
Chalcogen Bonding due to the Exo-Substitution of Icosahedral Dicarbaborane


Abstract
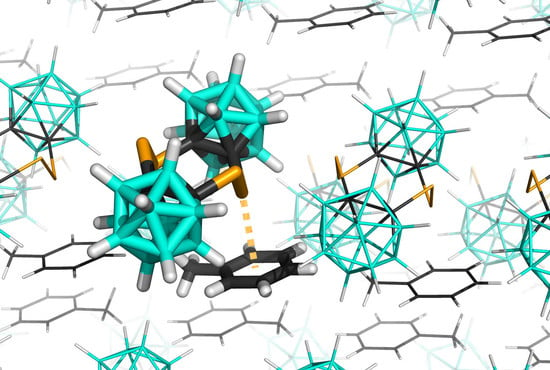
1. Introduction
2. Methods
3. Results and Discussion
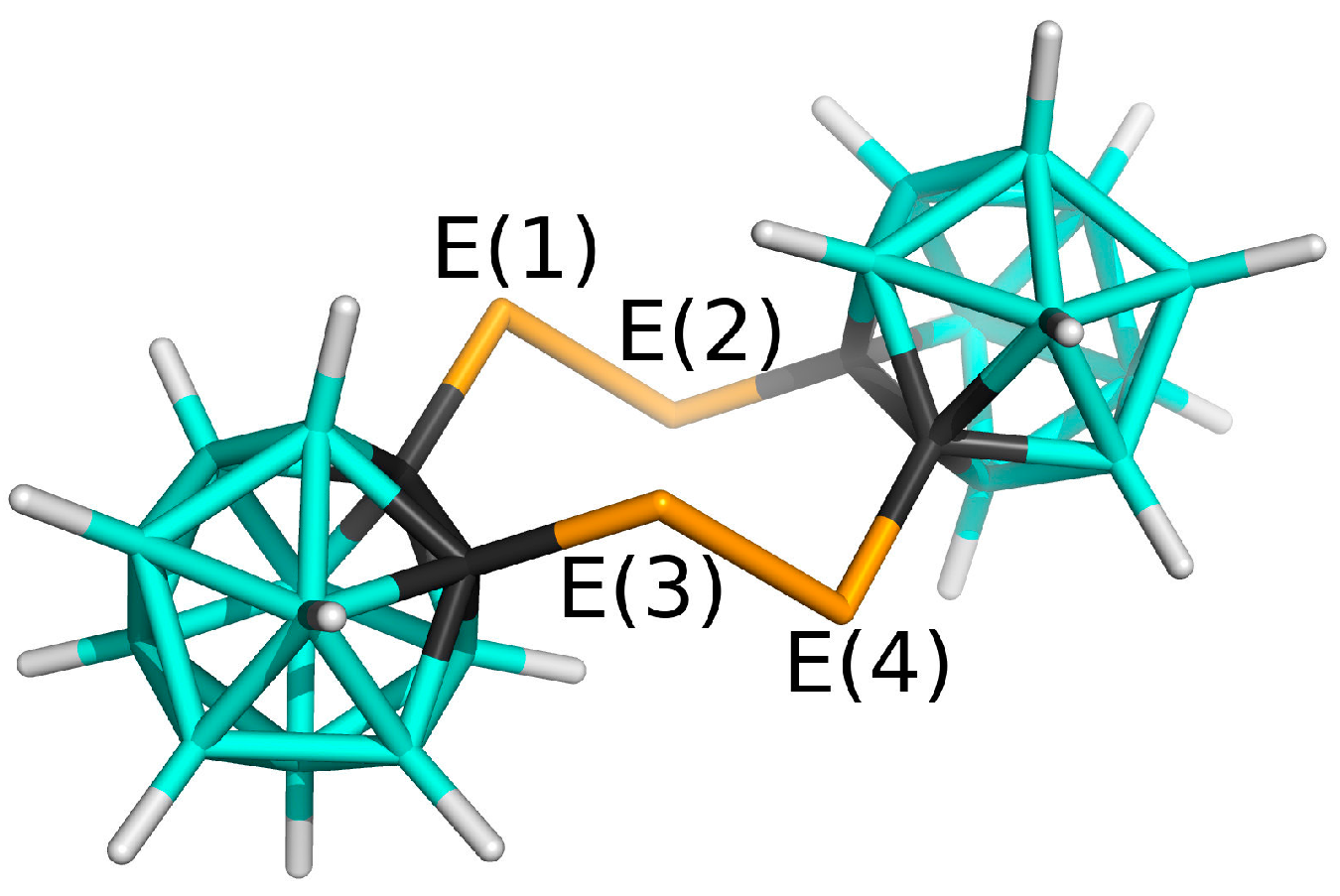
3.1. Charge Distribution of Isolated Molecules
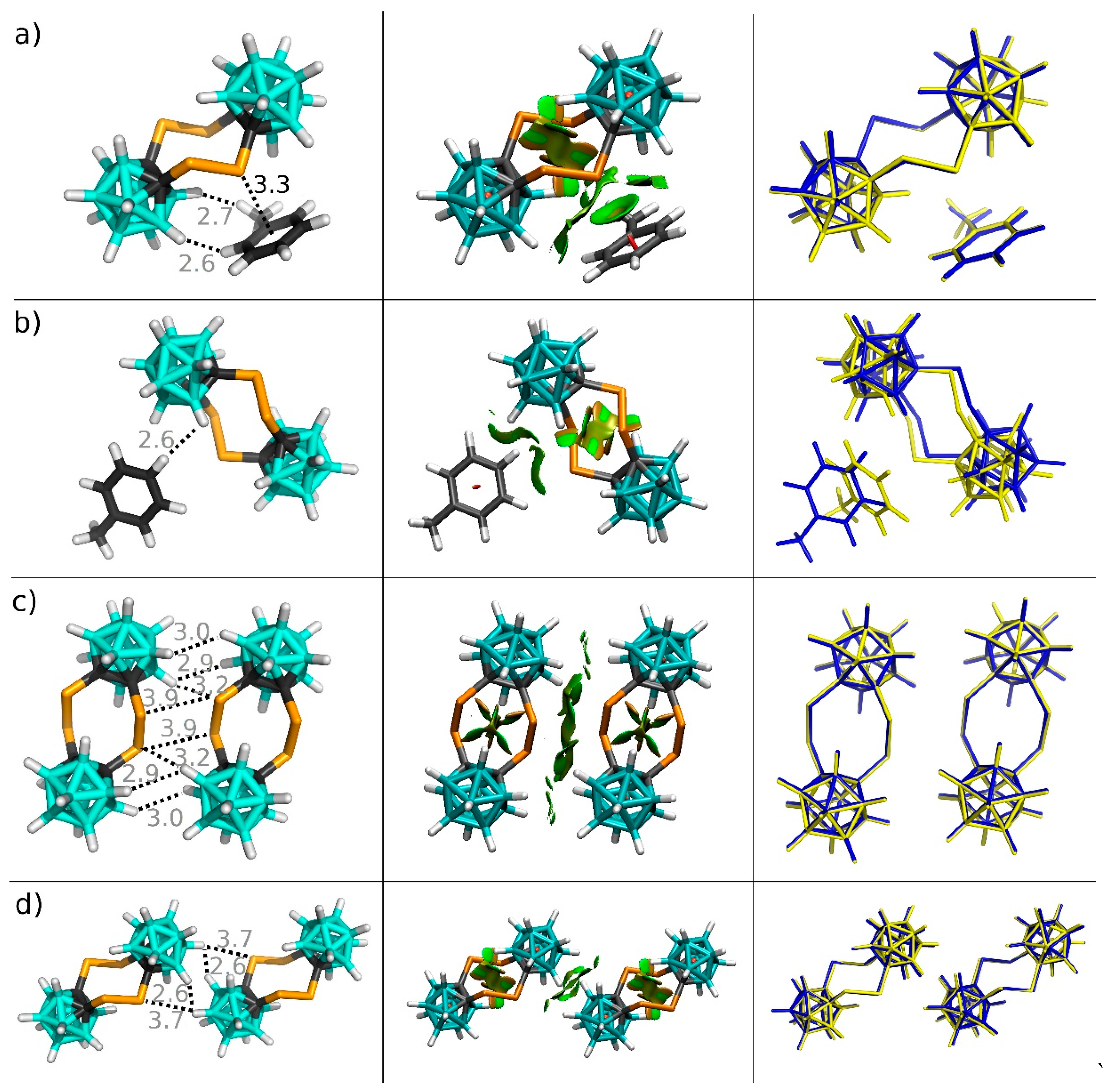
3.2. Interactions in the Crystal of Se4C4•toluene and in the Single Crystal of Te4C4
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Melichar, P.; Hnyk, D.; Fanfrlík, J. Systematic Examination of Classical and Multi-Center Bonding in Heteroborane Clusters. Phys. Chem. Chem. Phys. 2018, 20, 4666–4675. [Google Scholar] [CrossRef] [PubMed]
- Hnyk, D.; Wann, D.A. Molecular Structures of Free Boron Clusters. In Challenges and Advances in Computational Chemistry and Physics, (Boron: The Fifth Element); Hnyk, D.; Wann, D.A. Springer: Dordrecht, The Netherlands, 2016; Volume 20, pp. 17–48. [Google Scholar]
- Grimmes, R. Supercarboranes. Angew. Chem. Int. Ed. 2003, 42, 1198–1200. [Google Scholar] [CrossRef] [PubMed]
- Hnyk, D.; Všetečka, V.; Drož, L.; Exner, O. Charge Distribution within 1,2-Dicarba-closo-dodecaborane: Dipole Moments of its Phenyl Derivatives. Collect. Czech. Chem. Commun. 2001, 66, 1375–1379. [Google Scholar] [CrossRef]
- Schleyer, P.v.R.; Najafian, K. Stability and Three-Dimensional Aromaticity of closo-Monocarbaborane Anions, CBn-1Hn-, and closo-Dicarboranes, C2Bn-2Hn. Inorg. Chem. 1998, 37, 3454–3470. [Google Scholar] [CrossRef] [PubMed]
- Baše, T.; Holub, J.; Fanfrlík, J.; Hnyk, D.; Lane, P.D.; Wann, D.A.; Vishnevskiy, Y.V.; Tikhonov, D.; Reuter, C.G.; Mitzel, N.W. Icosahedral Carbaboranes with Peripheral Hydrogen-Chalcogenide Groups: Structures from Gas Electron Diffraction and Chemical Shielding in Solution. Chem. Eur. J. 2019, 25, 2313–2321. [Google Scholar] [CrossRef] [PubMed]
- Wrackmeyer, B.; García Hernández, Z.; Kempe, R.; Herbehold, M. Novel 1,2-Dicarba-closo-dodecaborane(12) Derivatives of Selenium. Eur. J. Inorg. Chem. 2007, 239–246. [Google Scholar] [CrossRef]
