3.1. Binary Systems
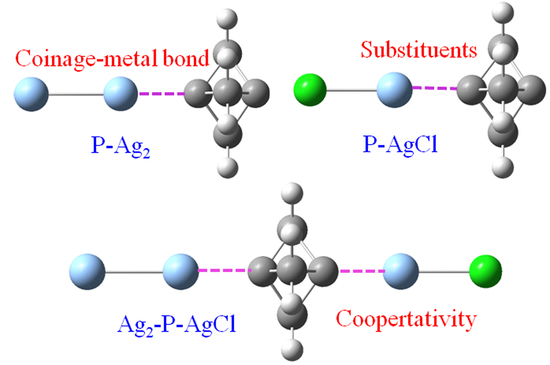
Figure 1 shows the optimized structures of P-M
2, P-MCl, and P-MCH
3 binary complexes at the wB97X-D/aug-cc-pVTZ(PP) level. All the geometries have C
3v symmetry with no imaginary frequencies. The M⋅⋅⋅C distance was much shorter than the sum of van der Waals (vdW) radii of the relevant atoms, indicating a strong attractive interaction between the two moieties. Though the vdW radius increased from Cu to Ag to Au, the M⋅⋅⋅C distance became longer from Cu⋅⋅⋅C to Au⋅⋅⋅C to Ag⋅⋅⋅C for each series of the complex. This inconsistency was primarily ascribed to the remarkably high first ionization energy and the electron affinity of Au. The M⋅⋅⋅C distance in P-MCl was shorter than that in the P-M
2 analogue, which can be explained with the positive MEP on both M
2 and MCl. The longer M⋅⋅⋅C distance in P-MCH
3 than that in P-MCl was due to the weaker electron-withdrawing ability of the methyl group compared to the Cl atom.
The attractive interaction between both molecules can be understood with the MEP maps as shown in
Figure 2. Obviously, there is a red region (σ-hole) along the M-M, M-Cl, and M-C bonds. The σ-hole was enlarged in sequence of Ag
2 < Cu
2 < Au
2, AgCl < AuCl <CuCl, but AuCH
3 < AgCH
3 < CuCH
3. The MEP of the σ-hole increased in the order M
2 < MCH
3 < MCl owing to the different electron-withdrawing ability among the Cl, CH
3, and M. The bridgehead carbon atoms in P have negative MEPs (see the blue area of P in
Figure 2). As a result, a coinage-metal bond formed between the bridgehead carbon atom of P and the M atom in M
2/MCl/MCH
3.
The interaction energy was computed with four methods including MP2/aug-cc-pVDZ(PP), MP2/aug-cc-pVTZ(PP), wB97X-D/aug-cc-pVTZ(PP), and CCSD(T)/aug-cc-pVTZ(PP), and the respective interaction energies were denoted as ∆E
MP2-pVDZ, ∆E
MP2-pVTZ, ∆E
wB97X-
D-pVTZ, and ∆E
CCSD(T)-pVTZ. As shown in
Table 1, the average deviation from the CCSD(T) results varied from 2.37 to 5.90 kcal/mol for the different systems. As expected, the larger basis set aug-cc-pVTZ(PP) brought out the larger interaction energy than the smaller basis set aug-cc-pVDZ(PP), and the difference was in a range of 1.8 to 5.7 kcal/mol, dependent on the different systems. Compared with ∆E
CCSD(T)-pVTZ, the MP2/aug-cc-pVTZ(PP) method overestimated the interaction energy and this overestimation was even larger than 10 kcal/mol in P-Au
2 and P-AuCl. The interaction energy calculated by the wB97X-D/aug-cc-pVTZ(PP) method was close to ∆E
CCSD(T)-pVTZ with comparison to the MP2 results. Furthermore, this method was successfully used to study coinage-metal bonds with small π molecules [
17]. Thus, the wB97X-D/aug-cc-pVTZ(PP) data were used for discussion.
In all cases, the interaction energy had a large range from −16.34 kcal/mol to −47.30 kcal/mol. The interaction energy was closely related to the nature of the coinage metal atom. The Au complexes had the largest interaction energy and the Ag complexes had the smallest interaction energy. The MEP on the M atom can be partly responsible for the change of interaction energy in P-M
2 since both terms linearly correlate. The MCl complexes show the stronger coinage-metal bond than the M
2 analogues with a similar reason. The methyl group in MCH
3 weakens the coinage-metal bond relative to MCl. The interaction energy in P-M
2 approaches that in NH
3-M
2 [
12], indicating [1.1.1]propellane is a good electron donor in coinage-metal bonding. It should be noted that the the oxidation state of M was different in M
2 and MCl/MCH
3, with 0 and +1, respectively [
14].
