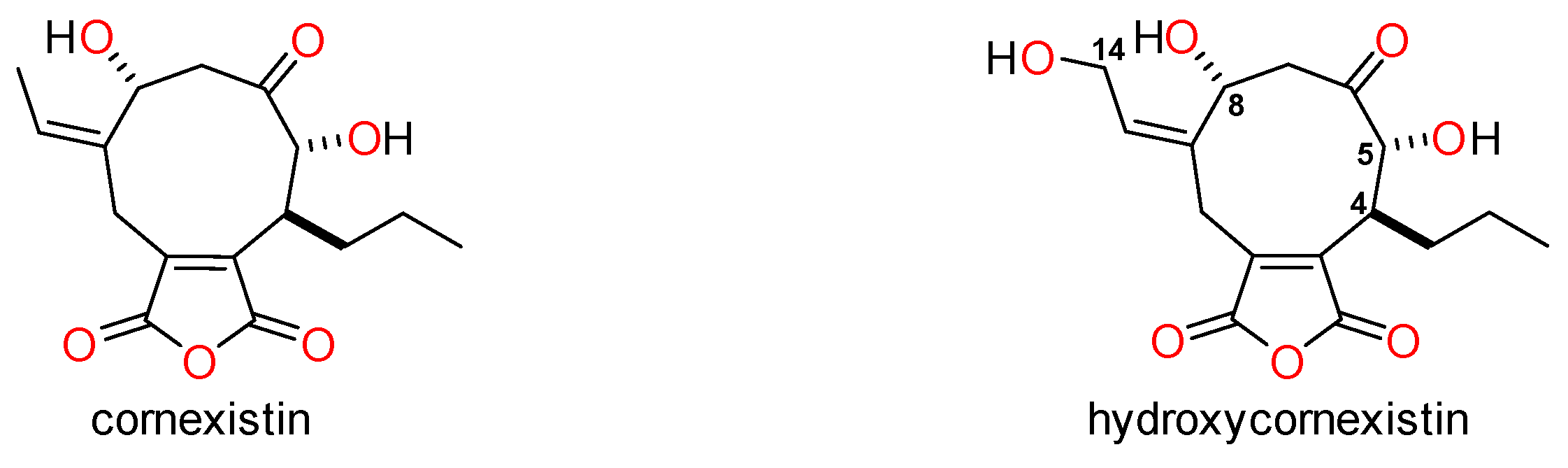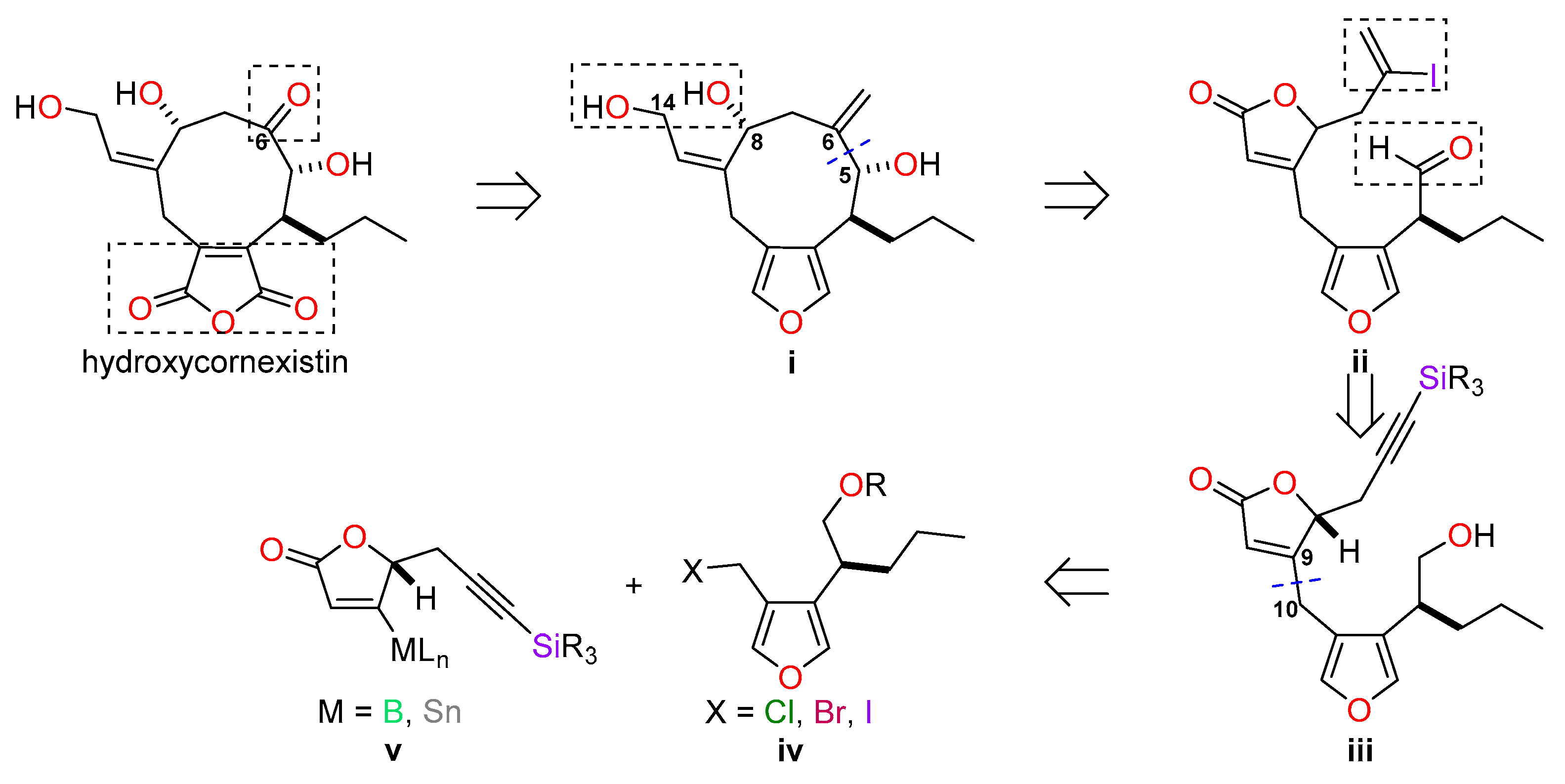4. Materials and Methods
Air and/or moisture sensitive reactions were performed under an atmosphere of Argon in flame-dried apparatus. When necessary, solvents were dried and purified using a Pure Solv™ solvent purification system (SPS). IR spectra were recorded using a type IIa diamond single reflection element on a Shimadzu FTIR-8400 instrument. The IR spectrum of the compound (solid or liquid) was obtained by analysis of a thin layer at ambient temperature. 1H and 13C-NMR spectra were recorded using either a Bruker 400 MHz or 500 MHz Spectrospin spectrometer at ambient temperature; 13C-NMR NMR spectra were recorded at 101 MHz or 126 MHz. Mass spectra were obtained by ionisation under EI, FAB, CI and ESI conditions on a Jeol MStation JMS-700 instrument. Elemental analyses were performed on an Exeter Analytical Elemental Analyser EA 440 by technical staff at the University of Glasgow. Melting points were recorded with an Electrothermal IA 9100 apparatus.
