
Amine-Catalyzed Decarboxylative Aldol Reaction of β-Ketocarboxylic Acids with Trifluoropyruvates


Abstract
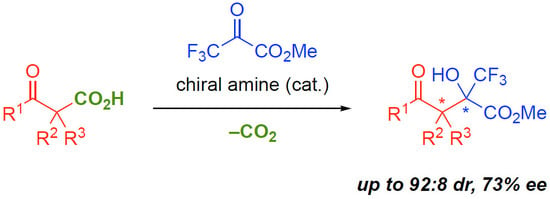
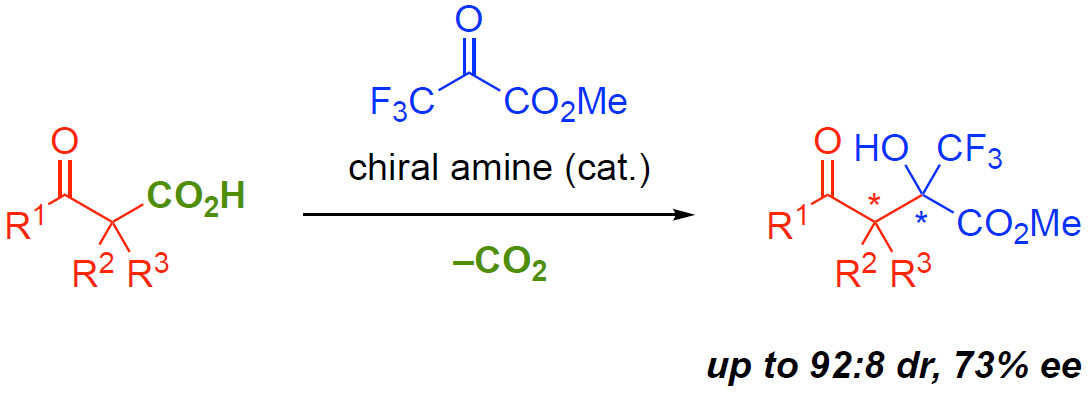
1. Introduction
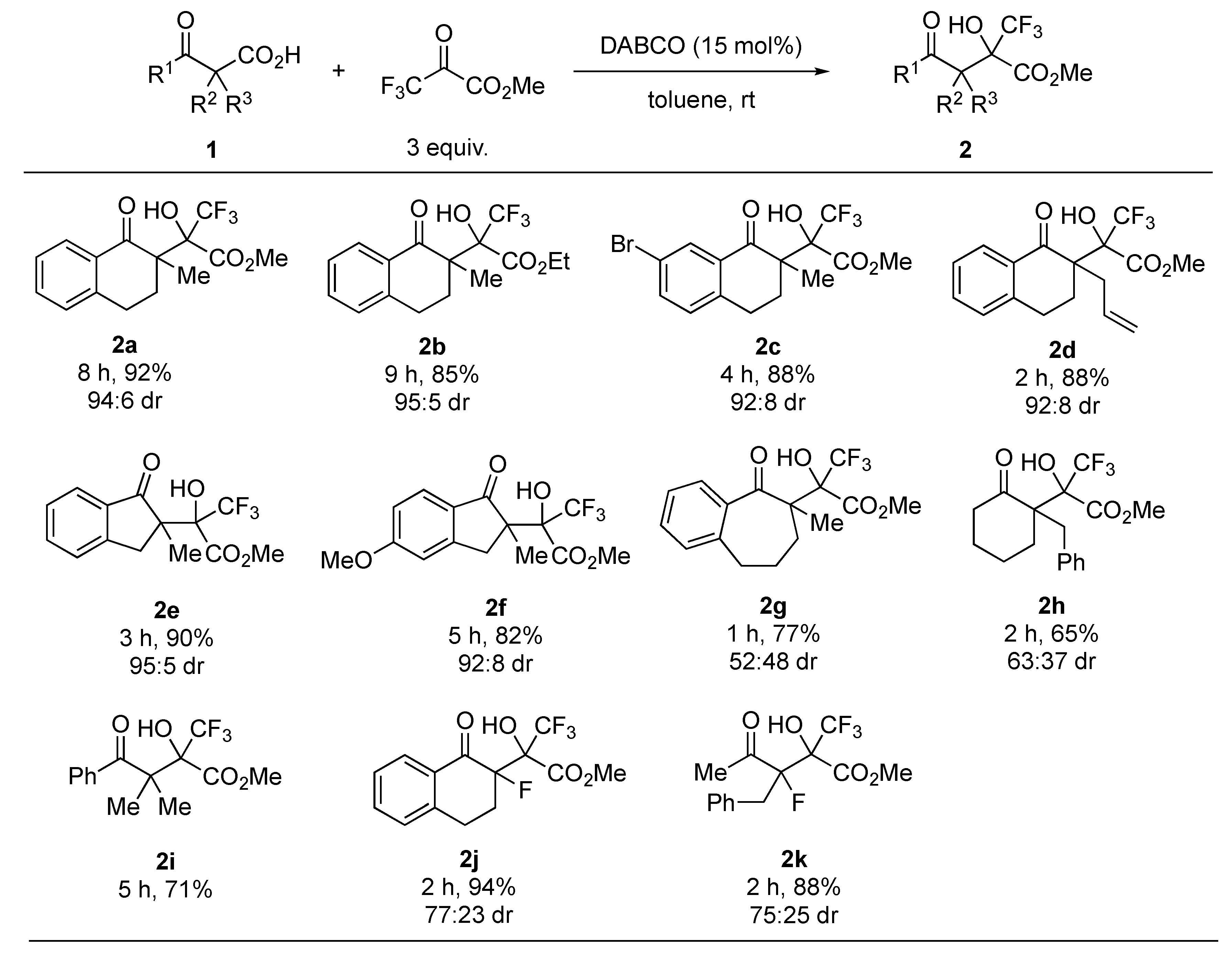
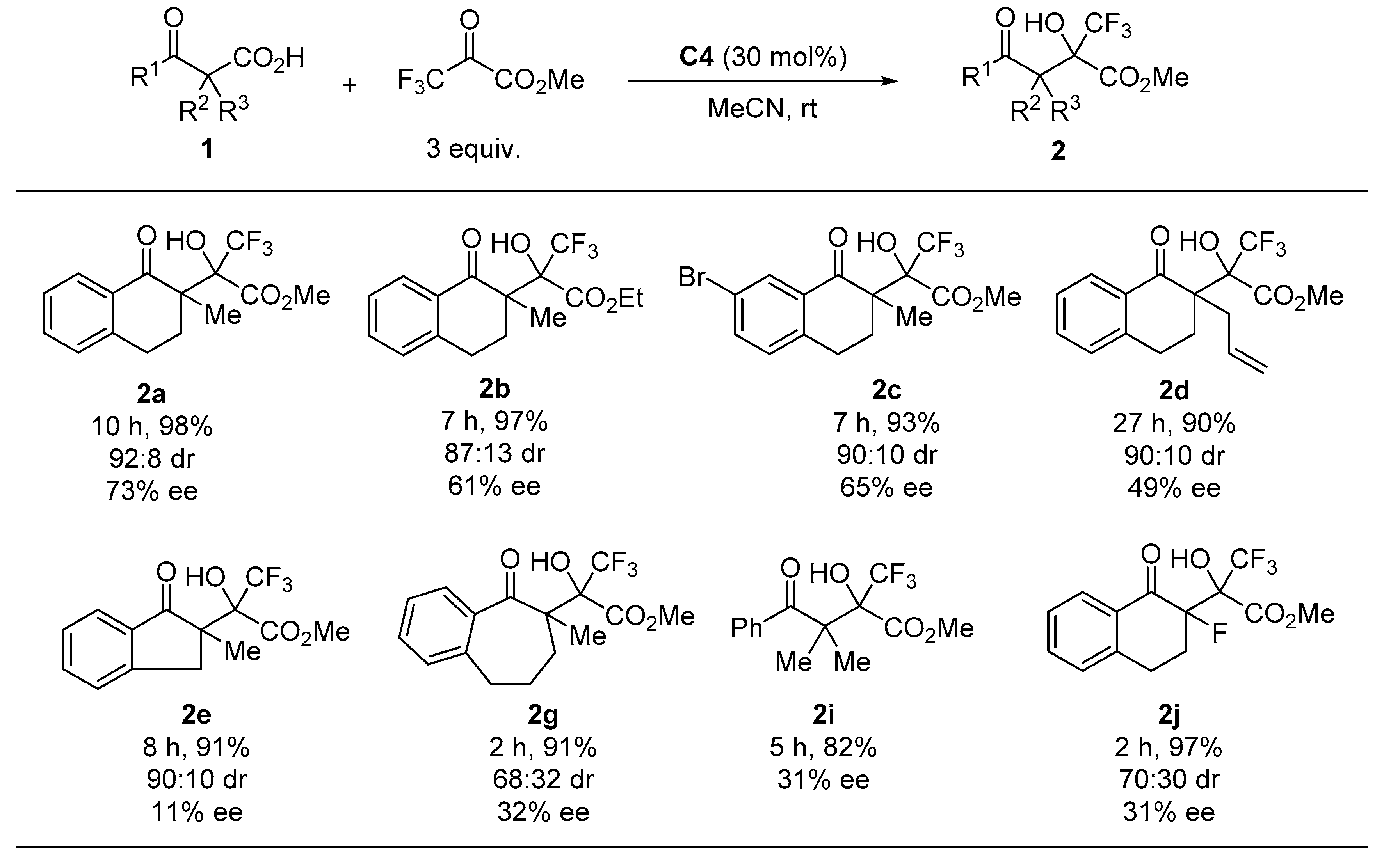
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. Materials
3.3. Synthesis of α,α-dialkyl-β-ketocarboxylic Acid 1c
3.4. General Procedure for the Decarboxylative Aldol Reaction
3.5. General Procedure of the Enantioselective Decarboxylative Aldol Reaction
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rodríguez, N.; Goossen, L.J. Decarboxylative coupling reactions: A modern strategy for C-C-bond formation. Chem. Soc. Rev. 2011, 40, 5030–5048. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Tan, C.-H. Catalytic decarboxylative reactions: Biomimetic approaches inspired by polyketide biosynthesis. Synthesis 2011, 13, 2044–2053. [Google Scholar] [CrossRef]
- Dzik, W.I.; Lange, P.P.; Gooßen, L.J. Carboxylates as sources of carbon nucleophiles and electrophiles: Comparison of decarboxylative and decarbonylative pathways. Chem. Sci. 2012, 3, 2671–2678. [Google Scholar] [CrossRef]
- Cornella, J.; Larrosa, I. Decarboxylative carbon-carbon bond-forming transformations of (Hetero)aromatic carboxylic acids. Synthesis 2012, 44, 653–676. [Google Scholar] [CrossRef]
- Wang, Z.-L. Recent advances in catalytic asymmetric decarboxylative addition reactions. Adv. Synth. Catal. 2013, 355, 2745–2755. [Google Scholar] [CrossRef]
- Nakamura, S. Catalytic enantioselective decarboxylative reactions using organocatalysts. Org. Biomol. Chem. 2014, 12, 394–405. [Google Scholar] [CrossRef]
- Xuan, J.; Zhang, Z.-G.; Xiao, W.J. Visible-light-induced decarboxylative functionalization of carboxylic acids and their derivatives. Angew. Chem. Int. Ed. 2015, 54, 15632–15641. [Google Scholar] [CrossRef]
- Liu, P.; Zhang, G.; Sun, P. Transition metal-free decarboxylative alkylation reactions. Org. Biomol. Chem. 2016, 14, 10763–10777. [Google Scholar] [CrossRef]
- Wei, Y.; Hu, P.; Zhang, M.; Su, W. Metal-catalyzed decarboxylative C-H functionalization. Chem. Rev. 2017, 117, 8864–8907. [Google Scholar] [CrossRef]
