
A New Heterogeneous Catalyst Obtained via Supramolecular Decoration of Graphene with a Pd2+ Azamacrocyclic Complex

 , , ,
, , ,  , ,
, ,
Abstract
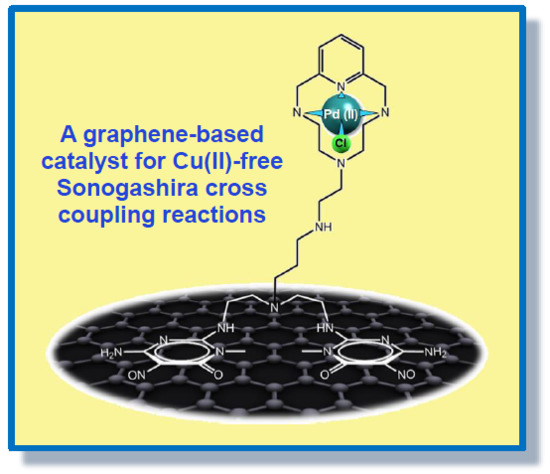
:
1. Introduction
2. Results and Discussion
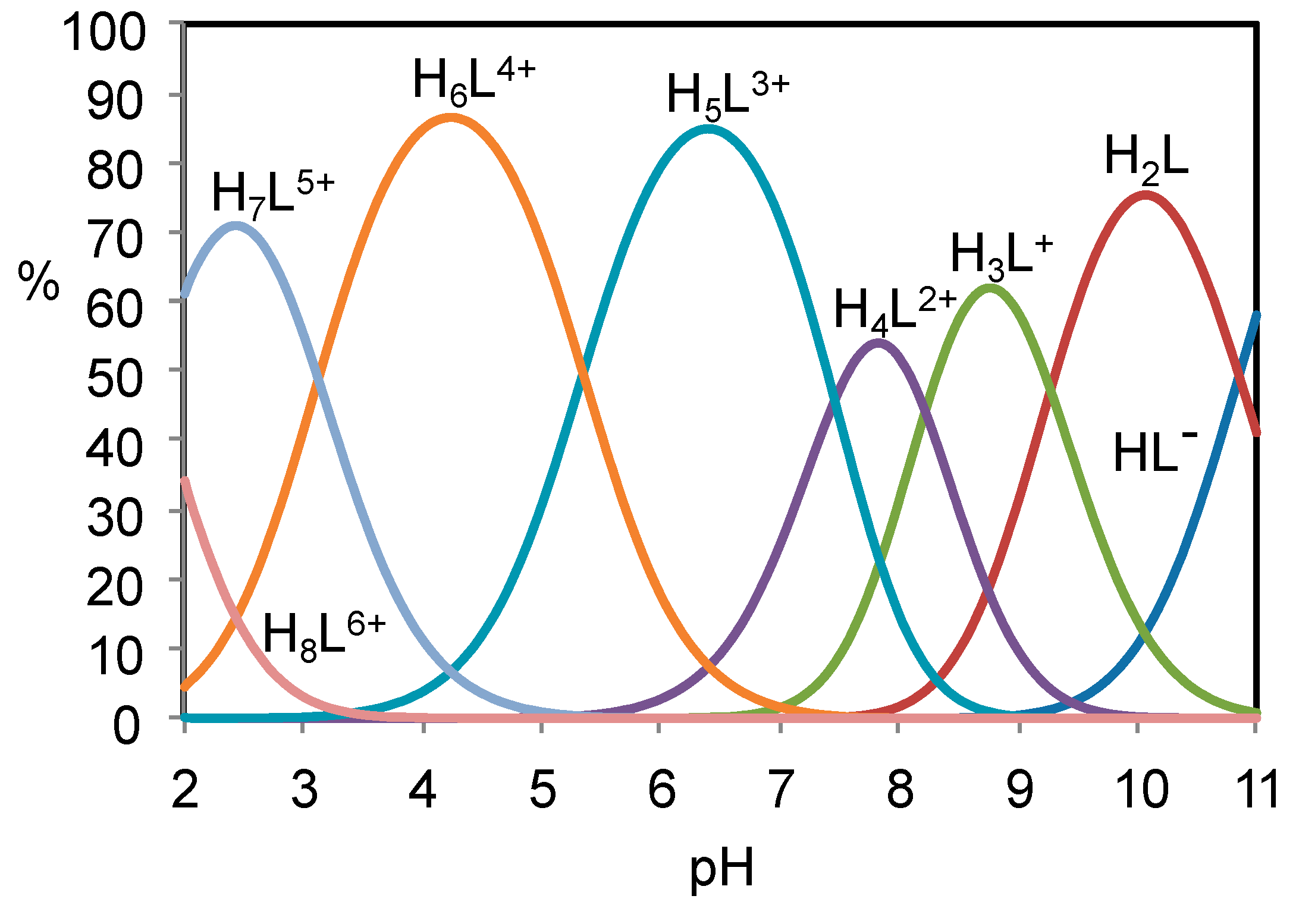
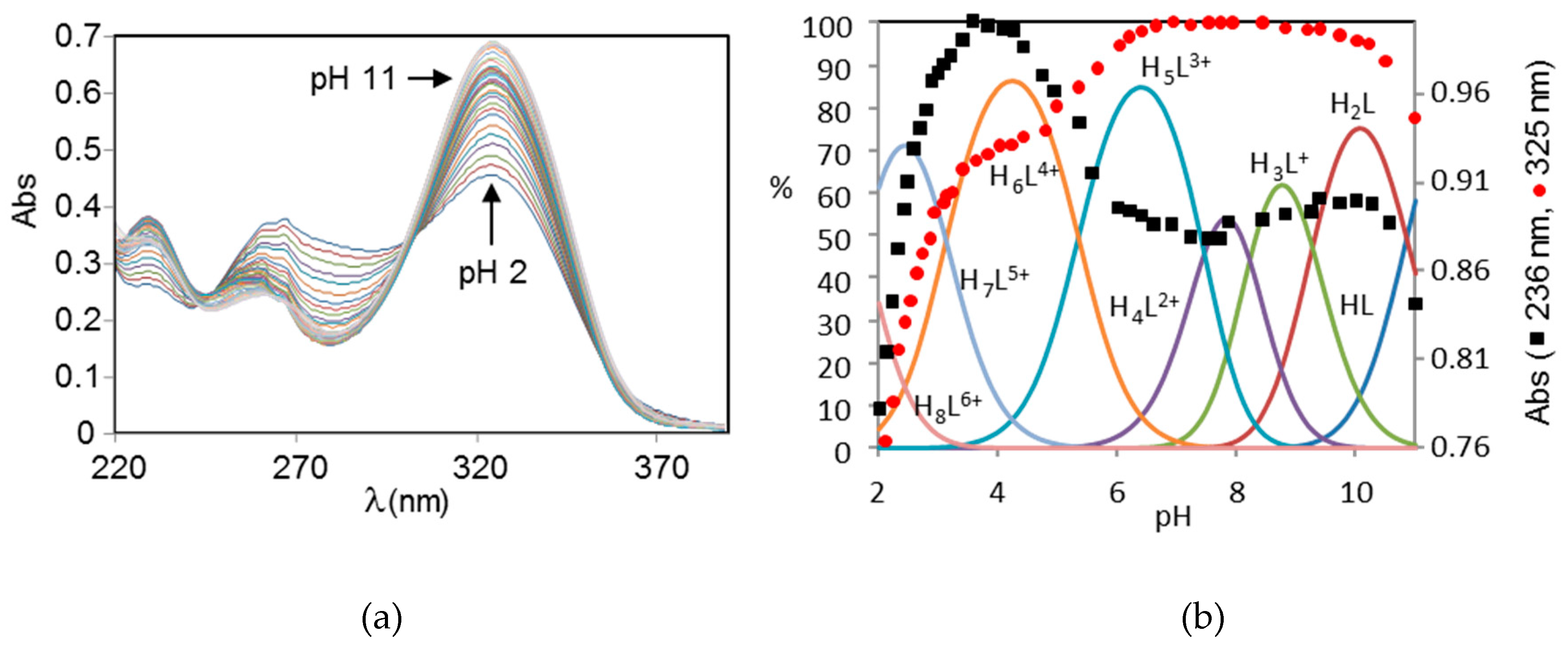
2.1. Acid-Base Properties of H2L
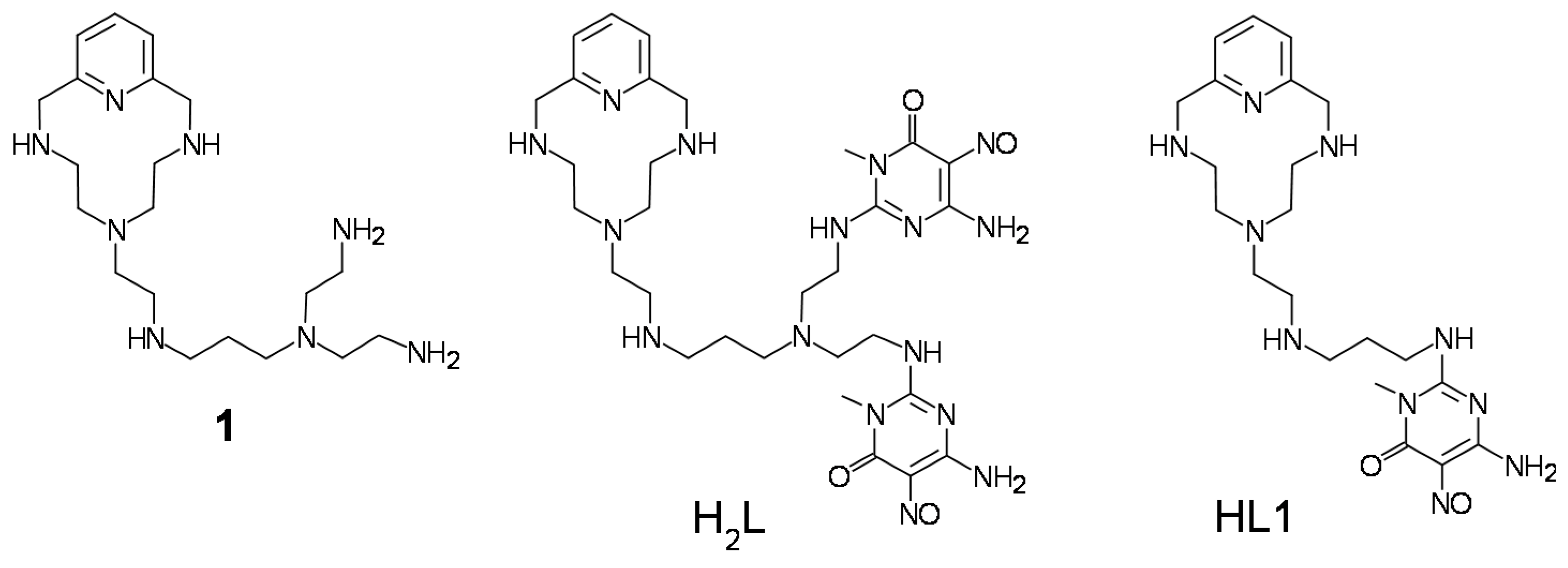
2.2. Formation of Pd(II) Complexes in Solution
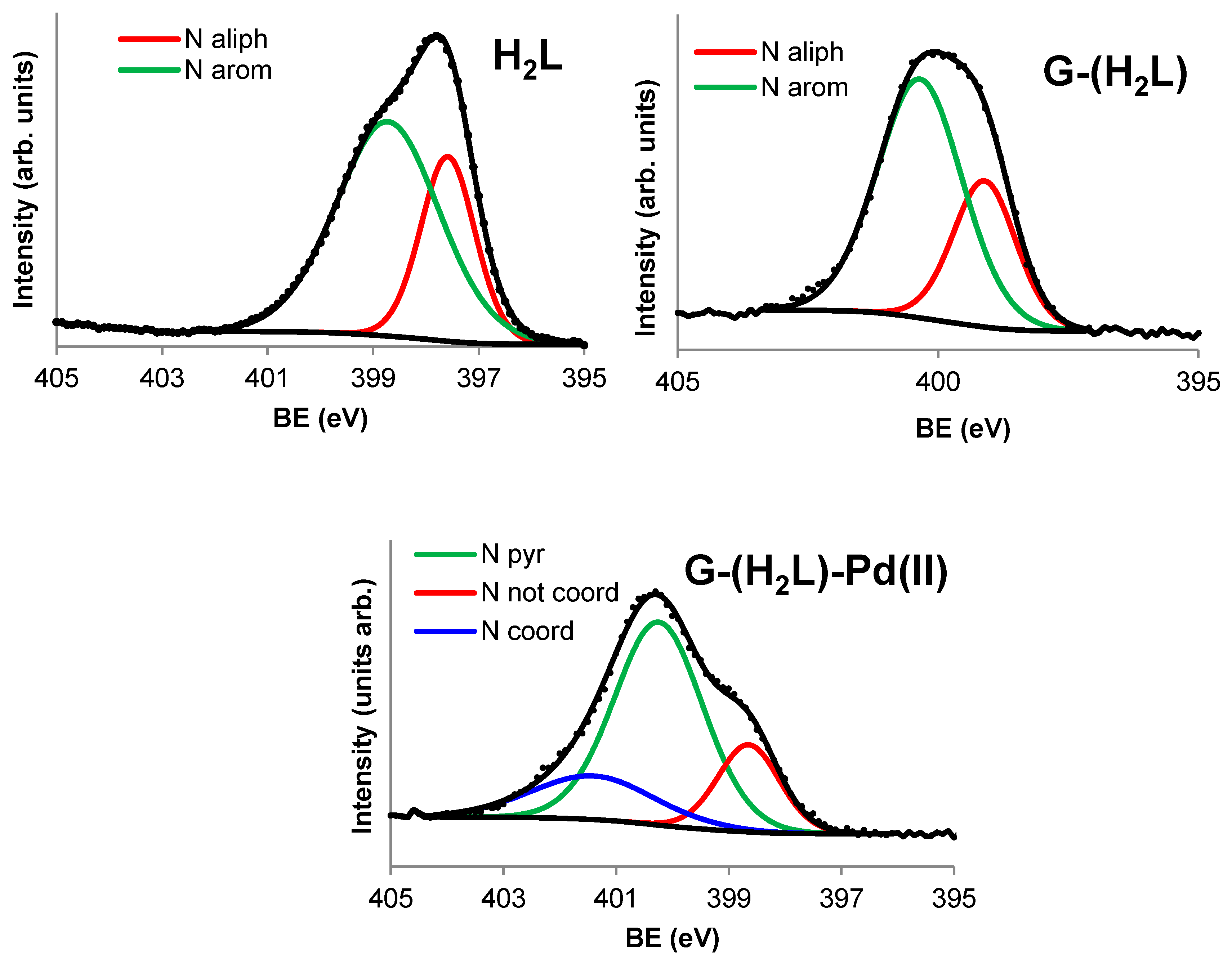
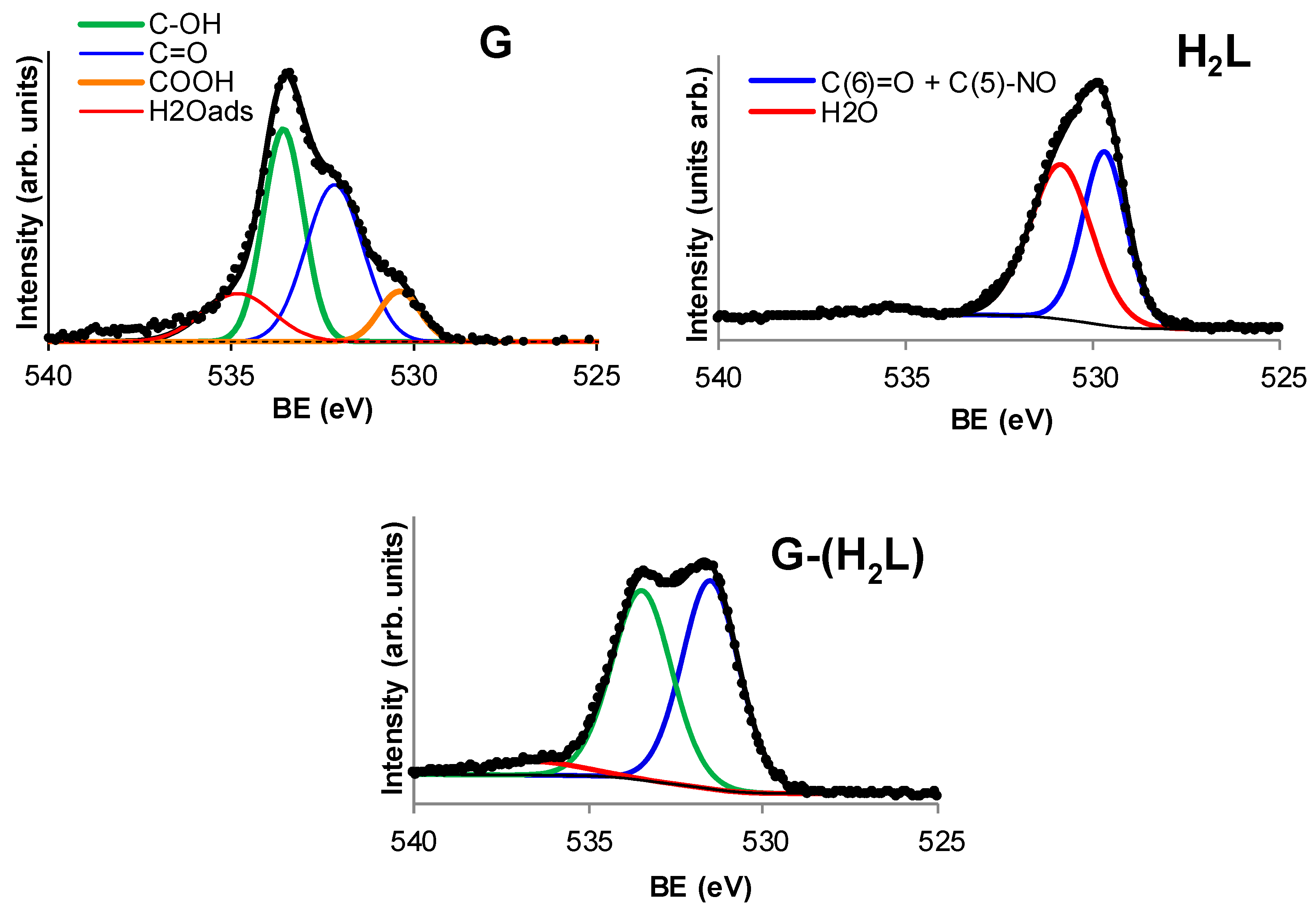
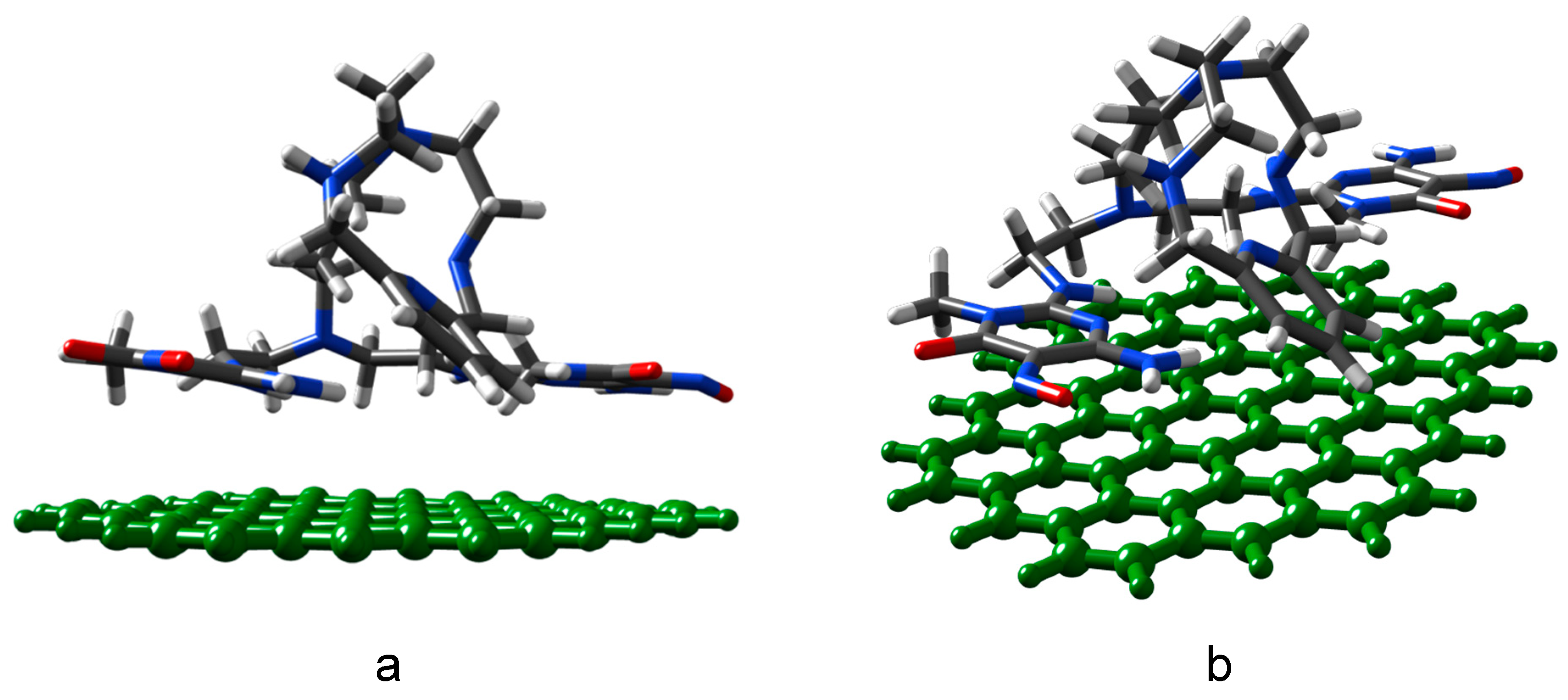
2.3. Preparation and Characterization of the G-(H2L) Hybrid
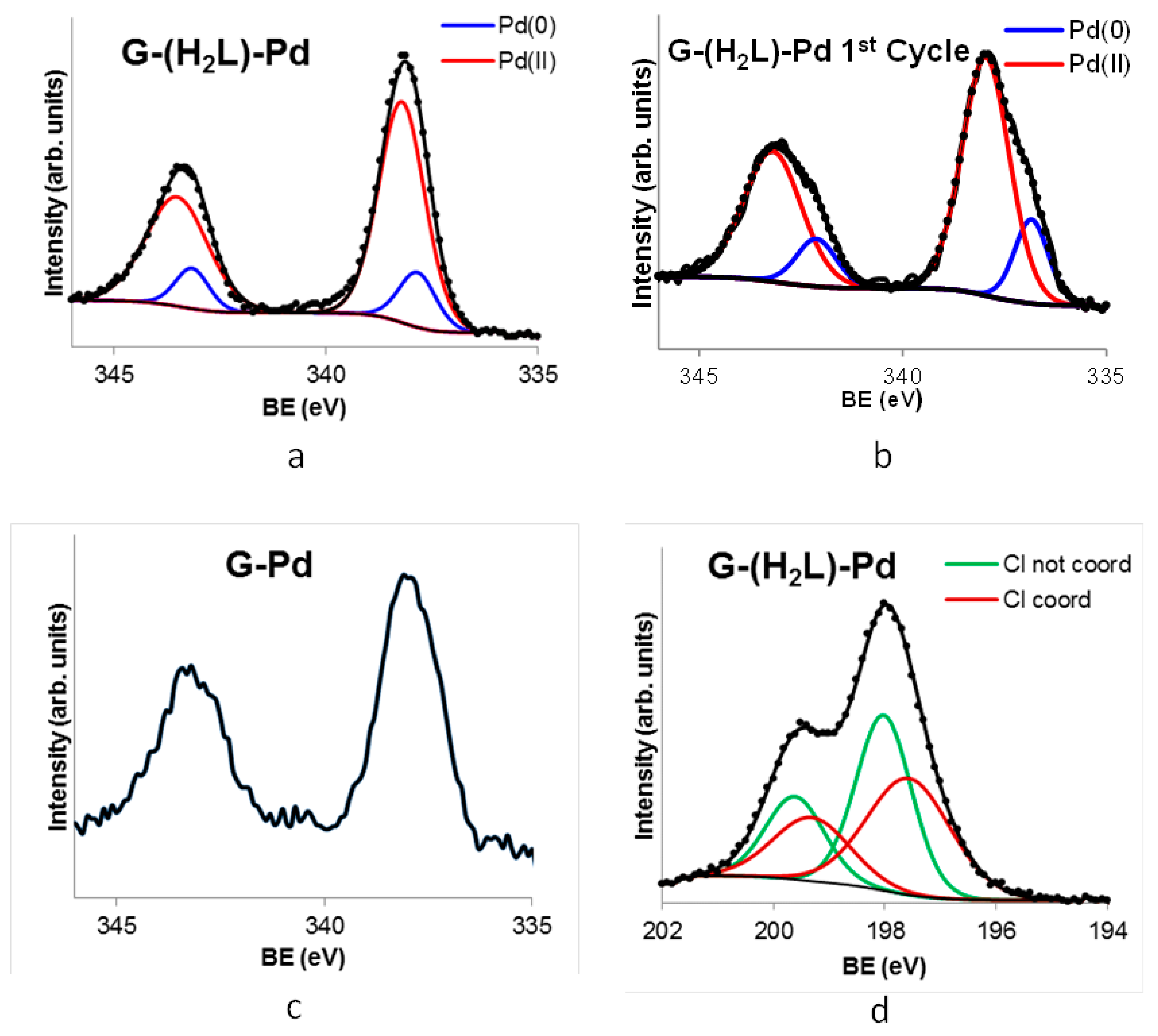
2.4. Preparation and Characterization of the G-(H2L)-Pd Hybrid
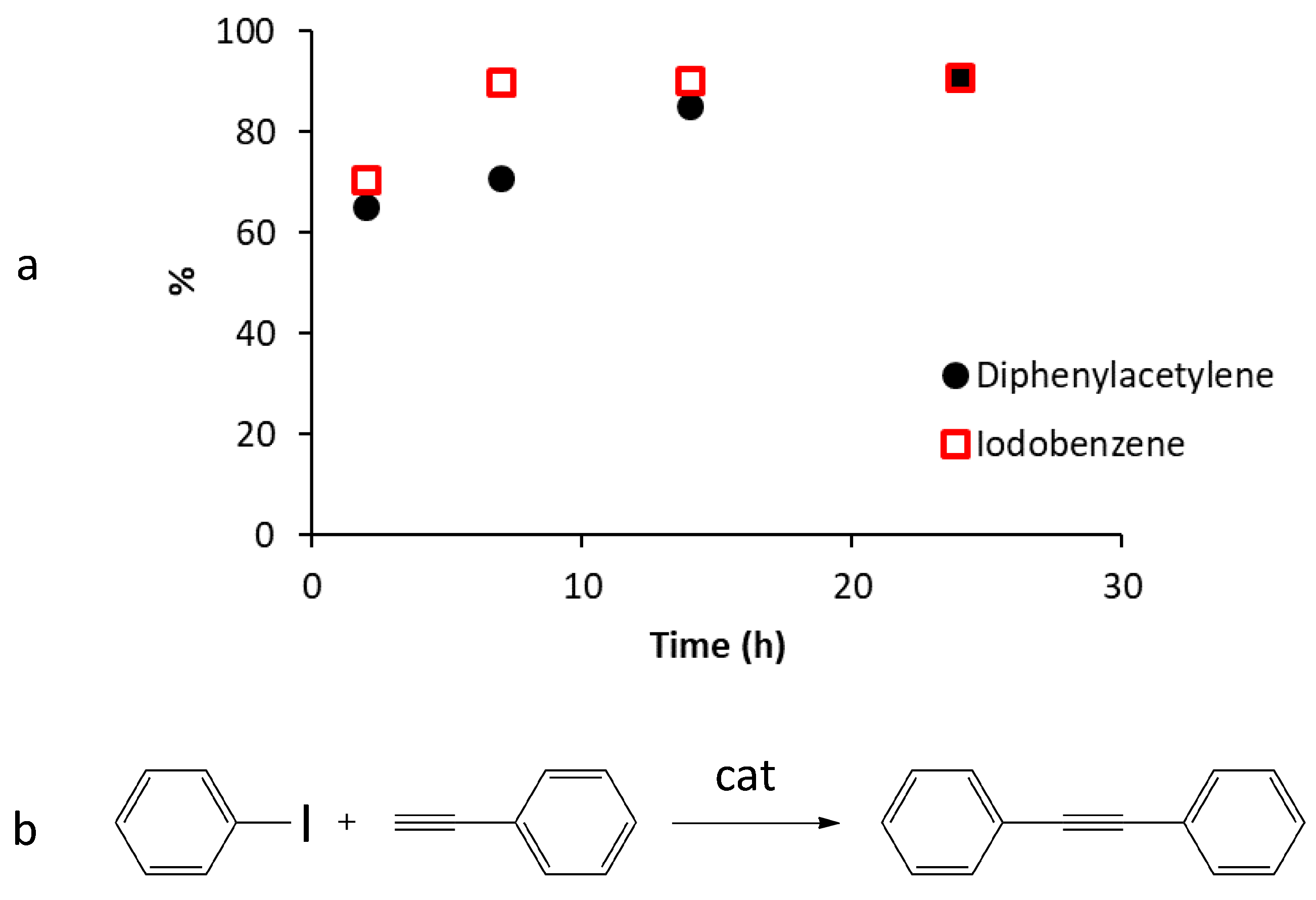
2.5. Catalytic Activity of G-(H2L)-Pd in the Sonogashira Carbon-Carbon Coupling Reaction
3. Materials and Methods
3.1. General
3.2. Synthesis of the Ligand
3.3. Synthesis of [Pd2(H2L)Cl4]∙(CH3OH)2∙2.5H2O
3.4. Potentiometric Measurements
3.5. Preparation of the Catalyst G-(H2L)-Pd
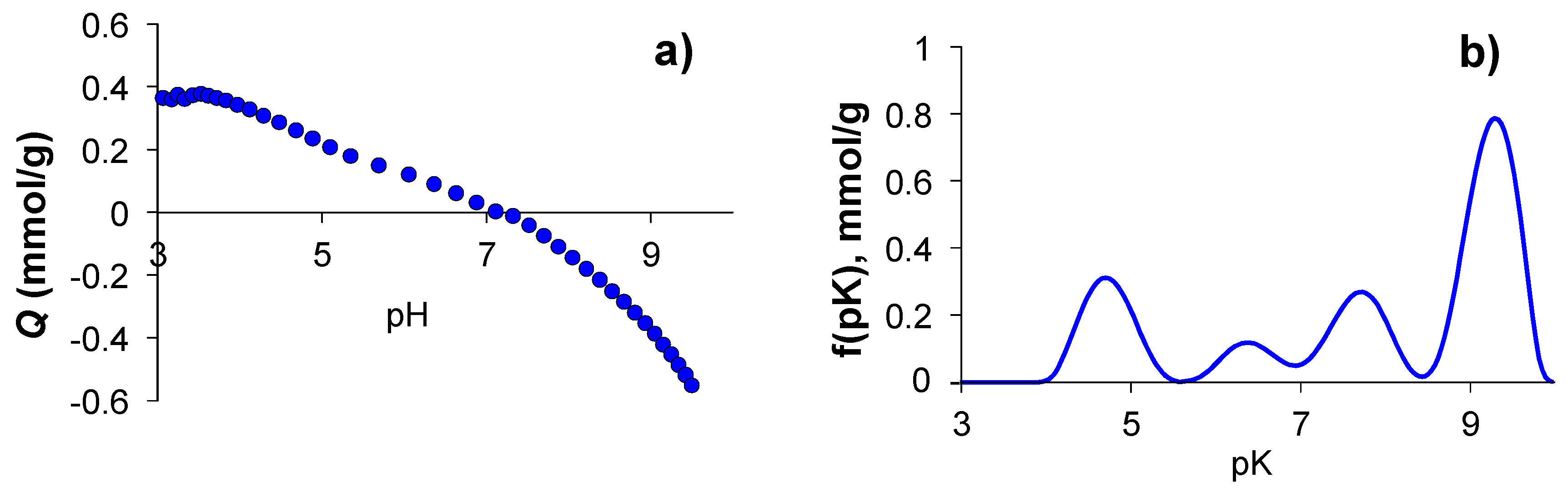
3.6. Determination of the Surface Charge Density of G-(H2L)
3.7. Spectroscopic Measurements
3.8. Elemental Analyses of G-Based Solids
3.9. General Procedure for the Sonogashira Reaction
3.10. TEM Micrographs
3.11. Theoretical Calculations of Optimum Geometry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Johansson Seechurn, C.C.C.; Kitching, M.O.; Colacot, T.J.; Snieckus, V. Palladium-catalyzed cross-coupling: A historical contextual perspective to the 2010 Nobel Prize. Angew. Chem. Int. Ed. 2012, 51, 5062–5085. [Google Scholar] [CrossRef] [PubMed]
