Minimal Structural Changes Determine Full and Partial Nicotinic Receptor Agonist Activity for Nicotine Analogues


 , , ,
, , ,
Abstract
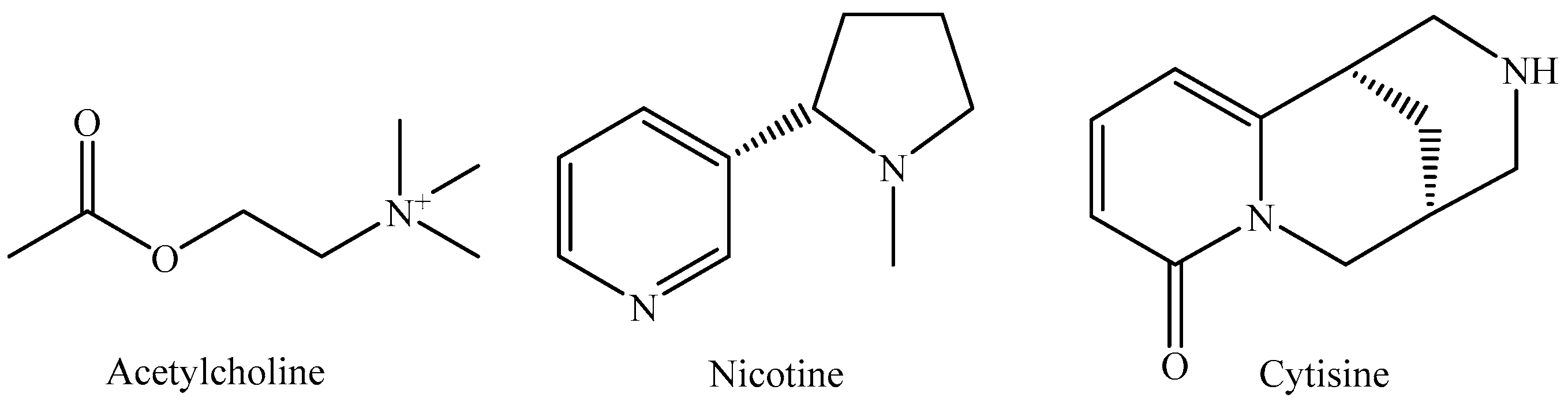
1. Introduction
2. Results and Discussion
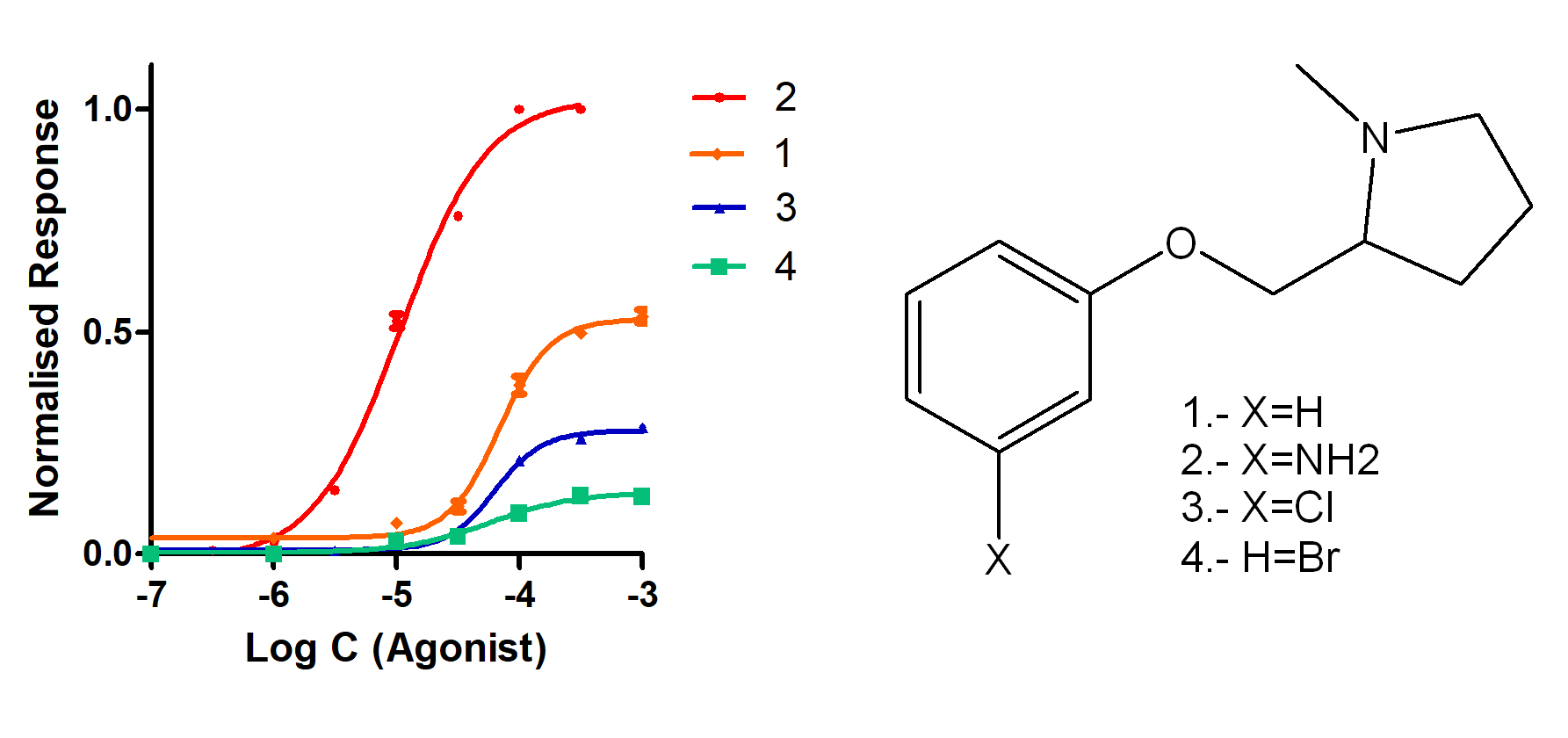
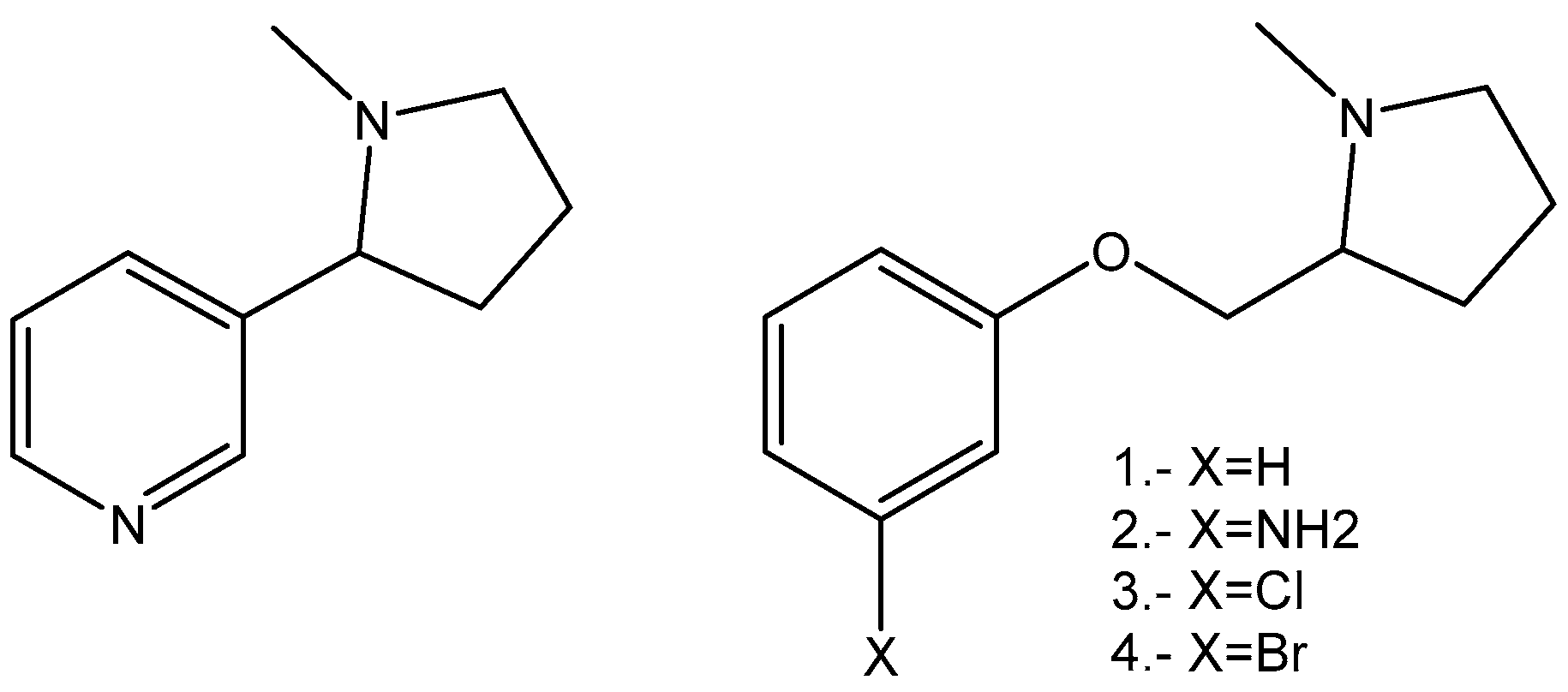
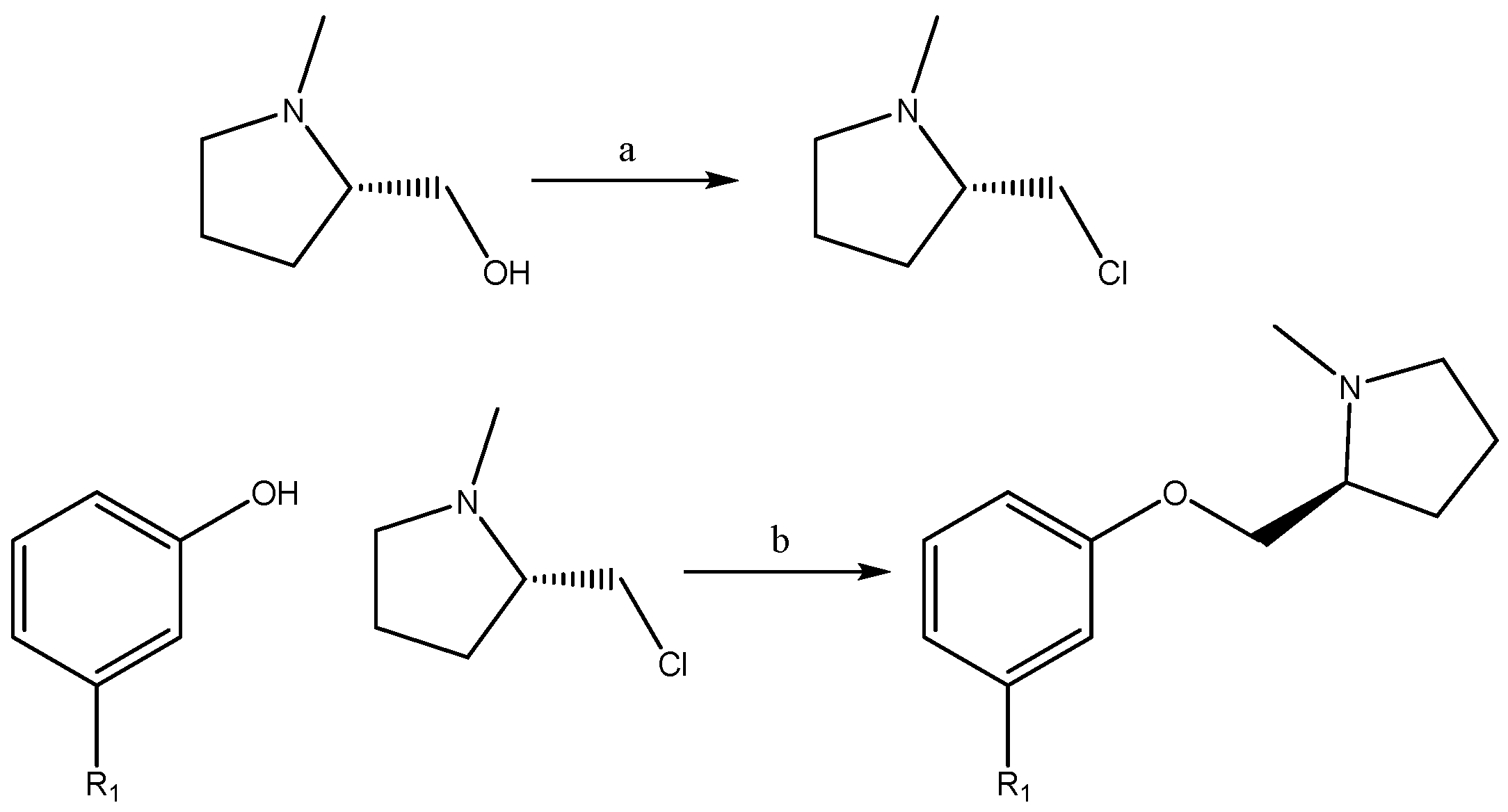
2.1. Synthesis
2.2. Biological Evaluation
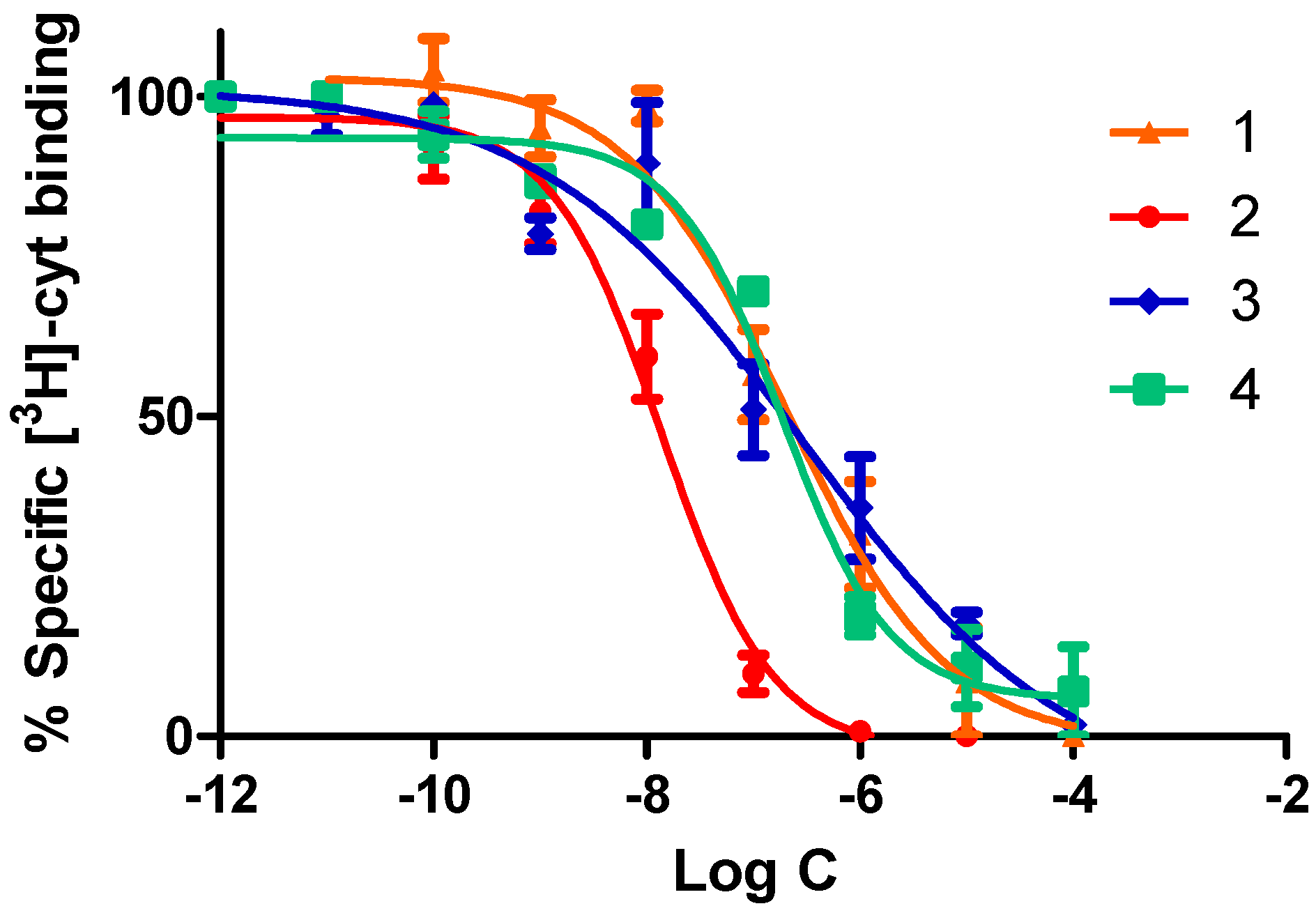
2.2.1. Competitive Radioligand Binding Studies
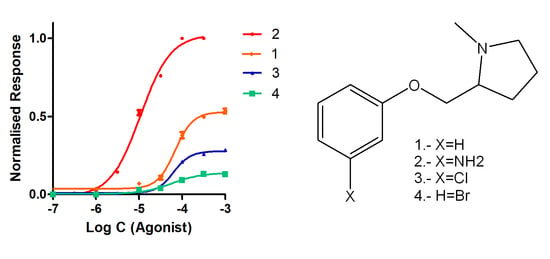
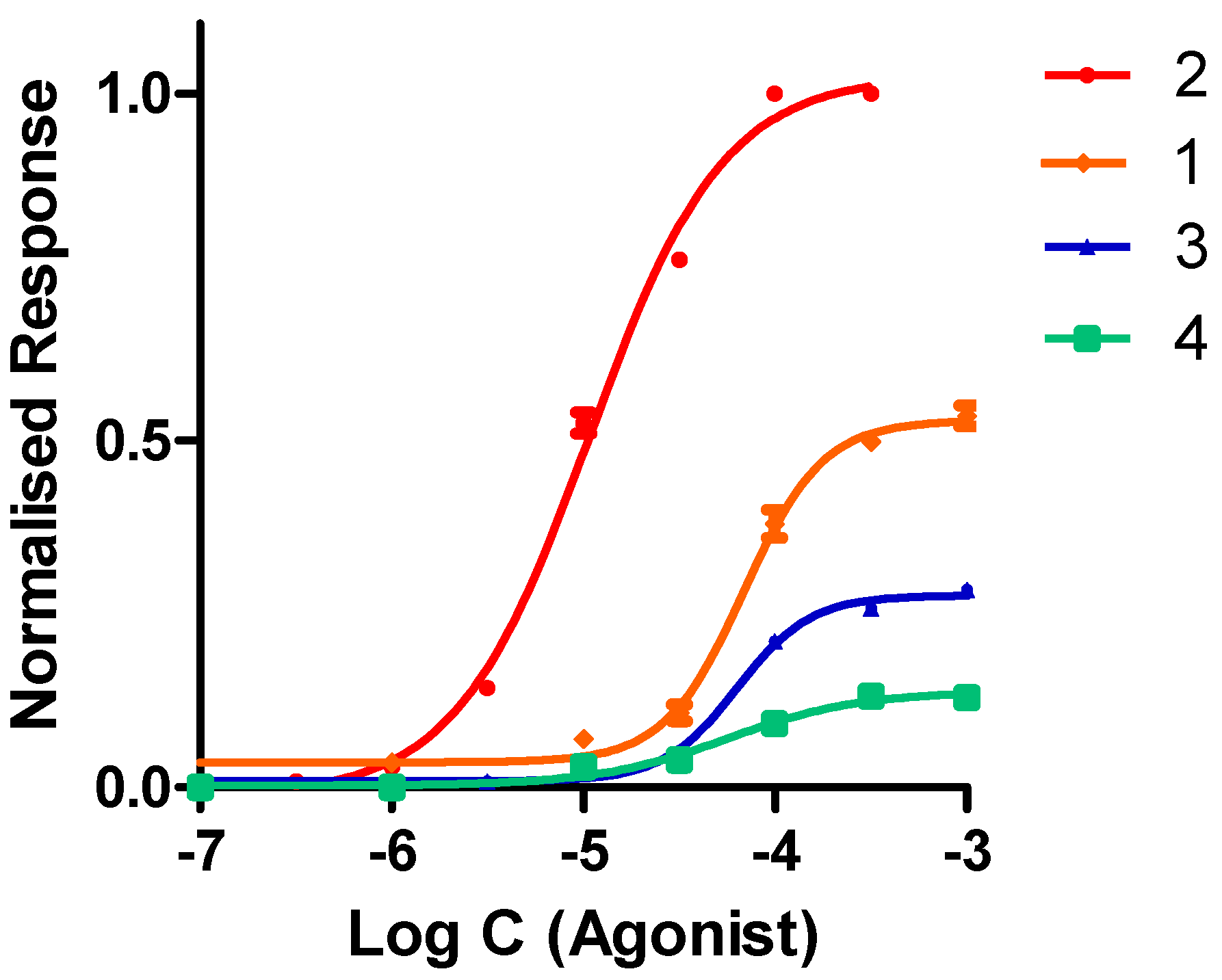
2.2.2. Functional Studies
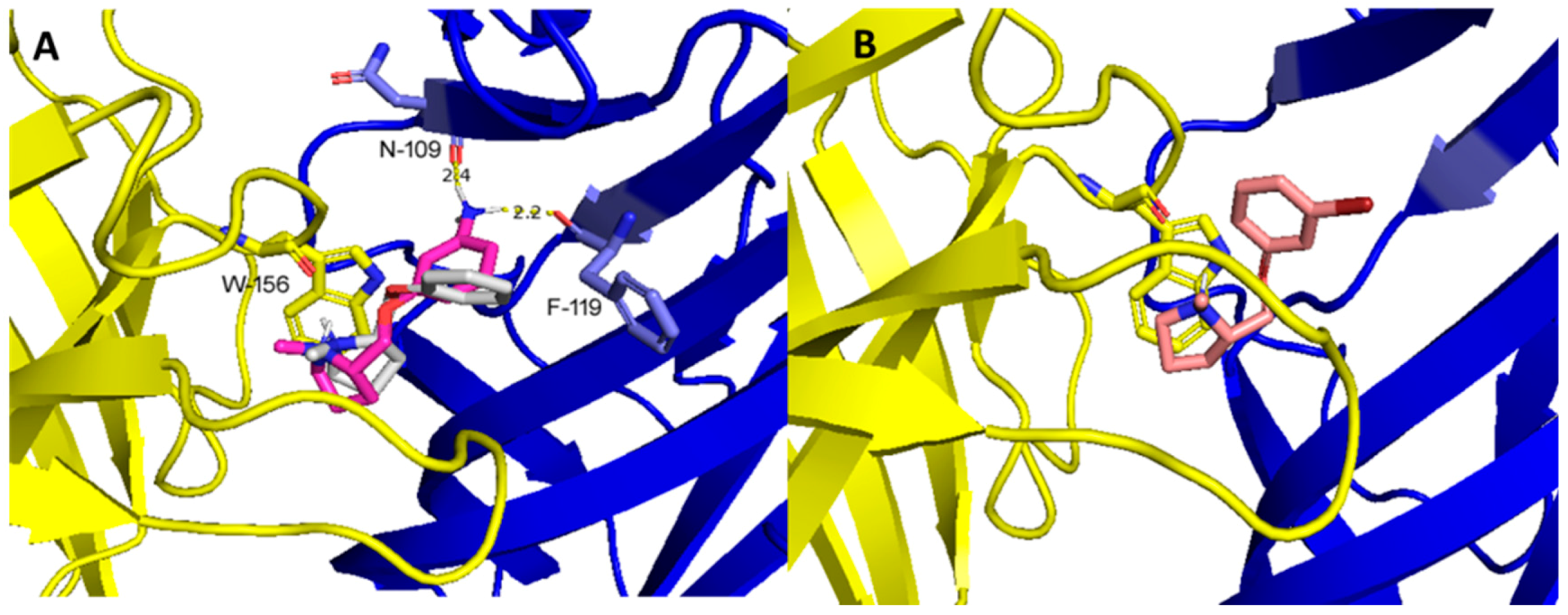
2.3. Molecular Docking Studies
3. Materials and Methods
3.1. Clonal Cell Lines
3.2. Ligand Binding Assays
3.3. Nicotinic Acetylcholine Receptors Expression in Xenopus laevis Oocytes
3.4. Electrophysiological Recordings
3.5. Molecular Docking Studies
4. Chemistry
4.1. General Procedures
4.1.1. Synthesis of (S)-2-(chloromethyl)-1-Methylpyrrolidine
4.1.2. Synthesis of (S)-1-methyl-2-(phenoxymethyl)pyrrolidine Derivatives
4.2. (S)-1-methyl-2-(phenoxymethyl)pyrrolidine Ether (1)
4.3. (S)-1-methyl-2-(3′-aminophenoxymethyl)pyrrolidine Ether (2)
4.4. (S)-1-methyl-2-(3′-Chlorophenoxymethyl)pyrrolidine Ether (3)
4.5. (S)-1-methyl-2-(3′-bromophenoxymethyl)pyrrolidine Ether (4)