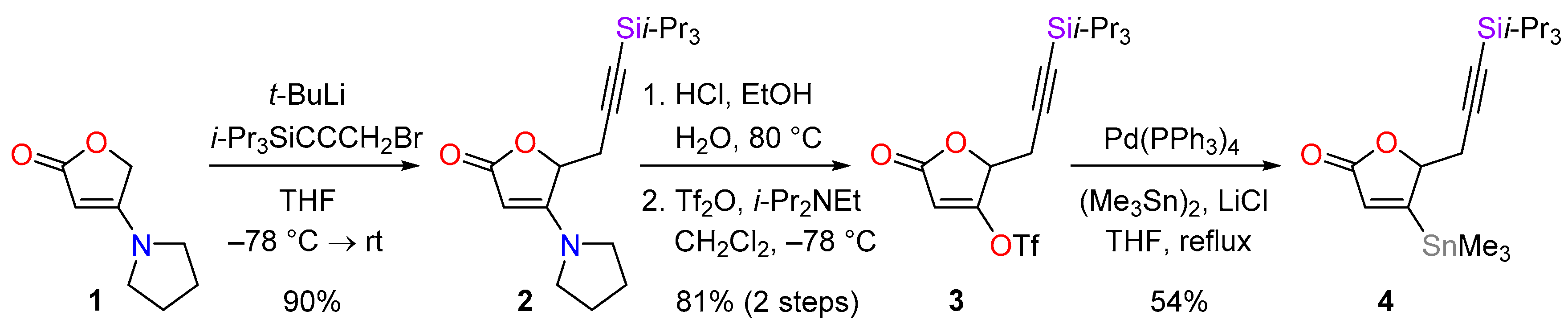
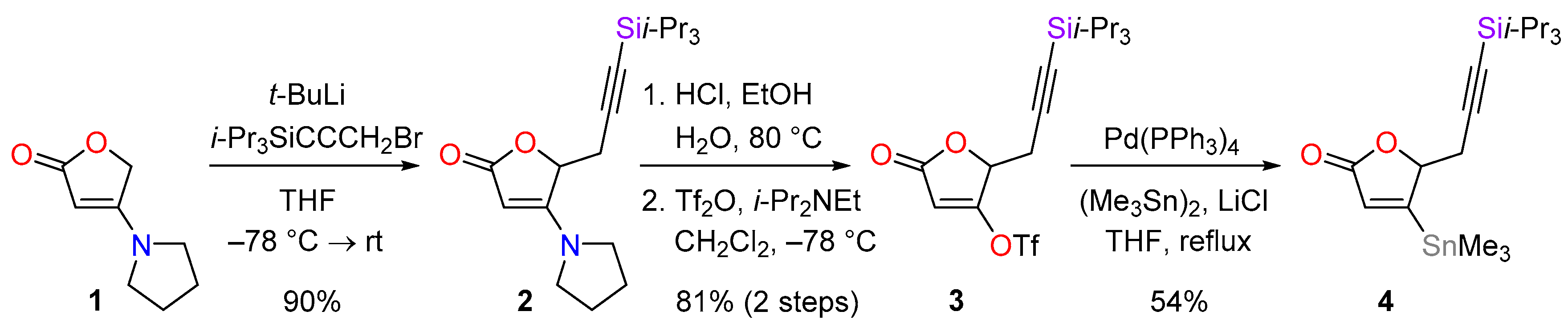
4.1. Synthesis of 4-(pyrrolidin-1-yl)-5-(3-triisopropylsilylprop-2-ynyl)furan-2(5H)-one (2)
To a solution of 1 (653 mg, 4.26 mmol) in THF (6 mL) at −78 °C was added carefully a solution of t-BuLi (4.0 mL of a 1.6 m solution in hexane, 6.4 mmol, 1.5 equiv.). The mixture was stirred for 30 min before a solution of 3-bromo-1-(triisopropylsilyl)-1-propyne (5.88 g, 21.4 mmol, 5.0 equiv.) in THF (8 mL) cooled to −78 °C was added carefully. After 3 h, the mixture was allowed to warm to room temperature over 45 min, and the reaction was quenched by the addition of saturated aqueous NH4Cl solution (10 mL). The mixture was diluted with ethyl acetate (20 mL) and the phases were separated. The aqueous phase was extracted with ethyl acetate (2 × 10 mL) and the combined organic extracts were washed with brine (20 mL), then dried over MgSO4, filtered and concentrated in vacuo. The crude material was purified by flash column chromatography (PE-EtOAc, 7:3) to give the desired alkyne 2 (1.34 g, 90%) as a colourless solid. M.p. 122−125 °C; Rf = 0.23 (PE-EtOAc, 1:1); IR νmax 2942, 2928, 2888, 2865, 2180, 1722, 1610, 994, 920, 901, 882, 853, 839, 773 cm−1; 1H-NMR (400 MHz, CDCl3) δ 4.95 (1H, dd, J = 4.5, 3.5 Hz), 4.55 (1H, s), 3.32 (4H, br s), 3.00 (1H, dd, J = 17.8, 3.5 Hz), 2.80 (1H, dd, J = 17.8, 4.5 Hz), 2.12−2.02 (2H, m), 1.99−1.90 (2H, m), 1.03 (21H, s); 13C-NMR (101 MHz, CDCl3) δ 173.9, 167.1, 100.6, 84.5, 83.3, 76.0 (CH-C5), 24.2, 18.7, 11.3; LRMS (CI, isobutane): m/z (int) 348 (98), 137 (61), 121 (51), 89 (94); HRMS (CI, isobutane) calculated for C20H34NO2Si [M + H]+: 348.2359; found 348.2364. Anal. calculated for C20H33NO2Si: C, 69.11; H, 9.57; N, 4.03. Found: C, 69.07; H, 9.65; N, 4.09.
4.2. Synthesis of 5-oxo-2-(3-triisopropylsilylprop-2-ynyl)-2,5-dihydrofuran-3-yl trifluoromethanesulfonate (3)
A solution of the lactone 2 (1.83 g, 5.27 mmol) was added to a solution of hydrochloric acid in EtOH (30 mL of a 1.2 m solution, 38 mmol, 7.1 equiv.) at 0 °C followed by water (3.0 mL). The mixture was heated at 78 °C for 5 h, cooled to room temperature and diluted with water (10 mL) and Et2O (40 mL). The phases were separated and the aqueous phase was extracted with Et2O (2 × 20 mL). The organic extracts were combined and washed with brine (20 mL), then dried over MgSO4, filtered and concentrated in vacuo. Azeotropic removal of water with toluene (3 × 50 mL) provided an orange residue that was used directly in the next step.
To a solution of the crude acid in CH2Cl2 (53 mL) at −78 °C was added dropwise freshly distilled Hünig’s base (1.40 mL, 8.03 mmol, 1.5 equiv.), and after 5 min triflic anhydride (4.15 mL, 6.96 mmol, 1.3 equiv.). The dark red solution was stirred at −78 °C for 1 h, then diluted with CH2Cl2 (30 mL) and warmed to room temperature. The reaction was quenched by the addition of water (20 mL) and the phases were separated. The aqueous phase was extracted with CH2Cl2 (2 × 20 mL), the organic extracts were combined, washed with brine (50 mL), dried with Na2SO4, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (PE-Et2O, 9:1 → 4:1) to afford the triflate 3 (1.82 g, 81% over two steps) as a colourless solid. M.p. 42−43 °C; Rf = 0.81 (PE-Et2O, 5:5); IR νmax 2947, 2870, 2176, 1767, 1643, 995, 818 cm−1; 1H-NMR (400 MHz, CDCl3) δ 6.08 (1H, d, J = 1.2 Hz), 5.07 (1H, ddd, J = 4.8, 3.5, 1.2 Hz), 3.09 (1H, dd, J = 17.7, 4.8 Hz), 2.84 (1H, dd, J = 17.7, 3.5 Hz), 1.03 (21H, s); 13C-NMR (101 MHz, CDCl3) δ 167.8, 167.2, 118.6 (q, J = 322 Hz), 104.8, 97.3, 87.1, 76.3, 23.1, 18.6, 11.2; LRMS (CI, isobutane): m/z (int) 427 (26), 334 (11), 279 (32), 276 (21), 235 (15), 181 (11), 133 (27), 97 (35), 71 (100). HRMS (CI, isobutane) calculated for C17H26F3O5SSi [M + H]+: 427.1222; found 427.1223. Anal. calculated for C17H25F3O5SSi: C, 47.87; H, 5.91. Found: C, 47.85; H, 5.97.
4.3. Synthesis of 4-trimethylstannyl-5-(3-triisopropylsilylprop-2-yn-1-yl)-2,5-dihydrofuran-2-one (4)
In a 50 mL 3-necked flask containing thoroughly flame-dried LiCl (600 mg, 14.2 mmol, 8.0 equiv.) and Pd(PPh3)4 (80.1 mg, 0.0693 mmol, 3.9 mol %) was added THF (5 mL). A solution of triflate 3 (755 mg, 1.77 mmol) in THF (17 mL) was added. After 5 min hexamethylditin (480 µL, 2.31 mmol, 1.3 equiv.) was added and the mixture was heated at reflux for 1.5 h. Further Pd(PPh3)4 (67.8 mg, 0.0586 mmol, 3.3 mol %) was then added and the mixture was stirred for 1.5 h. The mixture was cooled to 0 °C and the reaction was quenched with saturated aqueous NaHCO3 (15 mL) and diluted with Et2O (40 mL). The phases were separated and the aqueous phase was extracted with Et2O (2 × 30 mL). The organic extracts were combined, washed with brine (40 mL), dried with Na2SO4, filtered and concentrated in vacuo. The crude product was purified by flash column chromatography (PE-Et2O, 19:1 → 4:1) to give the corresponding stannane 4 (419 mg, 54%) as a pale yellow solid. M.p. 69−72 °C; Rf = 0.21 (PE-Et2O, 4:1); IR νmax 2940, 2863, 2176, 1751, 918, 880, 779 cm−1; 1H-NMR (400 MHz, CDCl3) δ 6.22 (1H, d, J = 1.9 Hz), 5.21 (1H, ddd, J = 5.3, 3.6, 1.9 Hz), 3.04 (1H, dd, J = 17.3, 5.3 Hz), 2.65 (1H, dd, J = 17.3, 3.6 Hz), 1.01 (21H, br s), 0.36 (9H, s); 13C-NMR (101 MHz, CDCl3) δ 175.4, 172.4, 131.1, 100.4, 85.8, 85.5, 25.1, 18.7, 11.3, −9.2; LRMS (CI, isobutane): m/z (int) 443 (76), 441 (57), 337 (11), 279 (48), 257 (15), 235 (12), 113 (20), 69 (100). HRMS (CI, isobutane) calculated for C19H35O2Si120Sn [M + H]+: 443.1429; found 443.1424.