- Bernardi, L.; Fochi, M.; Franchini, M.; Ricci, A. Bioinspired organocatalytic asymmetric reactions. Org. Biomol. Chem. 2012, 10, 2911–2922. [Google Scholar] [CrossRef]
- Bae, H.Y. Malonic acid half thioesters (MAHTs) as efficient enolate precursors in biomimetic catalysis. Synlett 2015, 26, 705–706. [Google Scholar] [CrossRef][Green Version]
- Pedersen, K.J. The ketonic decomposition of beta-keto carboxylic acids. J. Am. Chem. Soc. 1929, 51, 2098–2107. [Google Scholar] [CrossRef]
- Pedersen, K.J. The decomposition of α-nitorocarboxylic acids. J. Phys. Chem. 1933, 38, 559–571. [Google Scholar] [CrossRef]
- Pedersen, K.J. Amine catalysis of the ketonic decomposition of α, α-dimethylacetoacetic acid. J. Am. Chem. Soc. 1938, 60, 595–601. [Google Scholar] [CrossRef]
- Westheimer, F.H.; Jones, W.A. The effect of solvent on some reaction rates. J. Am. Chem. Soc. 1941, 63, 3283–3286. [Google Scholar] [CrossRef]
- Brown, B.B.R.; Phil, D. The mechanism of thermal decarboxylation. Quartery Rev. Chem. Soc. 1950, 5, 131–146. [Google Scholar] [CrossRef]
- Swain, C.G.; Bader, R.F.W.; Esteve, R.M.; Griffine, R.N. Use of substituent effects on isotope effects to distinguish between proton and hydride transfers. part II. mechanism of decarboxylation of β-keto acids in benzene. J. Am. Chem. Soc. 1961, 83, 1951–1954. [Google Scholar] [CrossRef]
- Brower, K.R.; Gay, B.; Konkol, T.L. The volume of activation in unimolecular decomposition reactions. Decarboxylation and demercuration. J. Am. Chem. Soc. 1966, 88, 1681–1685. [Google Scholar] [CrossRef]
- Straub, T.S.; Bender, M.L.; Straub, T.S. Cycloamyloses as enzyme models. The decarboxylation of benzoylacetic acids. J. Am. Chem. Soc. 1972, 94, 8881–8888. [Google Scholar] [CrossRef]
- Louge, M.W.; Pollack, R.M.; Vitullo, V.P. The nature of the transition state for the decarboxylation of β-keto acids. J. Am. Chem. Soc. 1975, 97, 6868–6869. [Google Scholar] [CrossRef]
- Lalic, G.; Aloise, A.D.; Shair, M.D. An Exceptionally mild catalytic thioester aldol reaction inspired by polyketide biosynthesis. J. Am. Chem. Soc. 2003, 125, 2852–2853. [Google Scholar] [CrossRef] [PubMed]