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Le Novère, N.; Changeux, J.P. LGICdb: The ligand-gated ion channel database. Nucleic Acids Res. 1999, 27, 340–342. [Google Scholar] [CrossRef] [PubMed]
- Le Novère, N.; Corringer, P.J.; Changeux, J.P. The diversity of subunit composition in nAChRs: Evolutionary origins, physiologic and pharmacologic consequences. J. Neurobiol. 2002, 53, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, E.X.; Pereira, E.F.R.; Alkondon, M.; Rogers, S.W. Mammalian Nicotinic Acetylcholine Receptors: From Structure to Function. Physiol. Rev. 2009, 89, 73–120. [Google Scholar] [CrossRef] [PubMed]
- Chavez-noriega, L.E.; Crona, J.H.; Washburn, M.S.; Urrutia, A.; Elliott, K.J.; Johnson, E.C. Pharmacological Characterization of Recombinant Human Neuronal Nicotinic Acetylcholine Receptors hα2β2, hα2β4, hα3β4, hαβ2 and α7 Expressed in Xenopus Oocytes. JPET 1997, 280, 346–356. [Google Scholar]
- Moroni, M.; Zwart, R.; Sher, E.; Cassels, B.K.; Bermudez, I. α4β2 Nicotinic Receptors with High and Low Acetylcholine Sensitivity: Pharmacology, Stoichiometry, and Sensitivity to Long-Term Exposure to Nicotine. Mol. Pharmacol. 