- The Nobel Prize in Chemistry 2010—Advanced Information. Nobel Media AB 2014. Available online: https://www.nobelprize.org/ (accessed on 13 December 2016).
- Gholinejad, M.; Naghshbandi, Z.; Nájera, C. Carbon-derived supports for palladium nanoparticles as catalysts for carbon-carbon bonds formation. ChemCatChem 2019, 11, 1792–1823. [Google Scholar] [CrossRef]
- Campisciano, V.; Gruttadauria, M.; Giacalone, F. Modified nanocarbons for catalysis. ChemCatChem 2019, 11, 90–133. [Google Scholar] [CrossRef]
- Serp, P.; Machado, B. Nanostructured Carbon Materials for Catalysis; RSC Catalysis Series No. 23; The Royal Society of Chemistry: Cambridge, UK, 2015; pp. 370–375. [Google Scholar]
- Yin, L.; Liebscher, J. Carbon−carbon coupling reactions catalyzed by heterogeneous palladium catalysts. Chem. Rev. 2007, 107, 133–173. [Google Scholar] [CrossRef] [PubMed]
- Fihri, A.; Bouhrara, M.; Nekoueishahraki, B.; Basset, J.-M.; Polshettiwar, V. Nanocatalysts for Suzuki cross-coupling reactions. Chem. Soc. Rev. 2011, 40, 5181–5203. [Google Scholar] [CrossRef] [PubMed]
- Guerra, J.; Herrero, M.A. Hybrid materials based on Pd nanoparticles on carbon nanostructures for environmentally benign C–C coupling chemistry. Nanoscale 2010, 2, 1390–1400. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Zhu, Y.; Sun, Z.; Li, Y.; Zhang, G.; Zhang, F.; Fan, X. Combining palladium complex and organic amine on graphene oxide for promoted Tsuji–Trost allylation. J. Mater. Chem. A 2015, 3, 2609–2616. [Google Scholar] [CrossRef]