- Orlandi, S.; Benaglia, M.; Cozzi, F. Cu(II)-catalyzed enantioselective aldol condensation between malonic acid hemithioesters and aldehydes. Tetrahedron Lett. 2004, 45, 1747–1749. [Google Scholar] [CrossRef]
- Magdziak, D.; Lalic, G.; Lee, H.M.; Fortner, K.C.; Aloise, A.D.; Shair, M.D. Catalytic Enantioselective thioester aldol reactions that are compatible with protic functional groups. J. Am. Chem. Soc. 2005, 127, 7284–7285. [Google Scholar] [CrossRef] [PubMed]
- Baudoux, J.; Lefebvre, P.; Legay, R.; Lasne, M.C.; Rouden, J. Environmentally benign metal-free decarboxylative aldol and Mannich reactions. Green Chem. 2010, 12, 252–259. [Google Scholar] [CrossRef]
- Yuan, J.-W.; Liu, S.-N.; Mai, W.-P. Copper-catalysed difluoroalkylation of aromatic aldehydes via a decarboxylation/aldol reaction. Org. Biomol. Chem. 2017, 15, 7654–7659. [Google Scholar] [CrossRef]
- Huang, D.-K.; Lei, Z.-L.; Zhu, Y.-J.; Liu, Z.-J.; Hu, X.-J.; Mao, H.-F. An expedient synthesis of α, α-difluoro-β-hydroxy ketones via decarboxylative aldol reaction of α, α-difluoro-β-keto acids with aldehydes. Tetrahedron Lett. 2017, 58, 3394–3397. [Google Scholar] [CrossRef]
- Tarui, A.; Oduti, M.; Shinya, S.; Sato, K.; Omote, M. Decarboxylative aldol reaction of α, α-difluoro-β-ketocarboxylate salt: A facile method for generation of difluoroenolate. RSC Adv. 2018, 8, 20568–20575. [Google Scholar] [CrossRef]
- Li, Y.-L.; Wang, X.-L.; Xiao, D.; Liu, M.-Y.; Du, Y.; Deng, J. Organocatalytic biomimetic decarboxylative aldol reaction of fluorinated β-keto acids with unprotected isatins. Adv. Synth. Catal. 2018, 360, 4147–4152. [Google Scholar] [CrossRef]
- Shibatomi, K.; Kitahara, K.; Sasaki, N.; Kawasaki, Y.; Fujisawa, I.; Iwasa, S. Enantioselective decarboxylative chlorination of β-ketocarboxylic acids. Nat. Commun. 2017, 8, 15600. [Google Scholar] [CrossRef]
- Katada, M.; Kitahara, K.; Iwasa, S.; Shibatomi, K. Catalyst-free decarboxylative fluorination of tertiary β -keto carboxylic acids. Synlett 2018, 29, 2408–2411. [Google Scholar]