2006, 70, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Sine, S.M.; Engel, A.G. Recent advances in Cys-loop receptor structure and function. Nature 2006, 440, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.D. Nicotinic systems and cognitive function. Psychopharmacology 1992, 108, 417–431. [Google Scholar] [CrossRef]
- Marubio, L.M.; del Mar Arroyo-Jimenez, M.; Cordero-Erausquin, M.; Léna, C.; Le Novère, N.; de Kerchove d’Exaerde, V.; Huchet, M.; Damaj, M.I.; Changuex, J.P. Reduced antinociception in mice lacking neuronal nicotinic receptor subunits. Nature 1999, 398, 805–810. [Google Scholar] [CrossRef]
- Holladay, M.W.; Dart, M.J.; Lynch, J.K. Neuronal nicotinic acetylcholine receptors as targets for drug discovery. J. Med. Chem. 1997, 40, 4169–4194. [Google Scholar] [CrossRef]
- Laviolette, S.R.; Van Der Kooy, D. The neurobiology of nicotine addiction: Bridging the gap from molecules to behaviour. Nat. Rev. Neurosci. 2004, 5, 55–65. [Google Scholar] [CrossRef]
- Rollema, H.; Chambers, L.K.; Coe, J.W.; Glowa, J.; Hurst, R.S.; Lebel, L.A.; Lu, Y.; Mansbach, R.S.; Mather, R.J.; Rovetti, C.C.; et al. Pharmacological profile of the α4β2 nicotinic acetylcholine receptor partial agonist varenicline, an effective smoking cessation aid. Neuropharmacology 2007, 52, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Stober, S.T.; Abrams, C.F. Enhanced meta-analysis of acetylcholine