- Arai, K.; Takei, T.; Okumura, M.; Watanabe, S.; Amagai, Y.; Asahina, Y.; Moroder, L.; Hojo, H.; Inaba, K.; Iwaoka, M. Preparation of Selenoinsulin as a Long-Lasting Insulin Analogue. Angew. Chem. Int. Ed. Engl. 2017, 56, 5522–5526. [Google Scholar] [CrossRef] [PubMed]
- Herbehold, M.; Milius, W.; Guo-Xin, J.; Kremnitz, W.; Wrackmeyer, B. Molecular Structures of Some Tellurium Derivatives of 1,2-Dicarba-closo-dodecaborane(12). Z. Anorg. Allg. Chem. 2006, 632, 2031–2036. [Google Scholar] [CrossRef]
- Teixidor, F.; Rudolph, R.W. Synthesis of Cyclic and Polymeric Derivatives of 1,2-Dithiol-o-carborane. J. Organomet. Chem. 1983, 241, 301–312. [Google Scholar] [CrossRef]
- Gleiter, R.; Haberhauer, G.; Werz, D.B.; Roming, F.; Bleiholder, C. From noncovalent Chalcogen-chalcogen Interactions to Supramolecular Aggregates: Experiments and Calculations. Chem. Rev. 2018, 118, 2010–2014. [Google Scholar] [CrossRef]
- Werz, D.B.; Gleiter, R.; Roming, F. Nanotube Formation Favored by Chalcogen-Chalcogen Interactions. J. Am. Chem. Soc. 2002, 124, 10638–10639. [Google Scholar] [CrossRef] [PubMed]
- Werz, D.B.; Staeb, T.H.; Benisch, C.; Rausch, B.J.; Rominger, F.; Gleiter, R. Self-Organization of Chalcogen-Containing Cyclic Alkynes and Alkenes to Yield Columnar Structures. Org. Lett. 2002, 4, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Macháček, J.; Plešek, J.; Holub, J.; Hnyk, D.; Všetečka, V.; Císařová, I.; Kaupp, M.; Štíbr, B. New Route to 1-Thia-closo-dodecaborane(11), closo-1-SB11H11, and its Halogenation Reactions. The Effect of the Halogen on the Dipole Moments and the NMR Spectra and the Importance of Spin−Orbit Coupling for the 11B Chemical Shifts. Dalton Trans. 2006, 1024–1029. [Google Scholar] [CrossRef] [PubMed]