- Naruse, A.; Kitahara, K.; Iwasa, S.; Shibatomi, K. Synthesis of α-fluoroenones by elimination of α-chloro-α-fluoroketones. Asian J. Org. Chem. 2019, 8, 691–693. [Google Scholar] [CrossRef]
- Kawanishi, R.; Phongphane, L.; Iwasa, S.; Shibatomi, K. Decarboxylative fluorination of 2-pyridylacetates. Chem. Eur. J. 2019, 25, 7453–7456. [Google Scholar] [CrossRef] [PubMed]
- Gathergood, N.; Juhl, K.; Poulsen, T.B.; Thordrup, K.; Jørgensen, K.A. Direct catalytic asymmetric aldol reactions of pyruvates: Scope and mechanism. Org. Biomol. Chem. 2004, 2, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, W.; Poulsen, T.B.; Jørgensen, K.A. A versatile catalyst for asymmetric reactions of carbonyl groups working purely by activation through hydrogen bonding: Mukaiyama-aldol, hetero Diels-Alder and Friedel-Crafts reactions. Org. Biomol. Chem. 2005, 3, 3284–3289. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Shibata, N.; Inagaki, J.; Nakamura, S.; Toru, T.; Shiro, M. Cinchona-alkaloid-catalyzed enantioselective direct aldol-type reaction of oxindoles with ethyl trifluoropyruvate. Angew. Chem. Int. Ed. 2007, 46, 8666–8669. [Google Scholar] [CrossRef] [PubMed]
- Frings, M.; Atodiresei, I.; Runsink, J.; Raabe, G.; Bolm, C. Catalyzed vinylogous mukaiyama aldol reactions with controlled enantio-and diastereoselectivities. Chem. Eur. J. 2009, 15, 1566–1569. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.; Liu, X.; Zheng, K.; Li, W.; Hu, X.; Lin, L.; Feng, X. Catalytic asymmetric synthesis of 3-(α-hydroxy-β-carbonyl) oxindoles by a ScIII-catalyzed direct aldol-type reaction. Chem. Eur. J. 2010, 16, 3736–3742. [Google Scholar] [CrossRef]
- Dong, X.; Sun, J. Catalytic asymmetric α-aldol reaction of vinylogous N-heterocyclic carbene enolates: Formation of quaternary and labile tertiary stereocenters. Org. Lett. 2014, 16, 2450–2453. [Google Scholar] [CrossRef]
- Ma, J.-A.; Cahard, D.; Recherche, I.; Fine, O. Update 1 of asymmetric fluorination, trifluoromethylation, and perfluoroalkylation reactions. Chem. Rev. 2008, 108, PR1–PR43. [Google Scholar] [CrossRef]
- Nie, J.; Guo, H.; Cahard, D.; Ma, J.-A. Asymmetric construction of stereogenic carbon centers featuring a trifluoromethyl group from prochiral trifluoromethylated substrates. Chem. Rev. 2011, 111, 455–529. [Google Scholar] [CrossRef]