binding protein structures reveals conformational signatures of agonism in nicotinic receptors. Protein Sci. 2002, 21, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Morales-Perez, C.L.; Noviello, C.M.; Hibbs, R.E. X-ray structure of the human α4β2 nicotinic receptor. Nature 2016, 538, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.M.; Roh, S.H.; Gharpure, A.; Morales-Perez, C.L.; Teng, J.; Hibbs, R.E. Structural principles of distinct assemblies of the human α4β2 nicotinic recepto. Nature 2018, 557, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Unwin, N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J. Mol. Biol. 2005, 346, 967–989. [Google Scholar] [CrossRef] [PubMed]
- Mazzaferro, S.; Gasparri, F.; New, K.; Alcaino, C.; Faundez, M.; Iturriaga-Vasquez, P.; Vijayan, R.; Biggin, P.C.; Bermudez, I. Non-equivalent ligand selectivity of agonist sites in (α4β2)2α4 nicotinic acetylcholine receptors: A key determinant of agonist efficacy. J. Biol. Chem. 2014, 289, 21795–21806. [Google Scholar] [CrossRef] [PubMed]
- Gotti, C.; Moretti, M.; Gaimarri, A.; Zanardi, A.; Clementi, F.; Zoli, M. Heterogeneity and complexity of native brain nicotinic receptors. Biochem. Pharmacol. 2007, 74, 1102–1111. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, J.D.; Sharples, C.G.V.; Caldwell, W.S. Molecular recognition in nicotinic acetylcholine receptors: The importance of π-cation interactions. J. Med. Chem. 1999, 42, 3066–3074. [Google Scholar] [CrossRef] [PubMed]
- Corringer, P.; Le Novère, N.; Changeux, J.P. Nicotinic receptors at the amino acid level. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 431–458. [Google Scholar] [CrossRef]
- Cahill, K.; Lindson-Hawley, N.; Thomas, K.; Fanshawe, T.; Lancaster, T. Nicotine receptor partial agonists for smoking cessation (Review). Cochrane Database Syst. Rev. 2016, 5, 1–208. [Google Scholar]
- Tutka, P. Nicotinic receptor partial agonists as novel compounds for the treatment of smoking cessation. Expert Opin. Investig. Drugs 2008, 17, 1473–1485. [Google Scholar] [CrossRef] [PubMed]
- Elliott, R.; Kopecka, A.; Gunn, D.E.; Lin, N.; Garvey, D.S.; Ryther, K.B.; Holladay, M.W.; Anderson, D.J.; Campbell, J.E.; Sullivan, J.P.; et al. 2-(Aryloxymethyl) azacyclic analogues as novel nicotinic acetylcholine receptor (nAChR) ligands. Bioorg. Med. Chem. Lett. 1996, 6, 2283–2288. [Google Scholar] [CrossRef]