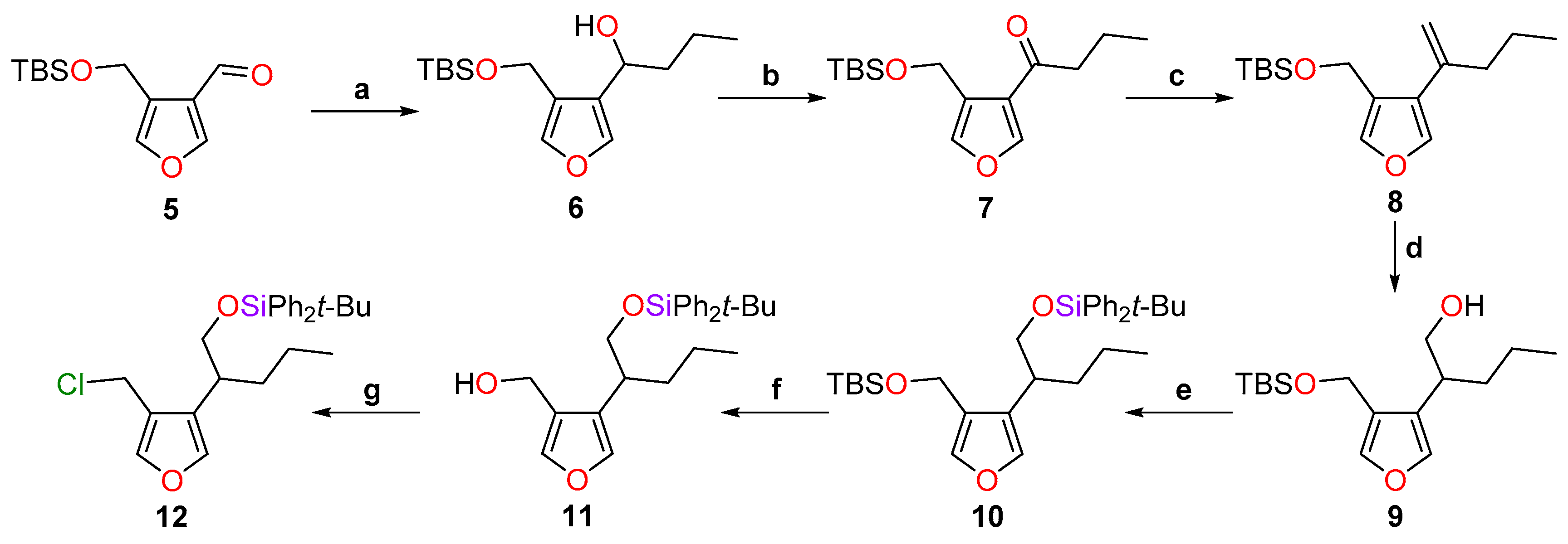
4.4. Synthesis of 1-[4-(tert-butyldimethylsilyloxy)methylfuran-3-yl]butan-1-ol (6)
To a suspension of lithium aluminium hydride (4.74 g, 125 mmol, 2.3 equiv.) in THF (200 mL) at −78 °C was added a solution of dimethyl 3,4-furandicarboxylate (10.0 g, 54.3 mmol) in THF (200 mL) over 20 min at −78 °C. The solution was warmed gently to room temperature over 2 h and stirred overnight. The reaction was cooled to 0 °C and quenched carefully by success addition of water (4.7 mL), aqueous NaOH (1 m, 4.7 mL) and water (13 mL). After warming to room temperatureand stirring for 1 h, a cloudy white suspension was formed. MgSO4 (~15 g) was added and the mixture was filtered through a pad of Celite and washed with ethyl acetate (1 L). After concentration in vacuo, the pale-yellow oil obtained was used directly for the next step.
To a solution of the crude diol obtained (6.95 g, ~54.2 mmol) in CH2Cl2 (450 mL) was added activated manganese(II) oxide (28.3 g, 326 mmol, 6.0 equiv.) at room temperature. The mixture was stirred vigorously for 2.5 h and further manganese(II) oxide (9.50 g, 108 mmol, 2.0 equiv.) was added three times at regular intervals. The black suspension was then filtered through a pad of Celite and washed with CH2Cl2 (1.5 L). After concentration in vacuo, the crude yellow oil was separated into two fractions—A (3.46 g) and B (3.72 g)—which were used in the subsequent steps without any further purification.
To a solution of the crude aldehyde A (3.46 g, ~27.5 mmol) in CH2Cl2 (250 mL), imidazole (2.24 g, 33.0 mmol, 1.2 equiv.), DMAP (336 mg, 2.75 mmol, 0.10 equiv.) and t-butyldimethylsilyl chloride (4.55 g, 30.2 mmol, 1.1 equiv.) were added sequentially. The solution was stirred for 20 min at room temperature and then water (60 mL) was added. The phases were separated and the aqueous phase was extracted with CH2Cl2 (2 × 60 mL). The organic extracts were combined, washed with brine (100 mL), dried over MgSO4, filtered and concentrated in vacuo. The resulting pale yellow oil was used immediately in the next step.
To a solution of the silylated aldehyde 5 (~27.5 mmol) in THF (250 mL) at −78 °C was added dropwise n-propylmagnesium chloride (23.3 mL of a 2.0 m solution in THF, 46.6 mmol, 1.7 equiv.). The solution was stirred at −78 °C for 1.5 h, warmed to 0 °C and the reaction was quenched by the addition of a saturated aqueous solution of NH4Cl (35 mL). Water (35 mL) and Et2O (60 mL) were added and the phases separated. The aqueous phase was extracted with Et2O (2 × 60 mL) and the organic extracts washed with brine (120 mL), dried over MgSO4, filtered and concentrated in vacuo. The residual material was purified by flash column chromatography (PE-Et2O, 9:1) to give the desired alcohol 6 as a colourless oil.
The same procedure was used to convert aldehyde B in the alcohol 6 and the batches were combined (10.7 g, 69% over 4 steps). Rf = 0.34 (PE-Et2O, 9:1); IR νmax 3349, 2955, 2929, 2858, 1749, 1669, 960, 877, 815, 777, 760, 742 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.30 (2H, s), 4.65 (1H, d, J = 12.2 Hz), 4.61 (1H, d, J = 12.2 Hz), 4.60 (1H, dt, J = 7.9, 5.6 Hz), 3.58 (1H, d, J = 5.6 Hz), 1.84 (1H, dddd, J = 13.4, 9.8, 7.9, 5.5 Hz), 1.73 (1H, dddd, J = 13.4, 9.8, 5.9, 5.6 Hz), 1.57−1.48 (1H, m), 1.43−1.34 (1H, m), 0.96 (3H, t, J = 7.4 Hz), 0.91 (9H, s), 0.12 (3H, s), 0.12 (3H, s); 13C-NMR (101 MHz, CDCl3) δ 140.6, 140.1, 128.5, 123.7, 65.8, 56.8, 38.5, 26.0, 19.5, 18.4, 14.1, −5.2; LRMS (CI, isobutane): m/z (int) 267 (31), 135 (49), 107 (9), 89 (100), 69 (20). HRMS (CI, isobutane) calculated for C15H27O2Si [M − OH]+: 267.1780; found 267.1775.
4.5. Synthesis of 1-[4-(tert-butyldimethylsilyloxy)methylfuran-3-yl]butan-1-one (7)
To a solution of alcohol 6 (522 mg, 1.94 mmol) in CH2Cl2 (20 mL) was added activated manganese(II) oxide (3.43 g, 39.5 mmol, 20 equiv.). The mixture was stirred at room temperature for 2 h, then heated at reflux for 2 h, and stirred overnight at room temperature. The suspension was filtered through a pad of Celite and washed with CH2Cl2 (500 mL). The filtrate was concentrated in vacuo and the residue was purified by flash column chromatography (PE-Et2O, 19:1), affording the desired ketone 7 (468 mg, 90%) as a colourless oil. Rf = 0.63 (PE-Et2O, 9:1); IR νmax 2957, 2930, 2886, 2859, 2361, 1676, 837, 814, 777 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.98 (1H, d, J = 1.7 Hz), 7.40 (1H, q, J = 1.7 Hz), 4.87 (2H, d, J = 1.7 Hz), 2.69 (2H, t, J = 7.3 Hz), 1.72 (2H, qt, J = 7.4, 7.3 Hz), 0.97 (3H, t, J = 7.4 Hz), 0.93 (9H, s), 0.10 (6H, s); 13C-NMR (101 MHz, CDCl3) δ 196.3, 148.5, 141.6, 127.1, 125.4, 58.8, 42.3, 26.1, 18.5, 17.9, 14.0, −5.3; LRMS (CI, isobutane): m/z (int) 283 (46), 225 (8), 89 (100), 69 (10). HRMS (CI, isobutane) calculated for C15H27O3Si [M + H]+: 283.1729; found 283.1732.
4.6. Synthesis of tert-butyldimethyl{[4-(pent-1-en-2-yl)furan-3-yl]methoxy}silane (8)
To a suspension of methyltriphenylphosphonium bromide (2.87 g, 8.03 mmol, 5.0 equiv.) in THF (7.5 mL) at 0 °C was added dropwise a solution of NaHMDS (6.4 mL of a 1.0 m solution in THF, 6.4 mmol, 4.0 equiv.). The bright yellow suspension was warmed to room temperature and stirred for 1 h, before being cooled to 0 °C. A solution of ketone 7 (453 mg, 1.60 mmol) in THF (7 mL) was added dropwise and the mixture was stirred for 1 h at room temperature. The reaction was quenched by the addition of a saturated aqueous solution of NH4Cl (10 mL) and the mixture was diluted with Et2O (30 mL). The phases were separated and the aqueous phase was extracted with Et2O (2 × 20 mL). The organic extracts were combined, washed with brine (40 mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (PE-Et2O, 99:1) to give the corresponding 1,1-disubstituted alkene 8 (426 mg, 95%) as a colourless oil. Rf = 0.91 (PE-Et2O, 19:1); IR νmax 2957, 2930, 2857, 1636, 874, 833, 814, 793, 773, 736 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.36 (1H, s), 7.34 (1H, s), 5.12 (1H, s), 5.00 (1H, d, J = 0.9 Hz), 4.63 (2H, s), 2.29 (2H, t, J = 7.4 Hz), 1.51 (2H, qt, J = 7.4, 7.4 Hz), 0.92 (3H, t, J = 7.4 Hz), 0.91 (9H, s), 0.08 (6H, s); 13C-NMR (101 MHz, CDCl3) δ 141.4, 140.5, 139.8, 125.7, 124.7, 112.6, 57.6, 38.6, 26.0, 21.5, 18.5, 14.0, −5.1; HRMS (ESI) calculated for C16H28NaO2Si [M + Na]+: 303.1751; found 303.1744.