Figure 3 shows the AIM diagrams of P-Cu
2. A bond critical point (BCP) was present between the Cu atom and the bridgehead C atom of P, confirming the existence of a coinage-metal bond. The topological parameters at the BCP, including electron density, Laplacian, and total energy density, were collected in
Table 2. The electron density ranging from 0.07 to 0.121 a.u. is partly out of the range suggested for non-covalent interactions [
39]. Although the M⋅⋅⋅C BCP is different due to the M atom, its electron density had a consistent change with the interaction energy for each series of the complex. For the same M⋅⋅⋅C BCP, the electron density in P-MCl was larger than that in the P-M
2 analogue. The electron density at the Au⋅⋅⋅C BCP in P-AuCH
3 was smaller than that in P-Au
2, while an opposite result is found for the electron density at the Ag⋅⋅⋅C BCP. Even so, both variations were consistent with the interaction energy. Therefore, the electron density could be used to estimate the strength of the coinage-metal bond. The value of Laplacian was in a range of 0.18 to 0.28 a.u., which was also out of the range suggested for non-covalent interactions [
39]. This further confirms the existence of a strong coinage-metal bond.
The sign of Laplacian and total energy density can give some useful information for the nature of a non-covalent interaction. For all complexes, Laplacian is positive and energy density is negative, indicating that coinage-metal bonding is a partially covalent interaction [
40]. The energy density was more negative for the stronger coinage-metal bond.
According to the NBO analyses, there are three main orbital interactions upon the formation of coinage-metal bonding: σ
C-C → σ*
M–M/σ
C-C → σ*
M-Cl/σ
C-C → σ*
M-C, σ
C-C → LP*
M, and LP
M → σ*
C-C. The former two orbital interactions are donation orbital interactions from the occupied C-C orbital into the empty metal orbitals, while the latter orbital interaction is a back-donation from the occupied
d orbital of metal into the empty C-C anti-bonding orbital. These orbital interactions are estimated with second-order perturbation; see
Table 3. Obviously, the donation orbital interactions are stronger than the back-donation interaction. For σ
C-C → σ*
M-Cl/σ
C-C → σ*
M-C, it is stronger with the increase of the coinage-metal mass, while σ
C-C → σ*
M–M has consistent change with the interaction energy. For σ
C-C → LP*
M, its change was consistent with the interaction energy only in the P-MCl complex. The back-donation orbital interaction showed an irregular change in the P-MCH
3 complex but had an enhancing tendency for the heavier metal in the P-M
2 and P-MCl complexes. For P-M
2, the σ
C-C → LP*
M orbital interaction was stronger than the σ
C-C → σ*
M–M one, while the former orbital interaction was weaker than σ
C-C → σ*
M–C in P-MCH
3. However, the relative magnitude of both donor orbital interactions was different for three complexes of P-MCl. Both σ*
M–M/σ*
M–Cl/σ*
M–C and LP*
M are related with the
d anti-bonding orbitals of M, but we did not give any explanation for the above variations since no rule was found.
Accompanied with these orbital interactions, charge transfer (CT) occurred from the P to the M2/MCl/MCH3. This quantity was calculated as the sum of the charge on all atoms of P. For each series of complex, the charge transfer was the smallest in the Ag complex but largest in the Au complex. This sequence was similar to that of the interaction energy for each series of P-M2, P-MCl, or P-MCH3 complex. For the same coinage-metal donor, CT increases from P-M2 to P-MCH3 to P-MCl, which was generally consistent with the interaction energy. Of course, there was one exception between them in P-Au2 and P-AuCH3. Namely, the former complex had larger interaction energy than the latter, while the smaller CT was found in the former. Even so, their difference was not large.
To unveil the origin of the coinage-metal bonding, the interaction energy was decomposed into five terms: Electrostatic energy (E
ele), exchange energy (E
ex), repulsion energy (E
rep), polarization energy (E
pol), and dispersion energy (E
disp), and the related results are shown in
Table 4. The largest attractive term was from E
ex and this term was often offset by E
rep, thus we only focused on other attractive terms of E
ele, E
pol, and E
disp. For each complex, E
disp was smaller than both E
ele and E
pol, but its contribution could not be ignored since its ratio to the sum of the three attractive energies was 14–22%. For most complexes, E
ele was larger than E
pol, and the reverse result was found in P-AuCl. This indicated that most coinage-metal bonded complexes were dominated by electrostatic interaction, with moderate contribution from polarization interaction. When the complex varied from Cu to Ag to Au, each term was smallest in the Ag complex and largest in the Au complex for most complexes. However, some exceptions were also found. For instance, E
disp was smallest in P-AgCH
3 but largest in P-CuCH
3.