- López-Garzón, R.; Godino-Salido, M.L.; Gutíerrez-Valero, M.D.; Arranz-Mascarós, P.; Melguizo, M.; García, C.; Domingo-García, M.; López-Garzón, F.J. Supramolecular assembling of molecular ion-ligands on graphite-based solid materials directed to specific binding of metal ions. Inorg. Chim. Acta 2014, 417, 208–221. [Google Scholar] [CrossRef]
- Godino-Salido, M.L.; Gutiérrez-Valero, M.D.; López-Garzón, R.; Arranz-Mascarós, P.; Santiago-Medina, A.; Melguizo, M.; Domingo-García, M.; López-Garzón, F.J.; Abdelkader-Fernández, V.K.; Salinas-Martínez De Lecea, C.; et al. New hybrid materials based on the grafting of Pd(II)-amino complexes on the graphitic surface of AC: Preparation, structures and catalytic properties. RSC Adv. 2016, 6, 58247–58259. [Google Scholar] [CrossRef]
- Godino-Salido, M.L.; Santiago-Medina, A.; López-Garzón, R.; Gutiérrez-Valero, M.D.; Arranz-Mascarós, P.; López de la Torre, M.D.; Domingo-García, M.; López-Garzón, F.J. Preparation and characterization of trihydroxamic acid functionalizedcarbon materials for the removal of Cu(II) ions from aqueous solution. Appl. Surf. Sci. 2016, 387, 128–138. [Google Scholar] [CrossRef]
- García-Martín, J.; Godino-Salido, M.L.; López-Garzón, R.; Gutiérrez-Valero, M.D.; Arranz-Mascarós, P.; Stoeckli-Evans, H. Adsorption of metal ions on an activated carbon/l-lysine derivative hybrid compouds. Eur. J. Inorg. Chem. 2008, 1095–1106. [Google Scholar] [CrossRef]