4.7. Synthesis of 2-[4-(tert-butyldimethylsilyloxymethyl)furan-3-yl]pentan-1-ol (9)
To a solution of 1,1-disubstituted alkene 8 (1.07 g, 3.81 mmol) in THF (4 mL), cooled to 0 °C, was added dropwise a solution of 9-BBN (23.0 mL of a 0.5 m solution in THF, 11.5 mmol, 3.0 equiv.). The mixture was warmed to 65 °C for 75 min and then cooled to 0 °C before careful addition of EtOH (18 mL) and aqueous 3 m NaOH (11.5 mL). After 15 min, aqueous hydrogen peroxide (30%, 18 mL) was added. The mixture was heated at reflux for 1 h, cooled to room temperature before the addition of Et2O (60 mL) and water (20 mL). The phases were separated and the aqueous phase was extracted with Et2O (2 × 50 mL). The organic extracts were combined and washed with brine, dried over MgSO4, filtered and concentrated in vacuo. The crude product was purified by flash column chromatography (PE-Et2O, 9:1) to give the desired primary alcohol 9 (1.01 g, 89%) as a colourless oil. Rf = 0.18 (PE-Et2O, 9:1); IR νmax 3381, 2955, 2930, 2895, 2858, 1602, 1541, 871, 837, 775 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.33 (1H, d, J = 1.6 Hz), 7.23 (1H, d, J = 1.6 Hz), 4.56 (1H, d, J = 12.5 Hz), 4.53 (1H, d, J = 12.5 Hz), 3.73 (1H, ddd, J = 10.8, 6.4, 4.9 Hz), 3.61 (1H, ddd, J = 10.8, 7.3, 6.0 Hz), 2.83−2.75 (1H, m), 2.17 (1H, dd, J = 6.4, 6.0 Hz), 1.67−1.57 (1H, m), 1.56−1.48 (1H, m), 1.38−1.27 (2H, m), 0.91 (9H, s), 0.89 (3H, t, J = 5.9 Hz), 0.10 (6H, s); 13C-NMR (126 MHz, CDCl3) δ 141.1, 140.6, 125.6, 124.9, 66.8, 56.5, 38.1, 33.8, 26.0, 20.8, 18.5, 14.2, −5.2; HRMS (ESI) calculated for C16H30NaO3Si [M + Na]+: 321.1856; found 321.1846.
4.8. Synthesis tert-butyl{[4-(1-tert-butyldiphenylsilyloxypentan-2-yl)furan-3-yl]methoxy}dimethylsilane (10)
To a solution of alcohol 9 (1.72 g, 5.76 mmol) in CH2Cl2 (58 mL), DMAP (203 mg, 1.66 mmol, 0.3 equiv.), t-butyldiphenylsilyl chloride (2.26 mL, 8.81 mmol, 1.5 equiv.) and Et3N (1.37 mL, 9.83 mmol, 1.7 equiv.) were added successively at 0 °C. The solution was stirred overnight at room temperature and the reaction was quenched with a saturated aqueous solution of NH4Cl (20 mL). The phases were separated and the aqueous phase was extracted with CH2Cl2 (2 × 30 mL). The organic extracts were combined and then washed with brine (40 mL), dried over MgSO4, filtered and concentrated in vacuo. The crude product was purified by flash column chromatography (PE-CH2Cl2, 9:1) to afford furan 10 (2.81 g, 91%) as a colourless oil. Rf = 0.50 (PE-CH2Cl2, 8:2); IR νmax 3073, 3050, 2955, 2930, 2857, 1589, 1541, 835, 775, 739, 700 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.61−5.56 (4H, m), 7.44−7.33 (6H, m), 7.27 (1H, d, J = 0.8 Hz), 7.17 (1H, d, J = 0.8 Hz), 4.45 (2H, s), 3.67 (1H, dd, J = 10.1, 5.8 Hz), 3.63 (1H, dd, J = 10.1, 6.4 Hz), 2.75 (1H, dddd, J = 8.6, 6.4, 6.1, 5.8 Hz), 1.78 (1H, app ddt, J = 13.3, 9.7, 6.1 Hz), 1.54−1.44 (1H, m), 1.35−1.20 (2H, m), 1.02 (9H, s), 0.90−0.84 (12H, m), 0.04 (3H, s), 0.03 (3H, s); 13C-NMR (101 MHz, CDCl3) δ 140.4, 139.9, 135.8, 135.7, 133.9, 129.7, 129.7, 127.7, 125.6, 125.5, 67.3, 56.9, 37.5, 34.0, 27.0, 26.0, 20.5, 19.4, 18.4, 14.4, −5.2; LRMS (CI, isobutane): m/z (int) 537 (5), 461 (27), 447 (12), 405 (100), 133 (13). HRMS (CI, isobutane) calculated for C32H49O3Si2 [M + H]+: 537.3220; found 537.3221.
4.9. Synthesis of [4-(1-tert-butyldiphenylsilyloxypentan-2-yl)furan-3-yl]methanol (11)
To a solution of furan 10 (2.81 g, 5.23 mmol) in ethanol (18 mL) was added PPTS (660 mg, 2.63 mmol, 0.5 equiv.) and the mixture was stirred overnight at 40 °C. The reaction was quenched with a saturated aqueous solution of NaHCO3 (10 mL) and the ethanol was removed in vacuo. The residue was diluted with Et2O (20 mL) and the phases were separated. The aqueous phase was extracted with Et2O (2 × 10 mL) and the combined organic extracts were washed with brine (20 mL), dried over MgSO4, filtered and concentrated in vacuo. Residual material was purified by flash column chromatography (PE-Et2O, 9:1) to give the desired alcohol 11 (2.09 g, 94%) as a colourless oil. Rf = 0.20 (PE-Et2O, 9:1); IR νmax 3366, 2042, 2893, 2866, 1757, 1600, 1541 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.62−7.53 (4H, m), 7.45−7.34 (7H, m), 7.19 (1H, d, J = 1.5 Hz), 4.42 (2H, d, J = 5.4 Hz), 3.72 (1H, dd, J = 9.8, 5.7 Hz), 3.63 (1H, dd, J = 9.8, 6.9 Hz), 2.81 (1H, ddt, J = 9.1, 6.9, 5.7 Hz), 1.95 (1H, t, J = 5.4 Hz), 1.69 (1H, dddd, J = 13.1, 9.5, 6.4, 5.7 Hz), 1.54−1.42 (1H, m), 1.37−1.20 (2H, m), 1.02 (9H, s), 0.87 (3H, t, J = 7.3 Hz); 13C-NMR (101 MHz, CDCl3) δ 140.5, 140.5, 135.8, 135.7, 133.4, 133.4, 129.8, 127.8, 127.7, 126.1, 125.4, 68.7, 55.5, 37.3, 34.0, 26.9, 20.6, 19.3, 14.3; LRMS (CI, isobutane): m/z (int) 405 (34), 365 (6), 341 (5), 265 (100), 237 (12), 217 (75) 135 (9). HRMS (CI, isobutane) calculated for C26H33O2Si [M − OH]+: 405.2250; found 405.2252.
4.10. Synthesis of tert-butyl[2-(4-chloromethylfuran-3-yl)pentyloxy]diphenylsilane (12)
To a solution of the alcohol 11 (1.47 g, 3.48 mmol) in CH2Cl2 (12 mL) cooled to 0 °C, Et3N (870 μL, 6.24 mmol, 1.8 equiv.) and MsCl (405 μL, 5.23 mmol, 1.5 equiv.), both freshly distilled, were added successively. The mixture was warmed to room temperature, stirred overnight and the reaction was quenched by the addition of a saturated aqueous solution of NH4Cl (10 mL). The phases were separated and the aqueous phase was extracted with CH2Cl2 (2 × 15 mL). The organic extracts were combined and dried over MgSO4, filtered and concentrated in vacuo. The crude product was purified by flash column chromatography (PE-CH2Cl2, 9:1) to deliver the chloride 12 (1.38 g, 90%) as a colourless oil. Rf = 0.40 (PE-CH2Cl2, 9:1); IR νmax 3071, 2943, 2910, 2862, 1589, 1543, 864, 702 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.61−7.57 (4H, m), 7.44−7.33 (7H, m), 7.20 (1H, d, J = 1.5 Hz), 4.35 (2H, s), 3.67 (2H, d, J = 5.8 Hz), 2.82 (1H, dtd, J = 9.0, 5.8, 5.7 Hz), 1.81 (1H, dddd, J = 13.2, 9.3, 6.5, 5.7 Hz), 1.59−1.46 (1H, m), 1.37−1.25 (2H, m), 1.03 (9H, s), 0.90 (3H, t, J = 7.3 Hz); 13C-NMR (101 MHz, CDCl3) δ 141.5, 140.9, 135.8, 135.7, 133.8, 133.7, 129.8, 129.7, 127.8, 126.1, 122.5, 67.8, 37.2, 36.4, 34.1, 27.0, 20.6, 19.4, 14.3; LRMS (CI, isobutane): m/z (int) 441 (60), 405 (98), 363 (72), 241 (61), 227 (77), 185 (100), 149 (45), 91 (31). HRMS (CI, isobutane) calculated for C26H34O235ClSi [M + H]+: 441.2017; found 441.2015. Anal. calculated for C26H33O2ClSi: C, 70.80; H, 7.54. Found: C, 70.93; H, 7.59.