3.2. Ternary Systems
Figure 4 shows the optimized structures of six ternary systems of Ag
2-P-Ag
2, AgCH
3-P-AgCH
3, AgCl-P-AgCl, Ag
2-P-AgCH
3, Ag
2-P-AgCl, and AgCH
3-P-AgCl, which were used to study the cooperativity of the coinage-metal bond. Only the Ag ternary systems are discussed since most Cu and Au counterparts have imaginary frequencies. The binding distance showed a similar change in the different series of ternary systems. Namely, the binding distance in the ternary system was longer than that in the binary analogue. If the coinage donor was the same in the ternary system, both interactions would have an equal elongation. Moreover, this elongation became larger from AgCl-P-AgCl to Ag
2-P-Ag
2 to AgCH
3-P-AgCH
3. When the coinage donor was different in the ternary system, both interactions had an unequal elongation. Furthermore, this elongation was larger for the weaker coinage-metal bond. For example, this elongation was 0.016 Å for the stronger P-AgCl interaction but 0.050 Å for the weaker P-AgCH
3 interaction.
The total interaction energy of the ternary system is listed in
Table 5. The stability of the ternary system is similar to that of the corresponding binary system. A ternary system composed of two binary systems with stronger coinage-metal bonds is expected to be more stable. For instance, the stability of the ternary system increases from Ag
2-P-Ag
2 to AgCH
3-P-AgCH
3 to AgCl-P-AgCl. Both interactions are weakened in the ternary system since their interaction energies decrease (positive
∆∆E). The stronger the coinage-metal bond is, the more it is weakened in the ternary system. The coinage-metal bond in Ag
2-P decreases from 4.69 kcal/mol in Ag
2-P-Ag
2 to 6.14 kcal/mol in Ag
2-P-AgCH
3 to 7.45 kcal/mol in Ag
2-P-AgCl. The similar weakening result is also found in other ternary systems. These results indicate that both coinage-metal bonds exhibit negative cooperativity in these Ag ternary systems. There is a repulsion force between two Ag donor molecules since their interaction energy is positive. This repulsion interaction increases from Ag
2 to AgCH
3 to AgCl.
The cooperativity was also estimated with cooperative energy (Ecoop), which was calculated with the formulas of Ecoop = ∆Etotal − ∆Eleft − ∆Eright − ∆Efar, where ∆Etotal was the total interaction energy of a ternary system, ∆Eleft the interaction energy of the optimized left dimer, ∆Eright the interaction energy of the optimized right dimer, and ∆Efar the interaction energy of two Ag donor molecules in the trimer. This term was positive, consistent with the negative cooperativity between both coinage-metal bonds. Similarly, Ecoop increased from Ag2-P-Ag2 to AgCH3-P-AgCH3 to AgCl-P-AgCl, depending on the strength of coinage-metal bonding.
The change of the coinage-metal bonding strength in the ternary system can also be estimated with the electron density at the intermolecular BCP since they are relevant. Compared with the binary system, the electron density at the intermolecular BCP decreases (see
Table 6), indicating the coinage-metal bond is weakened in the ternary system.
Considering the contribution of electrostatic interaction in the coinage-metal bond, the MEP on the free bridgehead carbon atom in the P binary complexes was examined. It was calculated to be 0.0445au in P-Ag2, 0.0469 a.u. in P-AgCH3 and 0.0590 a.u. in P-AgCl, respectively. It is of note that the negative MEP in the P monomer became positive in the P binary complexes. As a result, the second coinage-metal bond was unfavorable to be formed in views of electrostatic interaction.
It was interesting to find that the free bridgehead carbon atom with a positive MEP in the P binary complex still bound attractively with another coinage donor. This result can be partly attributed to the presence of orbital interactions between both molecules. These orbital interactions result in charge transfer from P to the coinage donor. This value decreased in the ternary system compared with that in the binary system (
Table 7), consistent with the weakening of the coinage-metal bond. For the ternary complexes with two same coinage-metal bonds, the decrease of charge transfer grows up with the strengthening of the coinage-metal bond.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}