
Structural and Biological Investigations for a Series of N-5 Substituted Pyrrolo[3,2-d]pyrimidines as Potential Anti-Cancer Therapeutics



Abstract
1. Introduction
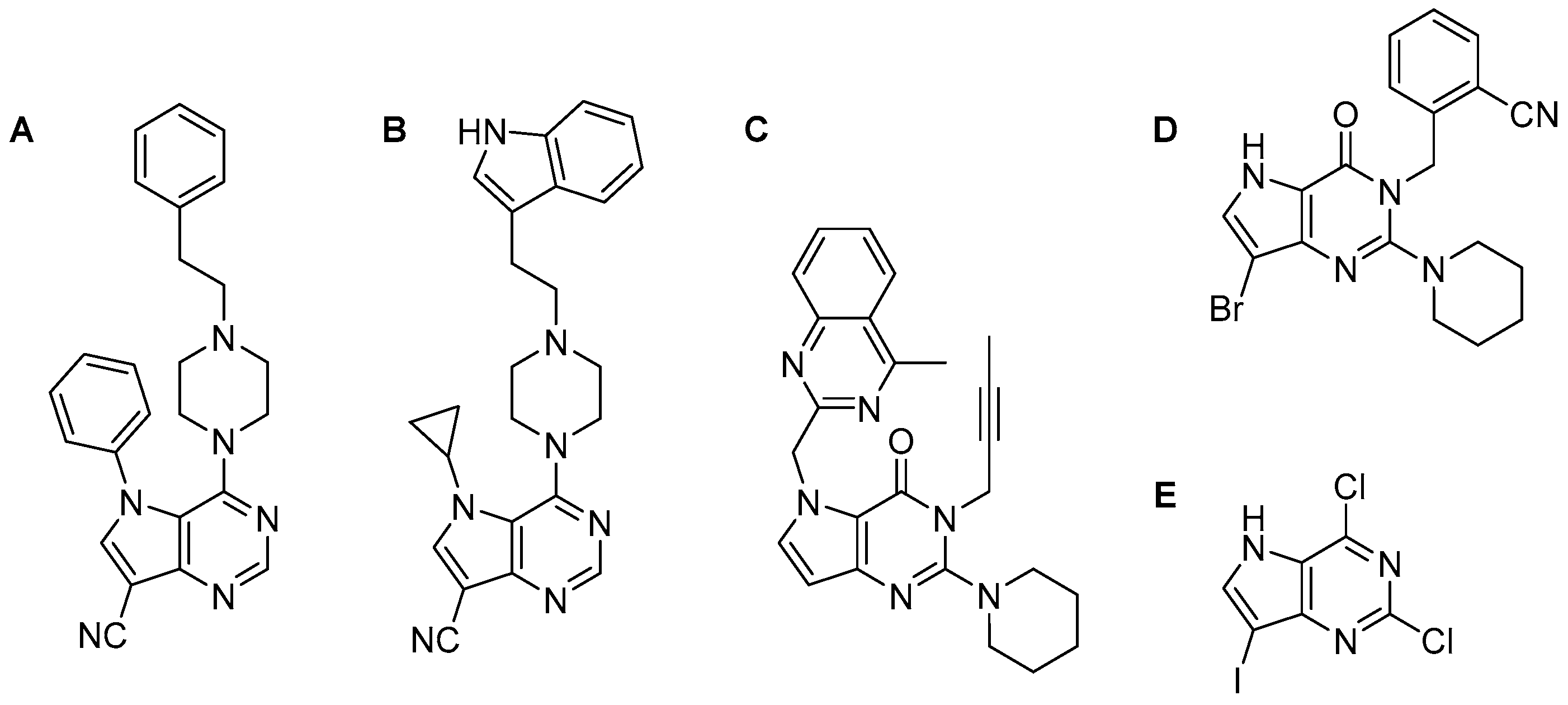
2. Results
2.1. National Cancer Institute-60 Human Tumor Cell Line Screen and COMPARE Analysis
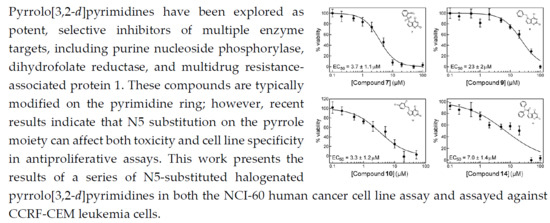
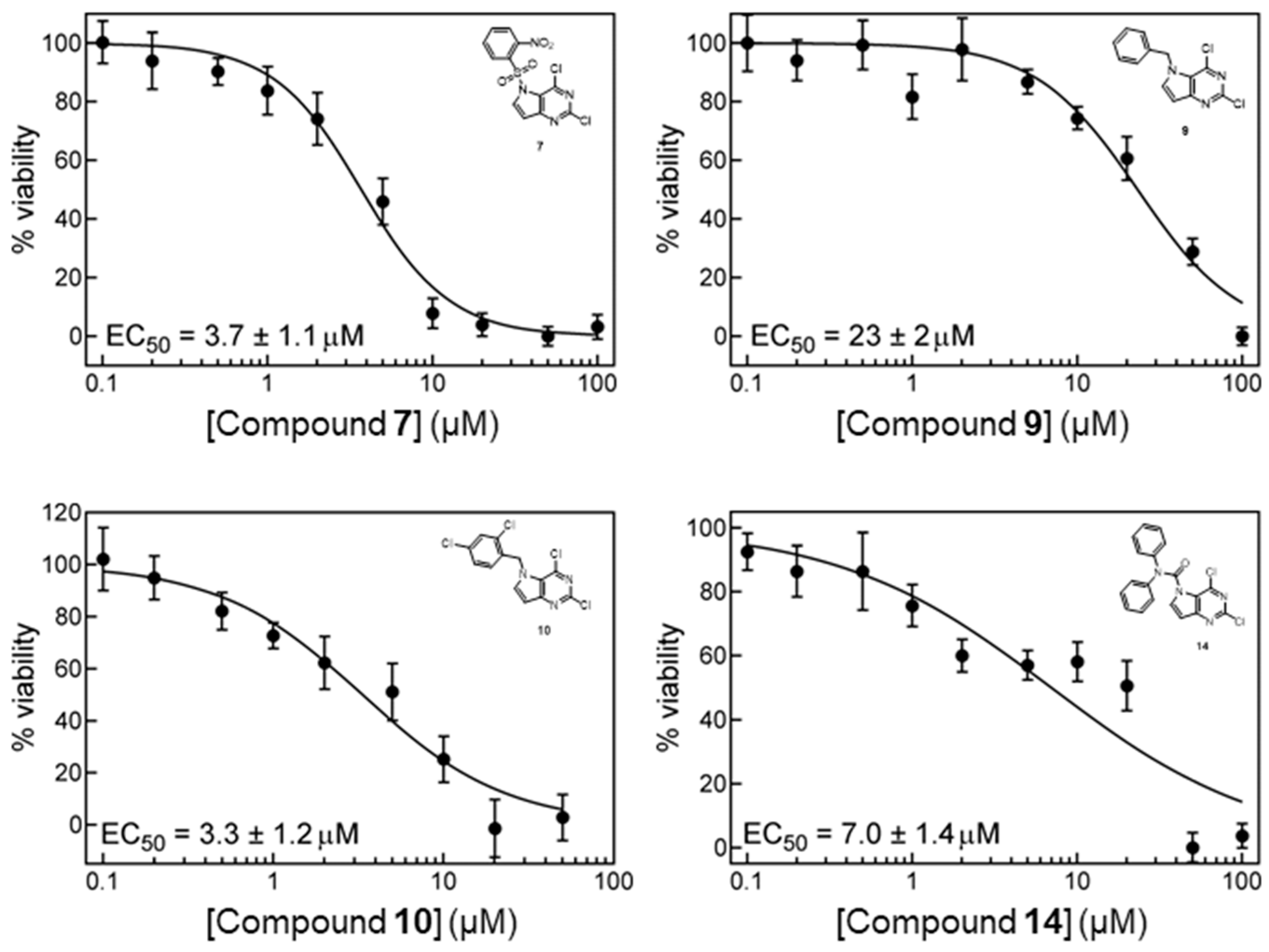
2.2. In Vitro Cell Growth Inhibition
3. Discussion
4. Materials and Methods
4.1. NCI-60 Human Tumor Cell Lines Screen and COMPARE Analysis
4.2. In Vitro Cell Growth Inhibition
4.3. Chemistry
4.3.1. General Experimental
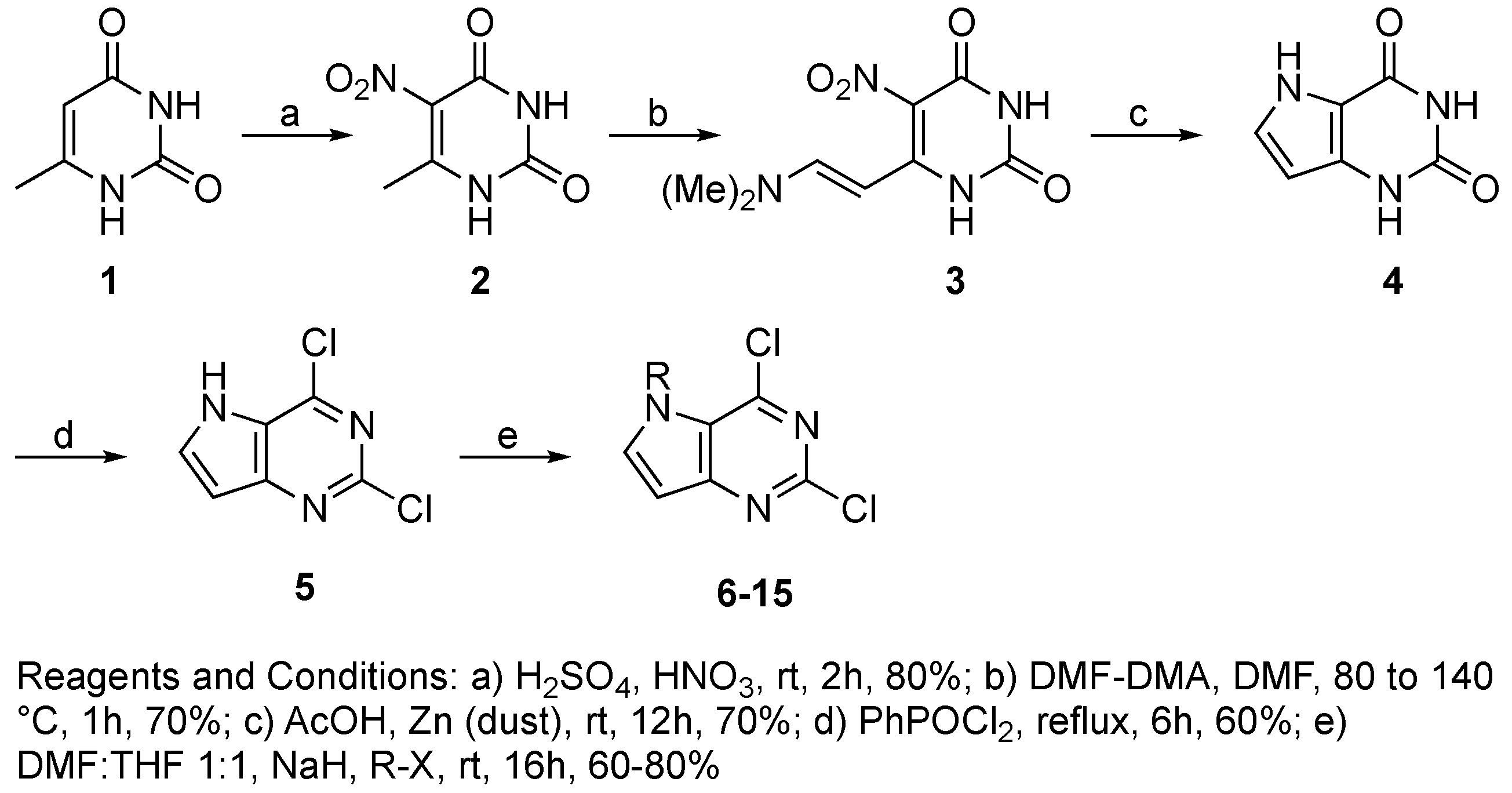
4.3.2. Synthesis of Target Compound 5
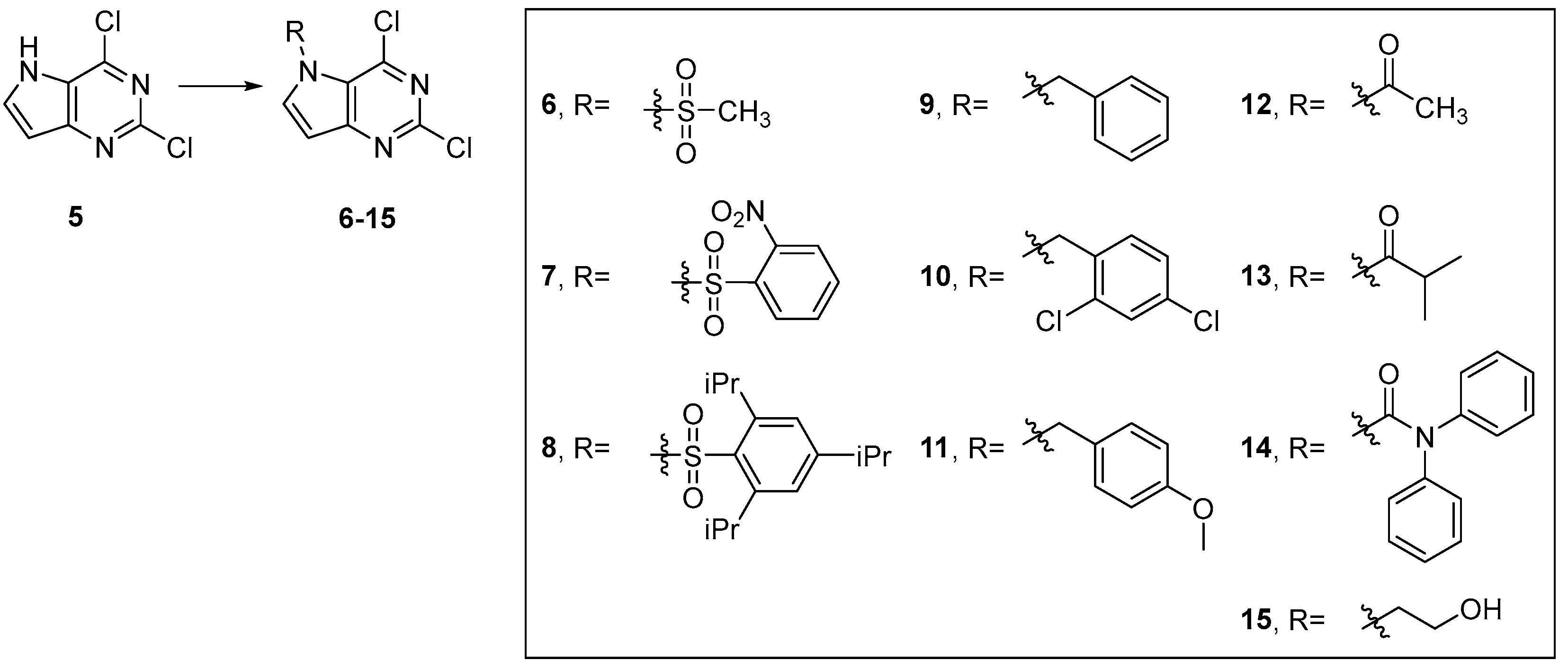
4.3.3. General Procedure for Synthesis of Compounds 6–15
4.3.4. Characterization
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Stefan, K.; Schmitt, S.M.; Wiese, M. 9-Deazapurines as Broad-Spectrum Inhibitors of the ABC Transport Proteins P-Glycoprotein, Multidrug Resistance-Associated Protein 1, and Breast Cancer Resistance Protein. J. Med. Chem. 2017, 60, 8758–8780. [Google Scholar] [CrossRef] [PubMed]
- de Coen, L.M.; Heugebaert, T.S.; García, D.; Stevens, C.V. Synthetic Entries to and Biological Activity of Pyrrolopyrimidines. Chem. Rev. 2016, 116, 80–139. [Google Scholar] [CrossRef] [PubMed]
- Gangjee, A.; Li, W.; Yang, J.; Kisliuk, R.L. Design, synthesis, and biological evaluation of classical and nonclassical 2-amino-4-oxo-5-substituted-6-methylpyrrolo[3,2-d]pyrimidines as dual thymidylate synthase and dihydrofolate reductase inhibitors. J. Med. Chem. 2008, 51, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Shih, H.; Cottam, H.B.; Carson, D.A. Facile synthesis of 9-substituted 9-deazapurines as potential purine nucleoside phosphorylase inhibitors. Chem. Pharm. Bull. 2002, 50, 364–367. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sircar, J.C.; Kostlan, C.R.; Pinter, G.W.; Suto, M.J.; Bobovski, T.P.; Capiris, T.; Schwender, C.F.; Dong, M.K.; Scott, M.E.; Bennett, M.K.; et al. 8-Amino-9-substituted guanines: Potent purine nucleoside phosphorylase (PNP) inhibitors. Agents Actions 1987, 21, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Sircar, J.C.; Kostlan, C.R.; Gilbertsen, R.B.; Bennett, M.K.; Dong, M.K.; Cetenko, W.J. Inhibitors of human purine nucleoside phosphorylase. Synthesis of pyrrolo[3,2-d]pyrimidines, a new class of purine nucleoside phosphorylase inhibitors as potentially T-cell selective immunosuppressive agents. Description of 2,6-diamino-3,5-dihydro-7-(3-thienylmethyl)-4H-pyrrolo[3,2-d]pyrimidin-4-one. J. Med. Chem. 1992, 35, 1605–1609. [Google Scholar] [PubMed]
- Montgomery, J.A.; Niwas, S.; Rose, J.D.; Secrist, J.A., III; Babu, Y.S.; Bugg, C.E.; Erion, M.D.; Guida, W.C.; Ealick, S.E. Structure-based design of inhibitors of purine nucleoside phosphorylase. 1. 9-(arylmethyl) derivatives of 9-deazaguanine. J. Med. Chem. 1993, 36, 55–69. [Google Scholar] [CrossRef]
- Secrist, J.A., III; Niwas, S.; Rose, J.D.; Babu, Y.S.; Bugg, C.E.; Erion, M.D.; Guida, W.C.; Ealick, S.E.; Montgomery, J.A. Structure-based design of inhibitors of purine nucleoside phosphorylase. 2. 9-Alicyclic and 9-heteroalicyclic derivatives of 9-deazaguanine. J. Med. Chem. 1993, 36, 1847–1854. [Google Scholar] [CrossRef]