- Bolchi, C.; Valoti, E.; Gotti, C.; Fasoli, F.; Ruggeri, P.; Fumagalli, L.; Binda, M.; Mucchietto, V.; Sciaccaluga, M.; Budriesi, R.; et al. Chemistry and Pharmacology of a Series of Unichiral Analogues of 2-(2-Pyrrolidinyl)-1,4-benzodioxane, Prolinol Phenyl Ether, and Prolinol 3-Pyridyl Ether Designed as α4β2-Nicotinic Acetylcholine Receptor Agonists. J. Med. Chem. 2015, 58, 6665–6677. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, H.; Ma, Q. QSAR study of a large set of 3-pyridyl ethers as ligands of the alpha4beta2 nicotinic acetylcholine receptor. J. Mol. Graph. Model. 2007, 26, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Gotti, C.; Zoli, M.; Clementi, F. Brain nicotinic acetylcholine receptors: Native subtypes and their relevance. Trends Pharmacol. Sci. 2006, 27, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Pabreza, L.; Dhawan, S.; Kellar, K. [3H] cytisine binding to nicotinic cholinergic receptors in brain. Mol. Pharmacol. 1991, 39, 9–12. [Google Scholar] [PubMed]
- Houlihan, L.M.; Slater, Y.; Guerra, D.L.; Peng, J.H.; Kuo, Y.P.; Lukas, R.J.; Cassels, B.K.; Bermudez, I. Activity of cytisine and its brominated isosteres on recombinant human α7, α4β2 and α4β4 nicotinic acetylcholine receptors. J. Neurochem. 2001, 78, 1029–1043. [Google Scholar] [CrossRef] [PubMed]