- Fanfrlík, J.; Přáda, A.; Padělková, Z.; Pecina, A.; Macháček, J.; Lepšík, M.; Holub, J.; Růžička, A.; Hnyk, D.; Hobza, P. The Dominant Role of Chalcogen Bonding in the Crystal Packing of 2D/3D Aromatics. Angew. Chem. Int. Ed. 2014, 53, 10139–10142. [Google Scholar] [CrossRef] [PubMed]
- Fanfrlík, J.; Hnyk, D. Dihalogen and Pnictogen Bonding in Crystalline Icosahedral Phosphaboranes. Crystals 2018, 8, 390–399. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Flűkiger, P.; Lűthi, H.P.; Portmann, S.; Weber, J. MOLEKEL 4.3; Swiss Center for Scientific Computing: Manno, Switzerland, 2000. [Google Scholar]
- Portmann, S.; Luthi, H.P. MOLEKEL: An Interactive Molecular Graphic Tool. CHIMIA Int. J. Chem. 2000, 54, 766–770. [Google Scholar]
- Hostaš, J.; Řezáč, J. Accurate DFT-D3 Calculations in a Small Basis Set. J. Chem. Theory Comput. 2017, 13, 3575–3585. [Google Scholar] [CrossRef]
- Řezáč, J.; Hobza, P. Advanced Corrections of Hydrogen Bonding and Dispersion for Semiempirical Quantum Mechanical Methods. J. Chem. Theory Comput. 2012, 8, 141–151. [Google Scholar] [CrossRef]
- Jeziorski, B.; Moszynski, R.; Szalewicz, K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem. Rev. 1994, 94, 1887–1930. [Google Scholar] [CrossRef]
- Parker, T.M.; Burns, L.A.; Parrish, R.M.; Ryno, A.G.; Sherrill, C.D. Levels of Symmetry Adapted Perturbation Theory (SAPT). I. Efficiency and Performance for Interaction Energies. J. Chem. Phys. 2014, 140, 094106. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalen Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Contreras-García, J.; Yang, W.; Johnson, E.R. Analysis of Hydrogen-Bond Interaction Potentials from Electron Density: Integration of Noncovalent Interaction Region. J. Phys. Chem. A 2011, 115, 12983–12990. [Google Scholar] [CrossRef] [PubMed]