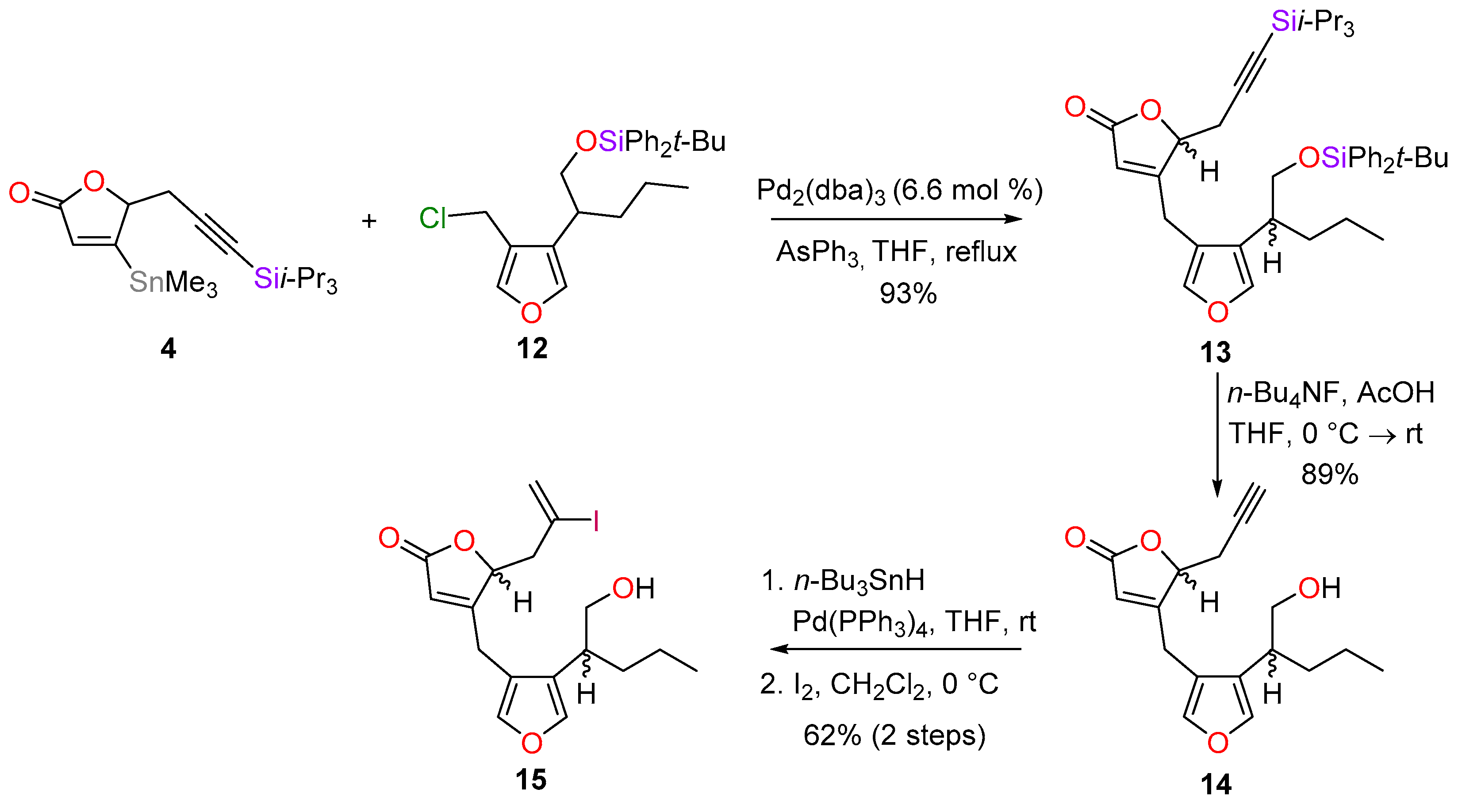
4.11. Synthesis of 4-[4-(1-tert-butyldiphenylsilyloxypentan-2-yl)furan-3-yl]methyl-5-[3-triisopropylsilyl-prop-2-yn-1-yl]-2,5-dihydrofuran-2-one (13)
To a solution of chloride 12 (449 mg, 1.02 mmol) in THF (2 mL) was added Pd2(dba)3 (57 mg, 0.062 mmol, 6.6 mol %) and triphenylarsine (106 mg, 0.346 mmol, 0.36 equiv.). The purple to yellow mixture was stirred for 5 min at room temperature before a solution of the stannane 4 (419 mg, 0.950 mmol) in THF (9 mL) was added. The mixture was heated at 65 °C overnight, cooled to room temperature and then diluted with Et2O (30 mL) and H2O (10 mL). The phases were separated and the aqueous phase was extracted with Et2O (2 × 20 mL). The organic extracts were combined, washed with brine (30 mL), dried over MgSO4, filtered and concentrated in vacuo. Residual material was purified by flash column chromatography (PE−Et2O, 95:5→92:8) to give the corresponding product 13 (605 mg, 93%) as a pale-yellow oil and an inseparable mixture (1:1) of diastereoisomers. Rf = 0.69 (PE−Et2O, 7:3); IR νmax 3071, 2932, 2862, 2175, 1759, 1643, 1589, 995, 926, 872, 802, 741, 702, 679 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.58−7.54 (8H, m), 7.45−7.33 (12H, m), 7.25 (1H, d, J = 1.6 Hz), 7.24−7.23 (2H, m), 7.22 (1H, d, J = 1.5 Hz), 5.72 (1H, d, J = 1.3 Hz), 5.67 (1H, d, J = 1.5 Hz), 4.82 (1H, app t, J = 5.0 Hz), 4.80 (1H, app t, J = 5.0 Hz), 3.62−3.59 (4H, m), 3.52 (1H, d, J = 17.7 Hz), 3.43 (1H, d, J = 18.4 Hz), 3.24 (1H, dd, J = 17.7, 1.5 Hz), 3.20 (1H, d, J = 18.4 Hz), 2.90 (1H, dd, J = 5.1, 2.0 Hz), 2.86 (1H, dd, J = 5.1, 2.0 Hz), 2.67 (1H, dd, J = 5.1, 4.0 Hz), 2.63 (1H, dd, J = 5.1, 3.9 Hz), 2.54 (1H, dq, J = 9.0, 5.8 Hz), 2.49 (1H, dq, J = 8.7, 5.8 Hz), 1.79−1.69 (2H, m), 1.51−1.38 (2H, m), 1.30−1.16 (4H, m), 1.05−0.98 (60H, m), 0.86 (3H, t, J = 7.3 Hz), 0.87 (3H, t, J = 7.3 Hz); 13C-NMR (101 MHz, CDCl3) δ 171.9, 169.7, 169.5, 140.8, 140.7, 140.4, 140.3, 135.7, 135.6, 133.6, 133.5, 129.9, 129.8, 127.8, 126.2, 119.4, 118.4, 99.7, 99.6, 85.4, 80.5, 80.4, 67.9, 67.8, 37.5, 34.2, 34.1, 27.0, 23.6, 23.5, 23.1, 23.0, 20.7, 19.4, 18.7, 14.4, 11.3; LRMS (FAB): m/z (int) 705 (100), 605 (67), 427 (9), 197 (40), 135 (73), 59 (42). HRMS (FAB) calculated for C42H58NaO4Si2 [M+Na]+: 705.3771; found 705.3763.