- Erion, M.D.; Niwas, S.; Rose, J.D.; Ananthan, S.; Allen, M.; Secrist, J.A., III; Babu, Y.S.; Bugg, C.E.; Guida, W.C. Structure-based design of inhibitors of purine nucleoside phosphorylase. 3. 9-Arylmethyl derivatives of 9-deazaguanine substituted on the methylene group. J. Med. Chem. 1993, 36, 3771–3783. [Google Scholar] [CrossRef]
- Baraldi, P.G.; Romagnoli, R.; Saponaro, G.; Tabrizi, M.A.; Baraldi, S.; Pedretti, P.; Fusi, C.; Nassini, R.; Materazzi, S.; Geppetti, P.; et al. 7-Substituted-pyrrolo[3,2-d]pyrimidine-2,4-dione derivatives as antagonists of the transient receptor potential ankyrin 1 (TRPA1) channel: A promising approach for treating pain and inflammation. Bioorg. Med. Chem. 2012, 20, 1690–1698. [Google Scholar] [CrossRef]
- Ishikawa, T.; Seto, M.; Banno, H.; Kawakita, Y.; Oorui, M.; Taniguchi, T.; Ohta, Y.; Tamura, T.; Nakayama, A.; Miki, H.; et al. Design and Synthesis of Novel Human Epidermal Growth Factor Receptor 2 (HER2)/Epidermal Growth Factor Receptor (EGFR) Dual Inhibitors Bearing a Pyrrolo 3,2-d pyrimidine Scaffold. J. Med. Chem. 2011, 54, 8030–8050. [Google Scholar] [CrossRef] [PubMed]
- Kawakita, Y.; Miwa, K.; Seto, M.; Banno, H.; Ohta, Y.; Tamura, T.; Yusa, T.; Miki, H.; Kamiguchi, H.; Ikeda, Y.; et al. Design and synthesis of pyrrolo[3,2-d]pyrimidine HER2/EGFR dual inhibitors: Improvement of the physicochemical and pharmacokinetic profiles for potent in vivo anti-tumor efficacy. Bioorg. Med. Chem. 2012, 20, 6171–6180. [Google Scholar] [CrossRef] [PubMed]