- Faundez-Parraguez, M.; Farias-Rabelo, N.; Gonzalez-Gutierrez, J.P.; Etcheverry-Berrios, A.; Alzate-Morales, J.; Adasme-Carreño, F.; Varas, R.; Bermudez, I.; Iturriaga-Vasquez, P. Neonicotinic analogues: Selective antagonists for α4β2 nicotinic acetylcholine receptors. Bioorg. Med. Chem. 2013, 21, 2687–2694. [Google Scholar] [CrossRef]
- Smulders, C.J.G.M.; Zwart, R.; Bermudez, I.; Van Kleef, R.G.D.M.; Groot-Kormelink, P.J.; Vijverberg, H.P.M. Cholinergic drugs potentiate human nicotinic α4β2 acetylcholine receptors by a competitive mechanism. Eur. J. Pharmacol. 2005, 509, 97–108. [Google Scholar] [CrossRef]
- Unwin, N. Nicotinic acetylcholine receptor and the structural basis of neuromuscular transmission: Insights from Torpedo postsynaptic membranes. Q. Rev. Biophys. 2014, 46, 283–322. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | α4β2 IC50 (nM) | α4β2 EC50 (μM) | Imax |
|---|---|---|---|
| Nicotine | 1.2 | 10 | 1.00 |
| 1 | 204.0 ± 6.0 | 68.8 ± 1.3 | 0.54 ± 0.01 |
| 2 | 13.8 ± 0.5 | 10.8 ± 0.7 | 1.00 ± 0.01 |
| 3 | 221.0 ± 10.0 | 63.3 ± 2.1 | 0.29 ± 0.01 |
| 4 | 188.0 ± 7.0 | 56.7 ± 1.7 | 0.13 ± 0.01 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalez-Gutierrez, J.P.; Hodar, M.; Viscarra, F.; Paillali, P.; Guerra-Díaz, N.; Pessoa-Mahana, H.; Hernández-Morantes, J.J.; Pérez-Sánchez, H.; Bermúdez, I.; Reyes-Parada, M.; et al. Minimal Structural Changes Determine Full and Partial Nicotinic Receptor Agonist Activity for Nicotine Analogues. Molecules 2019, 24, 2684. https://doi.org/10.3390/molecules24152684