4.12. Synthesis of 4-[4-(1-hydroxypentan-2-yl)furan-3-yl]methyl-5-(prop-2-yn-1-yl)-2,5-dihydrofuran-2-one (14)
To a solution of 13 (1.23 g, 1.80 mmol) in THF (31 mL) at 0 °C was added acetic acid (400 µL, 6.99 mmol, 3.88 equiv.) and TBAF (7.4 mL of a 1 m solution in THF, 7.4 mmol, 4.1 equiv.). The mixture was warmed slowly to room temperature, stirred for 24 h and then diluted with water (10 mL) and ethyl acetate (30 mL). The phases were separated and the aqueous phase was extracted with ethyl acetate (2 × 30 mL). The organic extracts were combined, washed with brine (50 mL), dried over MgSO4, filtered and concentrated in vacuo. Residual material was purified by flash column chromatography (PE−Et2O, 1:1→1:4) to give a diastereomeric mixture of the corresponding unprotected alcohols 14 (461 mg, 89%) as a pale-yellow oil and a partially separable 1:1 mixture of diastereoisomers. Rf = 0.44 and 0.42 (PE-Et2O, 1:4); IR νmax 3446, 2954, 2933, 2869, 2360, 1742, 1645, 924, 875, 850, 798 cm−1; Less polar diastereoisomer 1H-NMR (400 MHz, CDCl3) δ 7.30 (2H, s), 5.78 (1H, ddd, J = 1.5, 1.5, 1.2 Hz), 5.03 (1H, ddd, J = 5.6, 4.2, 1.2 Hz), 3.68−3.60 (2H, m), 3.59 (1H, dd, J = 18.3, 1.5 Hz), 3.45 (1H, dd, J = 18.3, 1.5 Hz), 2.85 (1H, ddd, J = 17.3, 5.6, 2.6 Hz), 2.73 (1H, ddd, J = 17.3, 4.2, 2.7 Hz), 2.59 (1H, dq, J = 8.6, 5.9 Hz), 2.07 (1H, dd, J = 2.7, 2.6 Hz), 1.69−1.59 (1H, m), 1.52−1.41 (1H, m), 1.36−1.23 (2H, m), 0.89 (3H, t, J = 7.3 Hz); Less polar diastereoisomer 13C-NMR (101 MHz, CDCl3) δ 172.0, 169.9, 141.0, 140.5, 126.0, 119.5, 118.5, 80.3, 76.6, 72.6, 66.7, 37.5, 34.5, 23.2, 22.6, 20.7, 14.3; More polar diastereoisomer 1H-NMR (400 MHz, CDCl3) δ 7.30 (2H, s), 5.81 (1H, ddd, J = 1.6, 1.6, 1.5 Hz), 5.00 (1H, ddd, J = 6.3, 4.6, 1.5 Hz), 3.70−3.59 (2H, m), 3.59 (1H, dd, J = 18.2, 1.6 Hz), 3.44 (1H, dd, J = 18.2, 1.6 Hz), 2.87 (1H, ddd, J = 17.3, 6.3, 2.5 Hz), 2.74 (1H, ddd, J = 17.3, 4.6, 2.5 Hz), 2.68−2.55 (1H, m), 2.08 (1H, t, J = 2.5 Hz), 1.69−1.58 (1H, m), 1.52−1.40 (1H, m), 1.36−1.27 (2H, m), 0.89 (3H, t, J = 7.3 Hz,); More polar diastereoisomer 13C-NMR (101 MHz, CDCl3) δ 172.0, 169.7, 141.1, 140.6, 126.0, 119.4, 118.5, 80.2, 76.5, 72.7, 66.9, 37.5, 34.2, 23.2, 22.6, 20.6, 14.3; LRMS (CI, isobutane): m/z (int) 289 (100), 71 (13). HRMS (CI, isobutane) calculated for C17H21O4 [M + H]+: 289.1440; found 289.1442.
4.13. Synthesis of (5S*)-4-{4-[(2S*)-1-Hydroxypentan-2-yl]furan-3-yl}methyl-5-(2-iodoprop-2-en-1-yl)-2,5-dihydrofuran-2-one (15a) and (5R*)-4-{4-[(2S*)-1-Hydroxypentan-2-yl]furan-3-yl}methyl-5-(2-iodoprop-2-en-1-yl)-2,5-dihydrofuran-2-one (15b)
To a solution of alkynes 14 (419 mg, 1.45 mmol) in THF (6.6 mL) at room temperature was added Pd(PPh3)4 (60.1 mg, 0.0520 mmol, 3.6 mol %) followed by tributyltin hydride (425 µL, 1.58 mmol, 1.1 equiv.). The mixture was stirred for 20 min and the reaction was quenched with saturated aqueous NaHCO3 (5 mL). The mixture was diluted with Et2O (10 mL) and the phases were then separated. The aqueous phase was extracted with Et2O (3 × 10 mL) and the organic extracts were combined, washed with brine (20 mL), dried with Na2SO4, filtered and concentrated in vacuo. The crude material was purified by flash column chromatography (PE-Et2O, 1:1→2:3) to give a partially separable regioisomeric mixture (3.2:1 ratio, estimated by 1H-NMR analysis) in favour of the required vinylic stannane. Most of the required stannane (572 mg) were isolated and taken straight to the next step for better characterisation. Rf = 0.68 and 0.76 (PE-Et2O, 1:4).
To a solution of the vinylic stannane (572 mg, 0.988 mmol) in CH2Cl2 (10 mL) at 0 °C was added I2 (289 mg, 1.14 mmol, 1.15 equiv.). The mixture was stirred for 20 min and the reaction was then quenched with a saturated aqueous solution of Na2S2O3 (15 mL). The mixture was diluted with CH2Cl2 (20 mL) and after 10 min, the phases were separated. The aqueous phase was extracted with CH2Cl2 (2 × 20 mL) and the organic extracts were combined, washed with brine (30 mL), dried with Na2SO4, filtered and concentrated in vacuo. The crude product was purified by flash column chromatography (PE-Et2O, 55:45) to give the separable diastereomeric vinyl iodides 15a and 15b (378 mg, 62% combined over two steps) as colourless oils. 15a (less polar diastereomer) Rf = 0.36 (PE-Et2O, 1:4); IR νmax 3446, 2955, 2929, 2870, 1743, 1637, 1618, 1541, 960, 912, 871, 857, 840, 799 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.30 (2H, s), 6.25 (1H, d, J = 1.7 Hz), 5.91 (1H, d, J = 1.7 Hz), 5.75 (1H, ddd, J = 1.4, 1.1, 1.1 Hz), 5.16 (1H, ddd, J = 8.7, 3.6, 1.4 Hz), 3.65 (1H, dd, J = 10.5, 5.6 Hz), 3.59 (1H, dd, J = 10.5, 6.8 Hz), 3.55 (1H, dd, J = 17.8, 1.1 Hz), 3.46 (1H, dd, J = 17.8, 1.1 Hz), 2.94 (1H, dd, J = 15.0, 3.6 Hz), 2.64 (1H, dd, J = 15.0, 8.7 Hz), 2.56 (1H, dddd, J = 8.5, 6.8, 5.9, 5.6 Hz), 1.69−1.58 (2H, m), 1.51−1.40 (1H, m), 1.36−1.23 (2H, m), 0.89 (3H, t, J = 7.3 Hz); 13C-NMR (101 MHz, CDCl3) δ 172.0, 170.2, 141.0, 140.5, 130.5, 126.0, 119.4, 118.3, 102.1, 81.9, 66.8, 48.2, 37.6, 34.4, 23.4, 20.7, 14.3; LRMS (EI+): m/z (int) 416 (100), 385 (10), 289 (56), 271 (20), 215 (33), 161 (35), 119 (21), 91 (45), 77 (20), 55 (10). HRMS (EI+) calculated for C17H21IO4 [M]+: 416.0485; found 416.0488. 15b (more polar diastereomer) Rf = 0.31 (PE-Et2O, 2:8); IR νmax 3446, 2955, 2928, 2869, 1746, 1638, 1618, 1539, 914, 870, 857, 840, 799 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.30 (2H, s), 6.26 (1H, dd, J = 1.7, 0.8 Hz), 5.91 (1H, d, J = 1.7 Hz), 5.78 (1H, ddd, J = 1.5, 1.4, 1.1 Hz), 5.13 (1H, ddd, J = 8.6, 3.6, 1.5 Hz), 3.67 (1H, dd, J = 10.5, 5.5 Hz), 3.60 (1H, dd, J = 18.1, 1.1 Hz), 3.59 (1H, dd, J = 10.5, 6.9 Hz), 3.44 (1H, dd, J = 18.1, 1.4 Hz), 2.95 (1H, dd, J = 15.0, 3.6 Hz), 2.63 (1H, ddd, J = 15.0, 8.6, 0.8 Hz), 2.57 (1H, dddd, J = 8.6, 6.9, 5.6, 5.5 Hz), 1.63 (1H, dddd, J = 13.1, 9.6, 6.1, 5.6 Hz), 1.55 (1H, br s), 1.51−1.41 (1H, m), 1.36−1.22 (2H, m), 0.89 (3H, t, J = 7.3 Hz); 13C-NMR (101 MHz, CDCl3) δ 172.0, 170.1, 141.0, 140.6, 130.5, 126.0, 119.4, 118.3, 102.2, 81.8, 67.0, 48.2, 37.6, 34.2, 23.4, 20.7, 14.3; LRMS (CI, isobutane): m/z (int) 417 (69), 291 (100), 273 (46), 251 (10), 97 (13), 71 (28). HRMS (CI, isobutane) calculated for C17H22IO4 [M + H]+: 417.0563; found 417.0564.