- Sogabe, S.; Kawakita, Y.; Igaki, S.; Iwata, H.; Miki, H.; Cary, D.R.; Takagi, T.; Takagi, S.; Ohta, Y.; Ishikawa, T. Structure-Based Approach for the Discovery of Pyrrolo[3,2-d]pyrimidine-Based EGFR T790M/L858R Mutant Inhibitors. ACS Med. Chem. Lett. 2013, 4, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Gangjee, A.; Pavana, R.K.; Li, W.; Hamel, E.; Westbrook, C.; Mooberry, S.L. Novel water-soluble substituted pyrrolo[3,2-d]pyrimidines: design, synthesis, and biological evaluation as antitubulin antitumor agents. Pharm. Res. 2012, 29, 3033–3039. [Google Scholar] [CrossRef] [PubMed]
- Cawrse, B.M.; Lapidus, R.S.; Cooper, B.; Choi, E.Y.; Seley-Radtke, K.L. Anticancer Properties of Halogenated Pyrrolo[3,2-d]pyrimidines with Decreased Toxicity via N5 Substitution. ChemMedChem 2018, 13, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Temburnikar, K.W.; Ross, C.R.; Wilson, G.M.; Balzarini, J.; Cawrse, B.M.; Seley-Radtke, K.L. Antiproliferative activities of halogenated pyrrolo[3,2-d]pyrimidines. Bioorg. Med. Chem 2015, 23, 4354–4363. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, S.M.; Stefan, K.; Wiese, M. Pyrrolopyrimidine derivatives and purine analogs as novel activators of Multidrug Resistance-associated Protein 1 (MRP1, ABCC1). Biochim. Biophys. Acta 2017, 1859, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.M.; Folkes, A.; Chuckowree, I.; Cockcroft, X.; Sohal, S.; Miller, W.; Milton, J.; Wren, S.P.; Vicker, N.; Depledge, P.; et al. Studies on pyrrolopyrimidines as selective inhibitors of multidrug-resistance-associated protein in multidrug resistance. J. Med. Chem. 2004, 47, 1329–1338. [Google Scholar] [CrossRef]