Gonzalez-Gutierrez JP, Hodar M, Viscarra F, Paillali P, Guerra-Díaz N, Pessoa-Mahana H, Hernández-Morantes JJ, Pérez-Sánchez H, Bermúdez I, Reyes-Parada M, et al. Minimal Structural Changes Determine Full and Partial Nicotinic Receptor Agonist Activity for Nicotine Analogues. Molecules. 2019; 24(15):2684. https://doi.org/10.3390/molecules24152684
Chicago/Turabian StyleGonzalez-Gutierrez, Juan Pablo, Martin Hodar, Franco Viscarra, Pablo Paillali, Nicolás Guerra-Díaz, Hernán Pessoa-Mahana, Juan José Hernández-Morantes, Horacio Pérez-Sánchez, Isabel Bermúdez, Miguel Reyes-Parada, and et al. 2019. "Minimal Structural Changes Determine Full and Partial Nicotinic Receptor Agonist Activity for Nicotine Analogues" Molecules 24, no. 15: 2684. https://doi.org/10.3390/molecules24152684
APA StyleGonzalez-Gutierrez, J. P., Hodar, M., Viscarra, F., Paillali, P., Guerra-Díaz, N., Pessoa-Mahana, H., Hernández-Morantes, J. J., Pérez-Sánchez, H., Bermúdez, I., Reyes-Parada, M., & Iturriaga-Vásquez, P. (2019). Minimal Structural Changes Determine Full and Partial Nicotinic Receptor Agonist Activity for Nicotine Analogues. Molecules, 24(15), 2684. https://doi.org/10.3390/molecules24152684