- Ahlrichs, R.; Bar, M.; Haser, M.; Horn, H.; Kolmel, C. Electronic Structure Calculations on Workstation Computers: The Program System Turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Turney, J.M.; Simmonett, A.C.; Parrish, R.M.; Hohenstein, E.G.; Evangelista, F.; Fermann, J.T.; Mintz, B.J.; Burns, L.A.; Wilke, J.J.; Abrams, M.L.; et al. Psi4: An Open-Source Ab Initio Electronic Structure Program. WIREs Comput. Mol. Sci. 2012, 2, 556–565. [Google Scholar] [CrossRef]
- Stewart, J.P. Optimization of parameters for semiempirical methods IV: Extension of MNDO, AM1, and PM3 to more main group elements. J. Mol. Model. 2004, 10, 155–164. [Google Scholar] [CrossRef]
- Řezáč, J. Cuby: An Integrative Framework for Computational Chemistry. J. Comput. Chem. 2016, 37, 1230–1237. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen Bonding: The σ-Hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Fanfrlík, J.; Hnyk, D. Chalcogens Act as Inner and Outer Heteroatoms in Borane Cages with Possible Consequences for σ-Hole Interactions. CrystEngComm 2016, 47, 8973–9162. [Google Scholar] [CrossRef]
- Fanfrlík, J.; Holub, J.; Růžičková, Z.; Řezáč, J.; Lane, P.D.; Wann, D.A.; Hnyk, D.; Růžička, A.; Hobza, P. Competition between Halogen, Hydrogen and Dihydrogen Bonding in Brominated Carboranes. ChemPhysChem 2016, 17, 3373–3376. [Google Scholar] [CrossRef]
- Mantina, M.; Chamberlin, A.C.; Valero, R.; Cramer, C.J.; Truhlar, D.G. Consistent van der Waals Radii for the Whole Main Group. J. Phys. Chem. A 2009, 113, 5806–5812. [Google Scholar] [CrossRef]
- Bhandary, S.; Sirohiwal, A.; Kadu, R.; Kumar, S.; Chopra, D. Dipsresion Stabilized Se/Te···π Double Chalcogen Bonding Synthons in in Situ Cryocrystallized Divalent Organochalcogen Liquids. Cryst. Growth Des. 2018, 18, 3734–3739. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Sato, N. Origin of Attraction in Chalcogen-nitrogen Interaction if 1,2,5-Chalcogenadiazole Dimers. J. Phys. Chem. A 2013, 117, 6849–6855. [Google Scholar] [CrossRef] [PubMed]