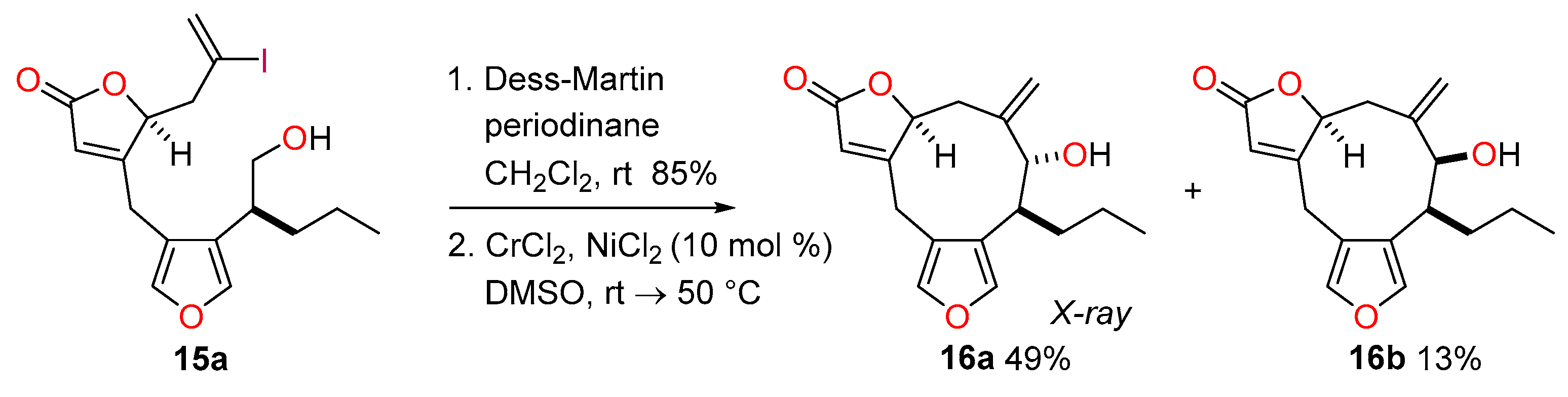
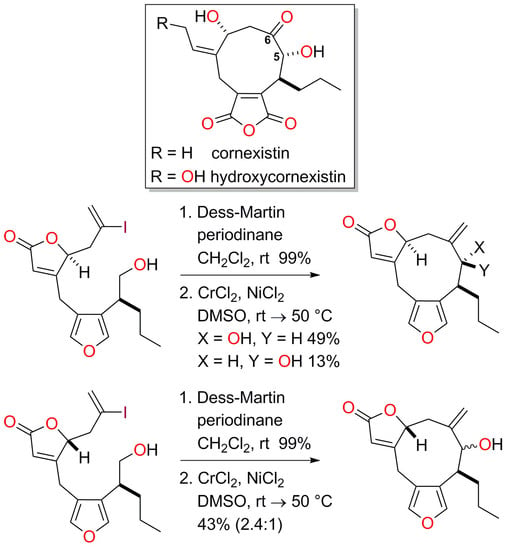
4.14. Synthesis of (8S*,9R*,12S*)-9-Hydroxy-10-methylidene-8-propyl-5,13-dioxatricyclo[10.3.0.03,7]penta-deca-1(15),3,6-trien-14-one (16a) and (8S*,9S*,12S*)-9-Hydroxy-10-methylidene-8-propyl-5,13-dioxa-tricyclo[10.3.0.03,7]pentadeca-1(15),3,6-trien-14-one (16b)
To a solution of the alcohol 15a (59.2 mg, 0.142 mmol) in CH2Cl2 (2.8 mL) was added Dess-Martin periodinane (132 mg, 0.311 mmol, 2.2 equiv.). The mixture was stirred at room temperature for 2 h and then cooled to 0 °C. The reaction was quenched by the addition of a saturated aqueous solution of Na2S2O3 (5 mL) and the mixture was diluted with water (5 mL) and CH2Cl2 (5 mL). After 10 min, the phases were separated and the aqueous phase was extracted with CH2Cl2 (3 × 5 mL). The organic extracts were combined and washed with brine (10 mL), then dried with Na2SO4, filtered and concentrated in vacuo. The crude product was purified quickly by passage through a small plug of silica (PE-Et2O, 4:1) to afford the corresponding aldehyde (50.1 mg, 85%) as a colourless oil.
To a solution of CrCl2 (200 mg, 1.63 mmol, 13.5 equiv.) and NiCl2 (1.6 mg, 0.012 mmol, 10 mol %) in previously degassed (three freeze-thaw cycles) DMSO (6 mL) was added a solution of aldehyde (50.1 mg) in degassed (three freeze-thaw cycles) DMSO (6 mL) at room temperature. The dark green mixture was stirred at room temperature for 24 h and at 50 °C for 16 h. The reaction was quenched by the addition of a saturated aqueous solution of NH4Cl (15 mL) and the mixture was diluted with EtOAc (30 mL). The biphasic mixture was stirred for 30 min, the phases were separated and the aqueous phase was extracted with EtOAc (3 × 30 mL). The organic extracts were combined, washed with brine (50 mL), dried over MgSO4, filtered and concentrated in vacuo. Residual material was purified by flash column chromatography (PE-Et2O, 9:11) allowing the separation of minor diastereomer 16b (5.5 mg, 13% over two steps) as a colourless oil and major diastereomer 16a (20.2 mg, 49% over two steps) as a colourless solid. The crystal structure of the major alcohol product 16a was obtained. 16b (less polar, minor diastereomer) M.p. 140−143 °C; Rf = 0.53 (PE-Et2O, 1:4); IR νmax 3454, 2955, 2930, 2870, 1743, 1636, 1537, 926, 868, 802, 766, 739 cm−1; 1H-NMR (500 MHz, CDCl3) δ 7.37 (1H, d, J = 1.5 Hz), 7.23 (1H, d, J = 1.5 Hz), 5.89 (1H, ddd, J = 1.6, 1.1, 1.1 Hz), 5.23 (1H, ddd, J = 3.6, 2.7, 1.6 Hz), 4.97 (1H, s), 4.94 (1H, s), 4.12 (1H, d, J = 1.5 Hz), 3.68 (1H, dd, J = 14.7, 1.1 Hz), 3.01 (1H, dd, J = 14.7, 1.1 Hz), 2.97 (1H, ddd, J = 8.6, 6.5, 1.5 Hz), 2.91 (1H, dd, J = 16.4, 2.7 Hz), 2.45 (1H, dd, J = 16.4, 3.6 Hz), 1.84−1.65 (2H, m, CH2-C7), 1.57 (1H, br s), 1.40−1.29 (2H, m), 0.92 (3H, t, J = 7.4 Hz); 13C-NMR (126 MHz, CDCl3) δ 172.6, 170.3, 143.2, 140.8, 140.5, 122.5, 122.0, 120.0, 113.5, 83.9, 74.8, 38.0, 30.5, 30.2, 21.5, 21.1, 14.1; LRMS (CI, isobutane): m/z (int) 289 (72), 273 (12), 137 (13), 113 (68), 97 (68), 81 (73), 71 (100). HRMS (CI, isobutane) calculated for C17H21O4 [M+H]+: 289.1440; found 289.1438. 16a (more polar, major diastereomer) M.p. 140−142 °C; Rf = 0.44 (PE-Et2O, 2:8); IR νmax 3400, 2957, 2928, 2872, 1736, 1636, 1537, 962, 939, 910, 870, 802, 777, 739 cm−1; 1H-NMR (500 MHz, CDCl3) δ 7.34 (1H, s), 7.23 (1H, s), 5.93 (1H, s), 5.15 (1H, s), 4.92 (1H, s), 4.87 (1H, s), 3.90 (1H, d, J = 4.3 Hz), 3.68 (1H, d, J = 16.0 Hz), 3.27 (1H, br s), 2.81−2.71 (2H, m), 2.55 (1H, br s), 1.85 (2H, br s), 1.55−1.44 (1H, m), 1.39−1.28 (1H, m), 1.27−1.14 (1H, m), 0.89 (3H, t, J = 7.4 Hz); 13C-NMR (126 MHz, CDCl3) δ 172.8, 170.4, 141.9, 141.5, 141.0, 123.3, 121.1, 119.7, 118.6, 83.2, 81.5, 39.7, 32.7, 28.9, 22.8, 20.9, 14.2; LRMS (EI+): m/z (int) 288 (100), 270 (43), 259 (36), 219 (64), 173 (51), 129 (39), 91 (81), 77 (47), 43 (47). HRMS (EI+) calculated for C17H20O4 [M]+: 288.1362; found 288.1360. X-ray crystal data (CCDC 1920025) for C17H20O4 (M = 288.34 g/mol): orthorhombic, space group P212121, a = 7.2695(5) Å, b = 11.8269(10) Å, c = 17.3278(12) Å, V = 1489.77(19) Å3, Z = 4, T = 100 K, μ(MoKα) = 0.091 mm–1, Dcalc. = 1.286 g/cm3, 37377 measured reflections, 2650 independent reflections (Rint = 0.074). Final R indices R1 = 0.0386 [for 2468 reflections, with I > 2σ(I)], wR2 = 0.0924 (all data).