- Sorochinsky, A.E.; Soloshonok, V.A. Recent advances in the asymmetric synthesis of α-(Trifluoromethyl) containing α-amino acids. Synthesis 2012, 44, 1592–1602. [Google Scholar]
- Bizet, V.; Besset, T.; Ma, J.A.; Cahard, D. Recent progress in asymmetric fluorination and trifluoromethylation reactions. Curr. Top. Med. Chem. 2014, 14, 901–940. [Google Scholar] [CrossRef] [PubMed]
- Shibatomi, K. Catalytic enantioselective constructions of trifluoromethylated chiral stereogenic centers via the conjugate addition to CF3-alkenes: Overview of current progress. Curr. Org. Chem. 2015, 19, 1619–1637. [Google Scholar] [CrossRef]
- Mei, H.; Xie, C.; Han, J.; Soloshonok, V.A. Ntert -Butylsulfinyl-3 3 3-trifluoroacetaldimine: Versatile reagent for asymmetric synthesis of trifluoromethyl containing amines and amino acids of pharmaceutical. Eur. J. Org. Chem. 2016, 2016, 5917–5932. [Google Scholar] [CrossRef]
- Kluger, R.; Brandl, M. β-Deuterium secondary isotope effects in heterolytic decarboxylation reactions. manifestations of negative hyperconjugation. J. Org. Chem. 1986, 51, 3964–3968. [Google Scholar] [CrossRef]
- Dummer, N.F.; Jenkins, R.; Li, X.; Bawaked, S.M.; McMorn, P.; Burrows, A.; Kiely, C.J.; Wells, R.P.K.; Willock, D.J.; Hutchings, G.J. Inversion of enantioselectivity for the hydrogenation of ethyl pyruvate in the gas-phase over Pt/SiO2 modified with derivatives of hydroquinidine. J. Catal. 2006, 243, 165–170. [Google Scholar] [CrossRef]
- Yoneda, N.; Fujii, Y.; Matsumoto, A.; Asano, K.; Matsubara, S. Organocatalytic enantio and diastereoselective cycloetherification via dynamic kinetic resolution of chiral cyanohydrins. Nat. Commun. 2017, 8, 1397. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Catalyst | Solvent | Time (h) | Yield (%) 2 | dr 3 | |
|---|---|---|---|---|---|---|
| 2a | 3a | |||||
| 1 | none | toluene | 4 | <1 | 20 | 66:33 |
| 2 | PhCH2CH2NH2 | toluene | 4 | 2 | 10 | 55:45 |
| 3 | (PhCH2)2NH | toluene | 4 | 58 | 10 | 92:8 |
| 4 |  | toluene | 4 | 19 | 12 | 88:12 |
| 5 | pyridine | toluene | 4 | 6 | 4 | 68:32 |