- Gutiérrez-Valero, M.D.; Godino-Salido, M.L.; Arranz-Mascarós, P.; López-Garzón, R.; Cuesta, R.; García-Martín, J. Adsorption of designed pyrimidine derivative ligands on an activated carbon for the removal of Cu(II) ions from aqueous solution. Langmuir 2007, 23, 5995–6003. [Google Scholar] [CrossRef] [PubMed]
- García-Martín, J.; López-Garzón, R.; Godino-Salido, M.L.; Gutiérrez-Valero, M.D.; Arranz-Mascarós, P.; Cuesta, R.; Carrasco-Marín, F. Ligand adsorption on an activated carbon for the removal of chromate ions from aqueous solutions. Langmuir 2005, 21, 6908–6914. [Google Scholar] [CrossRef] [PubMed]
- García-Martín, J.; López-Garzón, R.; Godino-Salido, M.L.; Cuesta-Martos, R.; Gutiérrez-Valero, M.D.; Arranz-Mascarós, P.; Stoeckli-Evans, H. Adsorption of Zn2+ and Cd2+ from aqueous solution onto a carbon sorbent containing a pyrimidine–polyamine conjugate as ion receptor. Eur. J. Inorg. Chem. 2005, 2005, 3093–3103. [Google Scholar] [CrossRef]
- Savastano, M.; Arranz-Mascarós, P.; Bazzicalupi, C.; Bianchi, A.; Giorgi, C.; Godino-Salido, M.L.; Gutiérrez-Valero, M.D.; López-Garzón, R. Binding and removal of octahedral, tetrahedral, square planar and linear anions in water by means of activated carbon functionalized with a pyrimidine-based anion receptor. RSC Adv. 2014, 4, 58505–58513. [Google Scholar] [CrossRef]