- Řezáč, J.; De la Lande, A. On the Role of Charge Transfer in Halogen Bonding. Phys. Chem. Chem. Phys. 2017, 19, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Fanfrlík, J.; Pecina, A.; Řezáč, J.; Sedlak, R.; Hnyk, D.; Lepšík, M.; Hobza, P. B-H···π: A Nonclassical Hydrogen Bond or Dispersion Contact? Phys. Chem. Chem. Phys. 2017, 19, 18194–18200. [Google Scholar] [CrossRef] [PubMed]
- Esterhuysen, C.; Heßelman, A.; Clark, T. Trifluoromethyl: An Amphiphilic Noncovalent Bonding Partner. ChemPhysChem 2017, 18, 772–784. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| μ | VS,max | VS,min | |
|---|---|---|---|
| Toluene | 0.4 | 14.6 | −19.8 |
| Se4C4 | 0.0 | 28.6 | −8.2 |
| Te4C4 | 0.0 | 35.6 | −10.1 |
| Interaction Motif | ΔE | |||||
|---|---|---|---|---|---|---|
| DFT-D3 | SAPT0 | |||||
| Total | Eelec | Eind | Edisp | Eexch | ||
| Se4C4···toluene (E-bonding) 1 | −9.22 | −9.28 | −5.47 (27%) | −1.81 (9%) | −12.66 (64%) | 10.66 |
| Se4C4···toluene (stacking) 2 | −4.07 | −3.62 | −1.32 (19%0 | −0.39 (5%) | −5.13 (75%) | 3.22 |
| Se4C4···Se4C4 (stacking_1) 3 | −10.23 | −10.27 | −5.06 (22%) | −1.53 (7%) | −16.04 (71%) | 12.36 |
| Se4C4···Se4C4 (stacking_2) 4 | −5.02 | −5.43 | −1.86 (19%) | −0.37 (4%) | −7.61 (77%) | 4.41 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fanfrlík, J.; Hnyk, D.; Hobza, P. Chalcogen Bonding due to the Exo-Substitution of Icosahedral Dicarbaborane. Molecules 2019, 24, 2657. https://doi.org/10.3390/molecules24142657
Fanfrlík J, Hnyk D, Hobza P. Chalcogen Bonding due to the Exo-Substitution of Icosahedral Dicarbaborane. Molecules. 2019; 24(14):2657. https://doi.org/10.3390/molecules24142657
Chicago/Turabian StyleFanfrlík, Jindřich, Drahomír Hnyk, and Pavel Hobza. 2019. "Chalcogen Bonding due to the Exo-Substitution of Icosahedral Dicarbaborane" Molecules 24, no. 14: 2657. https://doi.org/10.3390/molecules24142657
APA StyleFanfrlík, J., Hnyk, D., & Hobza, P. (2019). Chalcogen Bonding due to the Exo-Substitution of Icosahedral Dicarbaborane. Molecules, 24(14), 2657. https://doi.org/10.3390/molecules24142657