| 6 | DMAP | toluene | 4 | 39 | 9 | 82:18 |
| 7 | Et3N | toluene | 4 | 96 | 1 | 86:14 |
| 8 |  (quinuclidine) | toluene | 2 | 98 | 1 | 93:7 |
| 9 |  (DABCO) | toluene | 2 | 94 | 3 | 92:8 |
| 10 4 | DABCO | toluene | 8 | 92 | 3 | 94:6 |
| 11 4 | DABCO | MeCN | 2 | 62 | 10 | 71:29 |
| 12 4 | DABCO | Et2O | 2 | 47 | 48 | 74:26 |
| 13 4 | DABCO | CH2Cl2 | 2 | 57 | 9 | 84:16 |
| 14 4 | DABCO | MeOH | 2 | 0 | 55 | – |

| Entry | Catalyst | Solvent | Time (h) | Yield (%) 2 | dr 3 | %ee 4 (Major/Minor) | |
|---|---|---|---|---|---|---|---|
| 2a | 3a | ||||||
| 1 | C1 | toluene | 31 | 25 | 50 | 73:27 | 0/0 |
| 2 | C2 | toluene | 10 | 99 | 0 | 92:8 | –41/–28 |
| 3 | C3 | toluene | 7 | 91 | 6 | 93:7 | –33/–22 |
| 4 | C4 | toluene | 9 | 89 | 0 | 94:6 | 50/33 |
| 5 | C5 | toluene | 3 | 80 | 10 | 94:6 | 44/31 |
| 6 | C6 | toluene | 5 | 88 | 3 | 90:10 | –45/–63 |
| 7 | C7 | toluene | 33 | 40 | 53 | 74:26 | –2/0 |
| 8 | C8 | toluene | 1 | 83 | 10 | 92:8 | 5/44 |
| 9 | C9 | toluene | 0.5 | 96 | 4 | 94:6 | 33/58 |
| 10 | C4 | THF | 4 | 99 | 0 | 93:7 | 57/35 |
| 11 | C4 | CH2Cl2 | 7 | 89 | 8 | 92:8 | 65/32 |
| 12 | C4 | MeCN | 10 | 98 | 0 | 92:8 | 73/42 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawanishi, R.; Hattori, S.; Iwasa, S.; Shibatomi, K. Amine-Catalyzed Decarboxylative Aldol Reaction of β-Ketocarboxylic Acids with Trifluoropyruvates. Molecules 2019, 24, 2773. https://doi.org/10.3390/molecules24152773
Kawanishi R, Hattori S, Iwasa S, Shibatomi K. Amine-Catalyzed Decarboxylative Aldol Reaction of β-Ketocarboxylic Acids with Trifluoropyruvates. Molecules. 2019; 24(15):2773. https://doi.org/10.3390/molecules24152773
Chicago/Turabian StyleKawanishi, Ryouta, Shinya Hattori, Seiji Iwasa, and Kazutaka Shibatomi. 2019. "Amine-Catalyzed Decarboxylative Aldol Reaction of β-Ketocarboxylic Acids with Trifluoropyruvates" Molecules 24, no. 15: 2773. https://doi.org/10.3390/molecules24152773
APA StyleKawanishi, R., Hattori, S., Iwasa, S., & Shibatomi, K. (2019). Amine-Catalyzed Decarboxylative Aldol Reaction of β-Ketocarboxylic Acids with Trifluoropyruvates. Molecules, 24(15), 2773. https://doi.org/10.3390/molecules24152773