- Vaseghi, G.; Jafari, E.; Hassanzadeh, F.; Haghjooy-Javanmard, S.; Dana, N.; Rafieian-Kopaei, M. Cytotoxic Evaluation of Some Fused Pyridazino-and Pyrrolo-quinazolinones Derivatives on Melanoma and Prostate Cell Lines. Adv. Biomed. Res. 2017, 6, 76. [Google Scholar]
- Lee, B.D.; Li, Z.; French, K.J.; Zhuang, Y.; Xia, Z.; Smith, C.D. Synthesis and evaluation of dihydropyrroloquinolines that selectively antagonize P-glycoprotein. J. Med. Chem. 2004, 47, 1413–1422. [Google Scholar] [CrossRef]
- Tawari, N.R.; Bag, S.; Degani, M.S. Pharmacophore mapping of a series of pyrrolopyrimidines, indolopyrimidines and their congeners as multidrug-resistance-associated protein (MRP1) modulators. J. Mol. Model. 2008, 14, 911–921. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, S.M.; Stefan, K.; Wiese, M. Pyrrolopyrimidine Derivatives as Novel Inhibitors of Multidrug Resistance-Associated Protein 1 (MRP1, ABCC1). J. Med. Chem. 2016, 59, 3018–3033. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Xie, H.; Zeng, L.L.; Lu, X.; Zhao, X.; Zhang, G.C.; Tu, Z.C.; Xu, H.J.; Yang, L.; Zhang, X.Q.; et al. Discovery of potent dipeptidyl peptidase IV inhibitors through pharmacophore hybridization and hit-to-lead optimization. Bioorg. Med. Chem. 2013, 21, 1749–1755. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Zeng, L.; Zeng, S.; Lu, X.; Zhang, G.; Zhao, X.; Cheng, N.; Tu, Z.; Li, Z.; Xu, H.; et al. Novel pyrrolopyrimidine analogues as potent dipeptidyl peptidase IV inhibitors based on pharmacokinetic property-driven optimization. Eur. J. Med. Chem. 2012, 52, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Temburnikar, K.W.; Zimmermann, S.C.; Kim, N.T.; Ross, C.R.; Gelbmann, C.; Salomon, C.E.; Wilson, G.M.; Balzarini, J.; Seley-Radtke, K.L. Antiproliferative activities of halogenated thieno[3,2-d]pyrimidines. Bioorg. Med. Chem. 2014, 22, 2113–2122. [Google Scholar] [CrossRef] [PubMed]
- Radtke, K.L.; Temburnikar, K. Thieno-and Pyrrolopyrimidine Analogues as Anticancer Agents and Methods of Use Thereof. U.S. Patent 9,434,742 B1, 6 September 2016 and U.S. Patent 9,434,742 B2, 20 February 2018.
- Foley, G.E.; Lazarus, H.; Farber, S.; Uzman, B.G.; Boone, B.A.; McCarthy, R.E. Continuous Culture of Human Lymphoblasts from Peripheral Blood of a Child with Acute Leukemia. Cancer 1965, 18, 522–529. [Google Scholar] [CrossRef]
- Ntie-Kang, F. An in silico evaluation of the ADMET profile of the StreptomeDB database. Springerplus 2013, 2, 353. [Google Scholar] [CrossRef]
- Shoemaker, R.H.; Monks, A.; Alley, M.C.; Scudiero, D.A.; Fine, D.L.; McLemore, T.L.; Abbott, B.J.; Paull, K.D.; Mayo, J.G.; Boyd, M.R. Development of human tumor cell line panels for use in disease-oriented drug screening. Prog. Clin. Biol. Res. 1988, 276, 265–286. [Google Scholar]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef]
- Holbeck, S.L.; Collins, J.M.; Doroshow, J.H. Analysis of Food and Drug Administration-approved anticancer agents in the NCI60 panel of human tumor cell lines. Mol. Cancer Ther. 2010, 9, 1451–1460. [Google Scholar] [CrossRef]
- Paull, K.D.; Hamel, E.; Malspeis, L. COMPARE Analysis. Available online: https://dtp.cancer.gov/databases_tools/compare.htm (accessed on 3 February 2019).