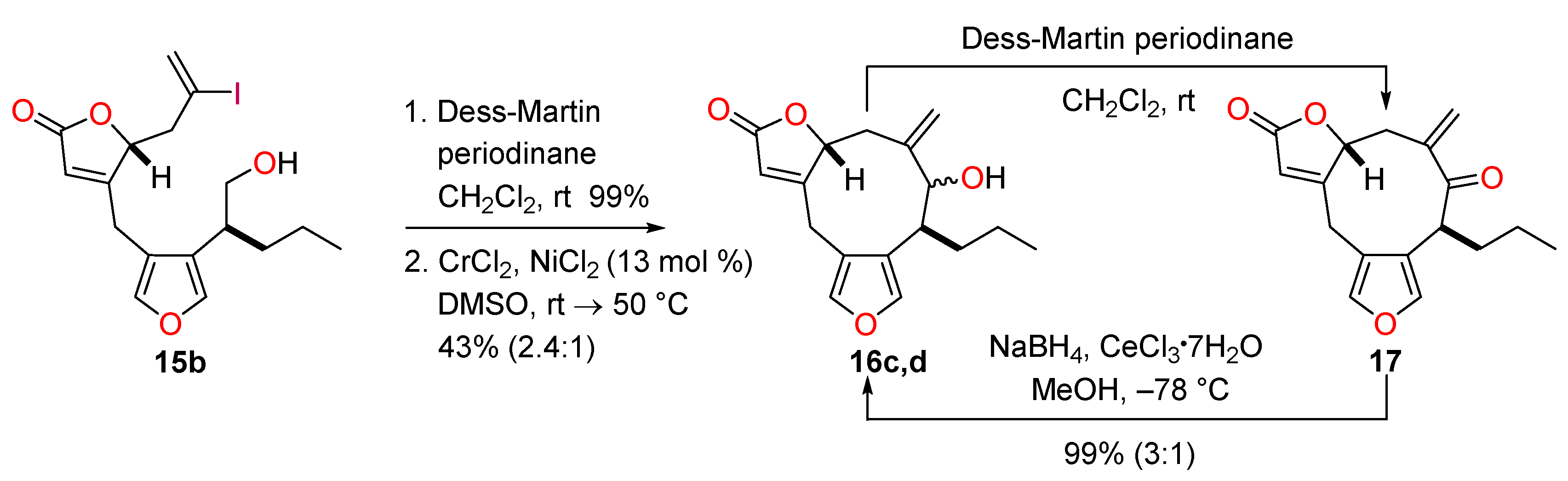
4.15. Synthesis of (8S*,9S*,12R*)-9-Hydroxy-10-methylidene-8-propyl-5,13-dioxatricyclo[10.3.0.03,7]penta-deca-1(15),3,6-trien-14-one and (8S*,9R*,12R*)-9-Hydroxy-10-methylidene-8-propyl-5,13-dioxatricyclo-[10.3.0.03,7]pentadeca-1(15),3,6-trien-14-one (16c, 16d)
To a solution of the alcohol 15b (59.2 mg, 0.142 mmol) in CH2Cl2 (2.5 mL) was added Dess-Martin periodinane (80.2 mg, 0.189 mmol, 1.54 equiv.). The mixture was stirred at room temperature for 40 min, cooled to 0 °C and the reaction was quenched with a saturated aqueous solution of Na2S2O3 (5 mL). The mixture was diluted with water (5 mL) and CH2Cl2 (5 mL) and after 10 min, the phases were separated. The aqueous phase was extracted with CH2Cl2 (3 × 5 mL) and the combined organic extracts were washed with brine (10 mL), dried with Na2SO4, filtered and concentrated in vacuo. The crude product was purified quickly by passage through a small plug of silica (PE-Et2O, 4:1) to afford the corresponding aldehyde (50.9 mg, 100%) as a colourless oil.
To a solution of CrCl2 (215 mg, 1.75 mmol, 12.4 equiv.) and NiCl2 (2.5 mg, 0.019 mmol, 13 mol %) in degassed (three freeze-thaw cycles) DMSO (6 mL) was added a solution of aldehyde (50.9 mg) in degassed (three freeze-thaw cycles) DMSO (6.5 mL) at room temperature. The dark green mixture was stirred for 48 h at 50 °C. The reaction was quenched with saturated aqueous NH4Cl (15 mL) and the mixture diluted with EtOAc (30 mL). The biphasic mixture was stirred for 30 min and the phases were separated. The aqueous phase was extracted with EtOAc (3 × 30 mL) and the combined organic extracts were washed with brine (50 mL), dried over MgSO4, filtered and concentrated in vacuo. The crude product was purified by flash column chromatography (PE-Et2O, 9:11) to give an inseparable mixture (1:2.4 / 2.4:1 based on 1H-NMR analysis) of the diastereoisomeric alcohols 16c and 16d (17.8 mg, 43%). Rf = 0.25 (PE-Et2O, 2:3); IR νmax 3448, 2957, 2928, 2872, 2360, 1748, 1633, 1541, 1465 cm−1; Minor diastereomer 1H-NMR (500 MHz, CDCl3) δ 7.34 (1H, d, J = 1.4 Hz), 7.32 (1H, d, J = 1.4 Hz), 6.02 (1H, br s), 5.14 (1H, s), 5.05 (1H, s), 4.98 (1H, ddd, J = 4.5, 3.6, 1.0 Hz), 4.14 (1H, s), 3.76 (1H, dd, J = 18.0, 0.8 Hz), 3.67 (1H, d, J = 18.0 Hz), 2.60−2.48 (2H, m), 2.05 (1H, app ddd, J = 15.2, 3.6, 1.3 Hz), 2.11 (1H, br s), 1.92 (1H, br s), 1.56−1.49 (1H, m), 1.37−1.24 (1H, m), 1.23−1.11 (1H, m), 0.87 (3H, t, J = 7.4 Hz); 13C-NMR (126 MHz, CDCl3) δ 172.6, 171.4, 146.4, 142.1, 140.4, 123.7, 119.5, 119.4, 119.2, 82.6, 77.8, 39.6, 35.2, 30.9, 24.2, 20.7, 14.0; Major diastereomer 1H-NMR (500 MHz, CDCl3) δ 7.28 (1H, s), 7.23 (1H, d, J = 1.5 Hz), 5.90 (1H, ddd, J = 1.5, 1.5, 1.5 Hz), 5.13 (1H, s), 5.05 (1H, s), 4.75 (1H, br s), 3.98 (1H, d, J = 7.4 Hz), 3.83−3.73 (1H, m), 3.61 (1H, d, J = 17.9 Hz), 2.60−2.48 (2H, m), 2.39 (1H, d, J = 15.4 Hz), 1.92 (1H, br s), 1.78−1.61 (2H, m), 1.37−1.24 (1H, m), 1.23−1.11 (1H, m), 0.87 (3H, t, J = 7.4 Hz); 13C-NMR (126 MHz, CDCl3) δ 172.5, 168.6, 143.4, 141.2, 140.4, 123.7, 119.1, 116.9, 116.4, 86.2, 81.5, 40.0, 37.1, 31.1, 22.5, 20.8, 14.2; LRMS (CI, isobutane): m/z (int) 289 (100), 271 (24), 137 (10), 71 (13). HRMS (CI, isobutane) calculated for C17H21O4 [M + H]+: 289.1440; found 289.1436.
4.16. Synthesis of (8S*,12R*)-10-Methylidene-8-propyl-5,13-dioxatricyclo[10.3.0.03,7]pentadeca-1(15),3,6-triene-9,14-dione (17)
To a solution of the diastereoisomeric alcohols 16c and 16d (7.1 mg, 0.025 mmol) in CH2Cl2 (1 mL) was added Dess-Martin periodinane (15.6 mg, 0.0368 mmol, 1.49 equiv.). The solution was stirred at room temperature for 1 h and then cooled to 0 °C. The reaction was quenched by the addition of a saturated aqueous solution of Na2S2O3 (5 mL) and the mixture was diluted with water (5 mL) and CH2Cl2 (5 mL). After 10 min, the phases were separated and the aqueous phase was extracted with CH2Cl2 (3 × 5 mL). The organic extracts were combined and washed with brine (10 mL), dried with Na2SO4, filtered and concentrated in vacuo. The crude product was purified by passage through a small plug of silica (PE-Et2O, 5:5) to give the enone 17 (7.0 mg, 99%) of a colourless oil. Rf = 0.50 (PE-Et2O, 1:4); IR νmax 3154, 3100, 2959, 2933, 2922, 2872, 1791, 1755, 1688, 1634, 1534, 981, 923, 898, 875, 859, 804, 765, 746 cm−1; 1H-NMR (500 MHz, CDCl3) δ 7.29 (1H, d, J = 1.2 Hz), 7.28 (1H, s), 5.88 (1H, td, J = 1.9, 1.3 Hz), 5.74 (1H, d, J = 1.2 Hz), 5.70 (1H, d, J = 1.2 Hz), 4.81 (1H, ddd, J = 8.6, 5.2, 1.3 Hz), 4.09 (1H, ddd, J = 10.3, 4.2, 1.2 Hz), 3.62 (2H, d, J = 1.9 Hz), 3.46 (1H, dd, J = 13.9, 5.2 Hz), 2.42 (1H, dd, J = 13.9, 8.6 Hz), 2.19−2.12 (1H, m), 1.75 (1H, dddd, J = 13.0, 8.5, 7.5, 4.2 Hz), 1.39 (2H, app tq, J = 7.5, 7.3 Hz), 0.99 (3H, t, J = 7.3 Hz).
4.17. Luche Reduction of (8S*,12R*)-10-Methylidene-8-propyl-5,13-dioxatricyclo[10.3.0.03,7]pentadeca-1(15), 3,6-triene-9,14-dione (17)
To a solution of the enone 17 (5.3 mg, 19 μmol) in MeOH (1.8 mL) was added CeCl3·7H2O (24 mg, 64 μmol, 3.4 equiv.). The reaction was cooled to −78 °C before addition of NaBH4 (1.6 mg, 42 μmol, 2.2 equiv.). The mixture was stirred for 1 h and the reaction was quenched by the addition of saturated aqueous NH4Cl (5 mL) and water (5mL). The mixture was warmed to room temperature and EtOAc (10 mL) was added. The phases were separated and the aqueous phase was extracted with EtOAc (3 × 10 mL). The organic extracts were combined, washed with brine (20 mL), dried over MgSO4, filtered and concentrated in vacuo. Analysis of the residue (5.3 mg, quant.) by 1H-NMR revealed a mixture of diastereoisomeric alcohols 16c and 16d with a slightly higher ratio (1:3 / 3:1 determined by 1H-NMR analysis) in favour of the major isomer produced by the original NHK reaction.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}