- Arranz, P.; Bianchi, A.; Cuesta, R.; Giorgi, C.; Godino, M.L.; Gutiérrez, M.D.; López, R.; Santiago, A. Binding and removal of sulfate, phosphate, arsenate, tetrachloromercurate and chromate in aqueous solution by means of an activated carbon functionalized with a pyrimidine-based anion receptor (HL). Crystal structures of [H3L(HgCl4)]·H2O and [H3L(HgBr4)]·H2O showing anion−π interactions. Inorg. Chem. 2010, 49, 9321–9332. [Google Scholar] [PubMed]
- Passaponti, M.; Savastano, M.; Clares, M.P.; Inclán, M.; Lavacchi, M.; Bianchi, A.; García-España, E.; Innocenti, M. MWCNTs-Supported Pd(II) complexes with high catalytic efficiency in oxygen reduction reaction in alkaline media. Inorg. Chem. 2018, 57, 14484–14488. [Google Scholar] [CrossRef] [PubMed]
- Savastano, M.; Arranz-Mascarós, P.; Bazzicalupi, C.; Clares, M.P.; Godino-Salido, M.L.; Gutiérrez-Valero, M.D.; Inclán, M.; Bianchi, A.; García-España, E.; López-Garzón, R. Construction of green nanostructured heterogeneous catalysts via non-covalent surface decoration of multi-walled carbon nanotubes with Pd(II) complexes of azamacrocycles. J. Catal. 2017, 353, 239–249. [Google Scholar] [CrossRef]
- Savastano, M.; Arranz-Mascarós, P.; Bazzicalupi, C.; Clares, M.P.; Godino-Salido, M.L.; Guijarro, L.; Gutiérrez-Valero, M.D.; Bianchi, A.; García-Espana, E.; López-Garzón, R. Polyfunctional Tetraaza-Macrocyclic Ligands: Zn(II), Cu(II) Binding and Formation of Hybrid Materials with Multiwalled Carbon Nanotubes. ACS Omega 2017, 2, 3868–3877. [Google Scholar] [CrossRef]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef]