- Paull, K.D.; Shoemaker, R.H.; Hodes, L.; Monks, A.; Scudiero, D.A.; Rubinstein, L.; Plowman, J.; Boyd, M.R. Display and analysis of patterns of differential activity of drugs against human tumor cell lines: development of mean graph and COMPARE algorithm. J. Natl. Cancer Inst. 1989, 81, 1088–1092. [Google Scholar] [CrossRef] [PubMed]
- Zaharevitz, D.W.; Holbeck, S.L.; Bowerman, C.; Svetlik, P.A. COMPARE: A web accessible tool for investigating mechanisms of cell growth inhibition. J. Mol. Graph. Model. 2002, 20, 297–303. [Google Scholar] [CrossRef]
- Sausville, E.A.; Johnson, J.I. Molecules for the millennium: how will they look? New drug discovery year 2000. Br. J. Cancer 2000, 83, 1401–1404. [Google Scholar] [CrossRef] [PubMed]
- Doroshow, J.H.; Juhasz, A.; Ge, Y.; Holbeck, S.; Lu, J.; Antony, S.; Wu, Y.; Jiang, G.; Roy, K. Antiproliferative mechanisms of action of the flavin dehydrogenase inhibitors diphenylene iodonium and di-2-thienyliodonium based on molecular profiling of the NCI-60 human tumor cell panel. Biochem. Pharmacol. 2012, 83, 1195–1207. [Google Scholar] [CrossRef] [PubMed]
- Schober, P.; Boer, C.; Schwarte, L.A. Correlation Coefficients: Appropriate Use and Interpretation. Anesth. Analg. 2018, 126, 1763–1768. [Google Scholar] [CrossRef] [PubMed]
- Monks, A.; Scudiero, D.A.; Johnson, G.S.; Paull, K.D.; Sausville, E.A. The NCI anti-cancer drug screen: A smart screen to identify effectors of novel targets. Anticancer Drug Des. 1997, 12, 533–541. [Google Scholar]
- Neben-Wittich, M.A.; Atherton, P.J.; Schwartz, D.J.; Sloan, J.A.; Griffin, P.C.; Deming, R.L.; Anders, J.C.; Loprinzi, C.L.; Burger, K.N.; Martenson, J.A.; et al. Comparison of provider-assessed and patient-reported outcome measures of acute skin toxicity during a Phase III trial of mometasone cream versus placebo during breast radiotherapy: the North Central Cancer Treatment Group (N06C4). Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.J.; Neuberg, D.S.; Rassenti, L.Z.; Hayes, G.; Redd, R.; Emson, C.; Li, K.; Brown, J.R.; Wierda, W.G.; Turner, S.; et al. Leukemia-cell proliferation and disease progression in patients with early stage chronic lymphocytic leukemia. Leukemia 2017, 31, 1348–1354. [Google Scholar] [CrossRef]
- Landau, D.A.; Carter, S.L.; Getz, G.; Wu, C.J. Clonal evolution in hematological malignancies and therapeutic implications. Leukemia 2014, 28, 34–43. [Google Scholar] [CrossRef]
- Civini, S.; Jin, P.; Ren, J.; Sabatino, M.; Castiello, L.; Jin, J.; Wang, H.; Zhao, Y.; Marincola, F.; Stroncek, D. Leukemia cells induce changes in human bone marrow stromal cells. J. Transl. Med. 2013, 11, 298. [Google Scholar] [CrossRef]
- Prada-Arismendy, J.; Arroyave, J.C.; Röthlisberger, S. Molecular biomarkers in acute myeloid leukemia. Blood Rev. 2017, 31, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.C.; Lin, Y.J.; Hsiao, Y.T.; Lin, M.L.; Yang, J.L.; Huang, Y.P.; Chu, Y.L.; Chung, J.G. Tetrandrine Induces Apoptosis in Human Nasopharyngeal Carcinoma NPC-TW 039 Cells by Endoplasmic Reticulum Stress and Ca. Anticancer Res. 2017, 37, 6107–6118. [Google Scholar] [PubMed]
- Patel, S.R.; Kvols, L.K.; Rubin, J.; O’Connell, M.J.; Edmonson, J.H.; Ames, M.M.; Kovach, J.S. Phase I-II study of pibenzimol hydrochloride (NSC 322921) in advanced pancreatic carcinoma. Investig. New Drugs 1991, 9, 53–57. [Google Scholar] [CrossRef]
- Ferguson, L.R.; Denny, W.A. Microbial mutagenic effects of the DNA minor groove binder pibenzimol (Hoechst 33258) and a series of mustard analogues. Mutat. Res. 1995, 329, 19–27. [Google Scholar] [CrossRef]
- Dubicki, H.; Zielinski, F.; Starks, F.r.W. Synthesis of 5-[(2-Fluoroethyl) (2-chloroethyl)amino]-6-methyl-uracil (Fluorodopan). J. Pharm. Sci. 1964, 53, 1422–1423. [Google Scholar] [CrossRef] [PubMed]
- Esma, F.; Salvini, M.; Troia, R.; Boccadoro, M.; Larocca, A.; Pautasso, C. Melphalan hydrochloride for the treatment of multiple myeloma. Expert Opin. Pharmacother. 2017, 18, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Roux, A.; Peeters, S.; Zanello, M.; Bou Nassif, R.; Abi Lahoud, G.; Dezamis, E.; Parraga, E.; Lechapt-Zalcmann, E.; Dhermain, F.; Dumont, S.; et al. Extent of resection and Carmustine wafer implantation safely improve survival in patients with a newly diagnosed glioblastoma: a single center experience of the current practice. J. Neurooncol. 2017, 135, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Danovich, D.; Apeloig, Y.; Shaik, S. A reliable and inexpensive method for calculating ionization potentials and electron affinities of radicals and molecules. J. Chem. Soc. Perkin Trans. 1993, 2, 321–330. [Google Scholar] [CrossRef]
- COMPARE Analysis. Available online: https://dtp.cancer.gov/databases_tools/compare.htm (accessed on 10 March 2018).
