 4.24 | 452.8 | 452.1 | di-oxygenation (M + 2O + H)+ |
| M3 | 9.33 | 5.02 | 436.7 | 436.1 | mono-oxygenation (M + O + H)+ |
| M4 | 9.86 | 7.03 | 406.6 | 406.1 | demethylation (M-CH3 + H)+ |
| M5 | 10.67 | 12.04 | 436.7 | 436.1 | mono-oxygenation (M + O + H)+ |
| M6 | not detected | 17.89 | 436.7 | 436.1 | mono-oxygenation (M + O + H)+ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ritawidya, R.; Ludwig, F.-A.; Briel, D.; Brust, P.; Scheunemann, M. Synthesis and In Vitro Evaluation of 8-Pyridinyl-Substituted Benzo[e]imidazo[2,1-c][1,2,4]triazines as Phosphodiesterase 2A Inhibitors. Molecules 2019, 24, 2791. https://doi.org/10.3390/molecules24152791
Ritawidya R, Ludwig F-A, Briel D, Brust P, Scheunemann M. Synthesis and In Vitro Evaluation of 8-Pyridinyl-Substituted Benzo[e]imidazo[2,1-c][1,2,4]triazines as Phosphodiesterase 2A Inhibitors. Molecules. 2019; 24(15):2791. https://doi.org/10.3390/molecules24152791
Chicago/Turabian StyleRitawidya, Rien, Friedrich-Alexander Ludwig, Detlef Briel, Peter Brust, and Matthias Scheunemann. 2019. "Synthesis and In Vitro Evaluation of 8-Pyridinyl-Substituted Benzo[e]imidazo[2,1-c][1,2,4]triazines as Phosphodiesterase 2A Inhibitors" Molecules 24, no. 15: 2791. https://doi.org/10.3390/molecules24152791
APA StyleRitawidya, R., Ludwig, F.-A., Briel, D., Brust, P., & Scheunemann, M. (2019). Synthesis and In Vitro Evaluation of 8-Pyridinyl-Substituted Benzo[e]imidazo[2,1-c][1,2,4]triazines as Phosphodiesterase 2A Inhibitors. Molecules, 24(15), 2791. https://doi.org/10.3390/molecules24152791