- Berger, C.; Song, Z.; Li, X.; Wu, X.; Brown, N.; Naud, C.; Mayou, D.; Li, T.; Hass, J.; Marchenkov, A.N.; et al. Electronic confinement and coherence in patterned epitaxial graphene. Science 2006, 312, 1191–1196. [Google Scholar] [CrossRef] [PubMed]
- Deng, D.; Novoselov, K.S.; Fu, Q.; Zheng, N.; Tian, Z.; Bao, X. Catalysis with two-dimensional materials and their heterostructures. Nat. Nanotechnol. 2016, 11, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Deng, D.; Yu, L.; Pan, X.; Wang, S.; Chen, X.; Hu, P.; Sun, L.; Bao, X. Size effect of graphene on electrocatalytic activation of oxygen. Chem. Commun. 2011, 47, 10016–10018. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, K.; Tao, F.; Stacchiola, D.J.; Hu, Y.H. 3D honeycomb-like structured graphene and its high efficiency as a counter-electrode catalyst for dye-sensitized solar cells. Angew. Chem. Int. Ed. 2013, 52, 9210–9214. [Google Scholar] [CrossRef] [PubMed]
- Parvulescu, V.I.; Garcia, H. Heterogeneous catalysis based on supramolecular association. Catal. Sci. Technol. 2018, 8, 4834–4857. [Google Scholar] [CrossRef]
- Navalon, S.; Herance, J.R.; Alvaro, M.; Garcia, H. Covalently modified graphenes in catalysis, electrocatalysis and photoresponsive materials. Chem. Eur. J. 2017, 23, 15244–15275. [Google Scholar] [CrossRef]
- Das, S.K.; Sarmah, M.; Bora, U. An ambient temperature Sonogashira cross-coupling protocol using 4-aminobenzoic acid as promoter under copper and amine free conditions. Tetrahedron Lett. 2017, 58, 2094–2097. [Google Scholar] [CrossRef]