- Ponticello, G.S.; Baldwin, J.J. Useful synthesis of 4-substituted indoles. J. Org. Chem. 1979, 44, 4003–4005. [Google Scholar] [CrossRef]
- Hirota, K.; Abe, Y.; Asao, T.; Senda, S.; Kitade, Y.; Maki, Y. Ring transformation of pyrimidines to pyridines. Synthesis of 4-alkylaminopyridin-2-ones by alkaline hydrolysis of 6-(2-dimethylaminovinyl)uracils. J. Heterocycl. Chem. 1988, 25, 985–990. [Google Scholar] [CrossRef]
- Girgis, N.S.; Cottam, H.B.; Larson, S.B.; Robins, R.K. 9-deazapurine nucleosides. The synthesis of certain N-5-2′-deoxy-β-d-erythropentofuranosyl and N-5-β-d-arabinofuranosylpyrrolo[3,2-d]pyrimidines. J. Heterocycl. Chem. 1987, 24, 821–827. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 5–15 are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Best Growth Percent * | Most Affected Cell Line |
|---|---|---|
| 5 | 60.77 | LOX-IMVI (Melanoma) |
| 6 | 84.49 | NCI-H522; NSC Lung |
| 7 | 49.57 | NCI-H522; NSC Lung |
| 8 | 93.16 | UO-31; Renal |
| 9 | 36.31 | CCRF-CEM; Leukemia |
| 10 | 22.88 | CCRF-CEM; Leukemia |
| 11 | 65.83 | CCRF-CEM; Leukemia |
| 12 | 81.36 | UO-31; Renal |
| 13 | 75.18 | SR, Leukemia |
| 14 | −0.95 | MDA-MB-468; Breast |
| 15 | 89.73 | NCI-H522; NSC Lung |
| Compound | Correlation (PCC) | Target Vector a | Common Cell Line Count b |
|---|---|---|---|
| 9 | 0.717 | D-Tetrandrine | 55 |
| 7 | 0.661 | Pibenzimol HCl | 57 |
| 9 | 0.640 | Fluorodopan | 58 |
| 9 | 0.622 | Melphalan | 58 |
| 9 | 0.622 | Carmustine (BCNU) | 59 |
| Compound | SASA a (Å2) | EA b (eV) | QPPCaco c (nm/sec) | QPlogS d (log[mol/dm3]) |
|---|---|---|---|---|
| 7 | 514.262 | 9.613 | 243.855 | −2.017 |
| 9 | 476.642 | 8.878 | 3989.857 | −4.405 |
| 10 | 509.662 | 8.947 | 4018.835 | −5.547 |
| 14 | 601.2 | 9.071 | 1349.875 | −4.892 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cawrse, B.M.; Robinson, N.M.; Lee, N.C.; Wilson, G.M.; Seley-Radtke, K.L. Structural and Biological Investigations for a Series of N-5 Substituted Pyrrolo[3,2-d]pyrimidines as Potential Anti-Cancer Therapeutics. Molecules 2019, 24, 2656. https://doi.org/10.3390/molecules24142656
Cawrse BM, Robinson NM, Lee NC, Wilson GM, Seley-Radtke KL. Structural and Biological Investigations for a Series of N-5 Substituted Pyrrolo[3,2-d]pyrimidines as Potential Anti-Cancer Therapeutics. Molecules. 2019; 24(14):2656. https://doi.org/10.3390/molecules24142656
Chicago/Turabian StyleCawrse, Brian M., Nia’mani M. Robinson, Nina C. Lee, Gerald M. Wilson, and Katherine L. Seley-Radtke. 2019. "Structural and Biological Investigations for a Series of N-5 Substituted Pyrrolo[3,2-d]pyrimidines as Potential Anti-Cancer Therapeutics" Molecules 24, no. 14: 2656. https://doi.org/10.3390/molecules24142656
APA StyleCawrse, B. M., Robinson, N. M., Lee, N. C., Wilson, G. M., & Seley-Radtke, K. L. (2019). Structural and Biological Investigations for a Series of N-5 Substituted Pyrrolo[3,2-d]pyrimidines as Potential Anti-Cancer Therapeutics. Molecules, 24(14), 2656. https://doi.org/10.3390/molecules24142656