- Karak, M.; Barbosa, L.C.; Hargaden, G.C. Recent mechanistic developments and next generation catalysts for the Sonogashira coupling reaction. RSC Adv. 2014, 4, 53442–53466. [Google Scholar] [CrossRef]
- Siemsen, P.; Livingston, R.C.; Diederich, F. Acetylenic coupling: A powerful tool in molecular construction. Angew. Chem. Int. Ed. 2000, 39, 2632–2657. [Google Scholar] [CrossRef]
- Gelman, D.; Buchwals, S. Efficient palladium-catalyzed coupling of aryl chlorides and tosylates with terminal alkynes: Use of a copper cocatalyst inhibits the reaction. Angew. Chem. Int. Ed. 2003, 42, 5993–5996. [Google Scholar] [CrossRef]
- Aufiero, M.; Proutiere, P.; Schoenebeck, F. Redox reactions in palladium catalysis: On the accelerating and/or inhibiting effects of copper and silver salt additives in cross-coupling chemistry involving electron-rich phosphine ligands. Angew. Chem. Int. Ed. 2012, 51, 7226–7230. [Google Scholar] [CrossRef] [PubMed]
- Chinchilla, R.; Nájera, C. Recent advances in Sonogashira reactions. Chem. Soc. Rev. 2011, 40, 5084–5121. [Google Scholar] [CrossRef] [PubMed]
- Arranz-Mascarós, P.; Godino-Salido, M.L.; Gutiérrez-Valero, M.D.; López-Garzón, R.; García-Gallarín, C.; Chamorro-Mena, I.; López-Garzón, F.J.; Fernández-García, E. Non-covalent functionalization of graphene to tune its band gap and to stabilize metal nanoparticles on its surface. Appl. Surf. Sci. Under review.
- Gazvoda, M.; Virant, M.; Pinter, B.; Kosmrlj, J. Mechanism of copper-free Sonogashira reaction operates through palladium-palladium transmetallation. Nat. Commun. 2018, 9, 4814. [Google Scholar] [CrossRef] [PubMed]
- Guijarro, L.; Inclán, M.; Pitarch-Jarque, J.; Doménech-Carbó, A.; Chicote, J.U.; Trefler, S.; García-España, E.; García-España, A.; Verdejo, B. Homo-and heterobinuclear Cu2+ and Zn2+ complexes of ditopic aza scorpiand ligands as superoxide dismutase mimics. Inorg. Chem. 2017, 56, 13748–13758. [Google Scholar] [CrossRef] [PubMed]
- Low, N.J.; López, M.D.; Arranz, P.; Cobo, J.; Godino, M.L.; López, R.; Gutiérrez, M.D.; Melguizo, M.; Ferguson, G.; Glidewell, C. N-(6-Amino-3, 4-dihydro-3-methyl-5-nitroso-4-oxopyrimidin-2-yl) derivatives of glycine, valine, serine, threonine and methionine: Interplay of molecular, molecular–electronic and supramolecular structures. Acta Cryst. B 2000, 56, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Bazzicalupi, C.; Bianchi, A.; Biver, T.; Giorgi, C.; Santarelli, S.; Savastano, M. Formation of double-strand dimetallic helicates with a terpyridine-based macrocycle. Inorg. Chem. 2014, 53, 12215–12224. [Google Scholar] [CrossRef] [PubMed]
- Savastano, M.; Bazzicalupi, C.; García-Gallarín, C.; Giorgi, C.; López de la Torre, M.D.; Pichierri, F.; Bianchi, A.; Melguizo, M. Halide and hydroxide anion binding in water. Dalton Trans. 2018, 47, 3329–3338. [Google Scholar] [CrossRef] [PubMed]
- Gran, G. Determination of the equivalence point in potentiometric titrations. Part II. Analyst 1952, 77, 661–671. [Google Scholar] [CrossRef]
- Gans, P.; Sabatini, A.; Vacca, A. Investigation of equilibria in solution. Determination of equilibrium constants with the HYPERQUAD suite of programs. Talanta 1996, 43, 1739–1753. [Google Scholar] [CrossRef]
- Bandosz, T.J.; Jagiello, J.; Contescu, C.; Schwarz, J.A. Characterization of the surfaces of activated carbons in terms of their acidity constant distributions. Carbon 1993, 31, 1193–1202. [Google Scholar] [CrossRef]
- Jagiełło, J.; Bandosz, T.J.; Schwarz, J.A. Carbon surface characterization in terms of its acidity constant distribution. Carbon 1994, 32, 1026–1028. [Google Scholar] [CrossRef]
- Particle Size Analysis. In Standard Operating Protocol v1.1; Dune Sciences, Inc.: Eugene, OR, USA, 2011.
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Yang, Z.; Wang, Z.; Tian, X.; Xiu, P.; Zhou, R. Amino acid analogues bind to carbon nanotube via π-π interactions: Comparison of molecular mechanical and quantum mechanical calculations. J. Chem. Phys. 2012, 136, 025103. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Log K | |
|---|---|
| HL- + H+ = H2L | 10.85(2) |
| H2L + H+ = H3L+ | 9.27(2) |
| H3L+ + H+ = H4L2+ | 8.22(2) |
| H4L2+ + H+ = H5L3+ | 7.46(3) |
| H5L3+ + H+ = H6L4+ | 5.35(1) |
| H6L4+ + H+ = H7L5+ | 3.13(1) |
| H7L5+ + H+ = H8L6+ | 1.75(1) |
| Pd(H2L)2+ + H+ = Pd(H3L)3+ | 7.30(8) |
| Pd(H3L)3+ + H+ = Pd(H4L)4+ | 5.1(1) |
| Pd(H4L)4+ + H+ = Pd(H5L)5+ | 2.7(1) |
| Pd(H5L)5+ + H+ = Pd(H6L)6+ | 2.4(1) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savastano, M.; Arranz-Mascarós, P.; Clares, M.P.; Cuesta, R.; Godino-Salido, M.L.; Guijarro, L.; Gutiérrez-Valero, M.D.; Inclán, M.; Bianchi, A.; García-España, E.; et al. A New Heterogeneous Catalyst Obtained via Supramolecular Decoration of Graphene with a Pd2+ Azamacrocyclic Complex. Molecules 2019, 24, 2714. https://doi.org/10.3390/molecules24152714
Savastano M, Arranz-Mascarós P, Clares MP, Cuesta R, Godino-Salido ML, Guijarro L, Gutiérrez-Valero MD, Inclán M, Bianchi A, García-España E, et al. A New Heterogeneous Catalyst Obtained via Supramolecular Decoration of Graphene with a Pd2+ Azamacrocyclic Complex. Molecules. 2019; 24(15):2714. https://doi.org/10.3390/molecules24152714
Chicago/Turabian StyleSavastano, Matteo, Paloma Arranz-Mascarós, Maria Paz Clares, Rafael Cuesta, Maria Luz Godino-Salido, Lluis Guijarro, Maria Dolores Gutiérrez-Valero, Mario Inclán, Antonio Bianchi, Enrique García-España, and et al. 2019. "A New Heterogeneous Catalyst Obtained via Supramolecular Decoration of Graphene with a Pd2+ Azamacrocyclic Complex" Molecules 24, no. 15: 2714. https://doi.org/10.3390/molecules24152714
APA StyleSavastano, M., Arranz-Mascarós, P., Clares, M. P., Cuesta, R., Godino-Salido, M. L., Guijarro, L., Gutiérrez-Valero, M. D., Inclán, M., Bianchi, A., García-España, E., & López-Garzón, R. (2019). A New Heterogeneous Catalyst Obtained via Supramolecular Decoration of Graphene with a Pd2+ Azamacrocyclic Complex. Molecules, 24(15), 2714. https://doi.org/10.3390/molecules24152714