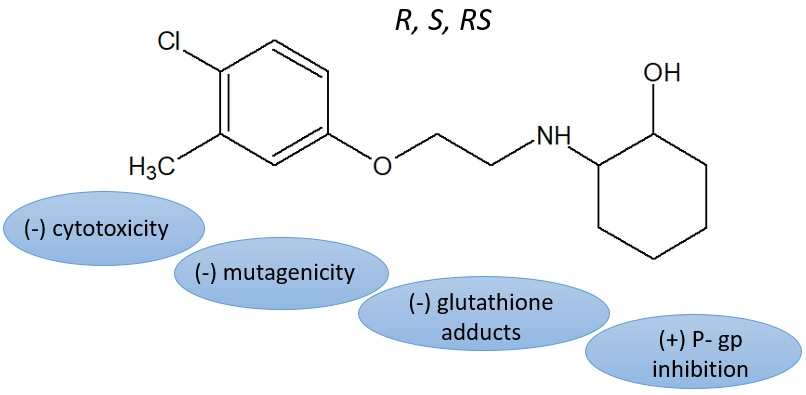
Similar Safety Profile of the Enantiomeric N-Aminoalkyl Derivatives of Trans-2-Aminocyclohexan-1-ol Demonstrating Anticonvulsant Activity

 ,
,
Abstract

1. Introduction
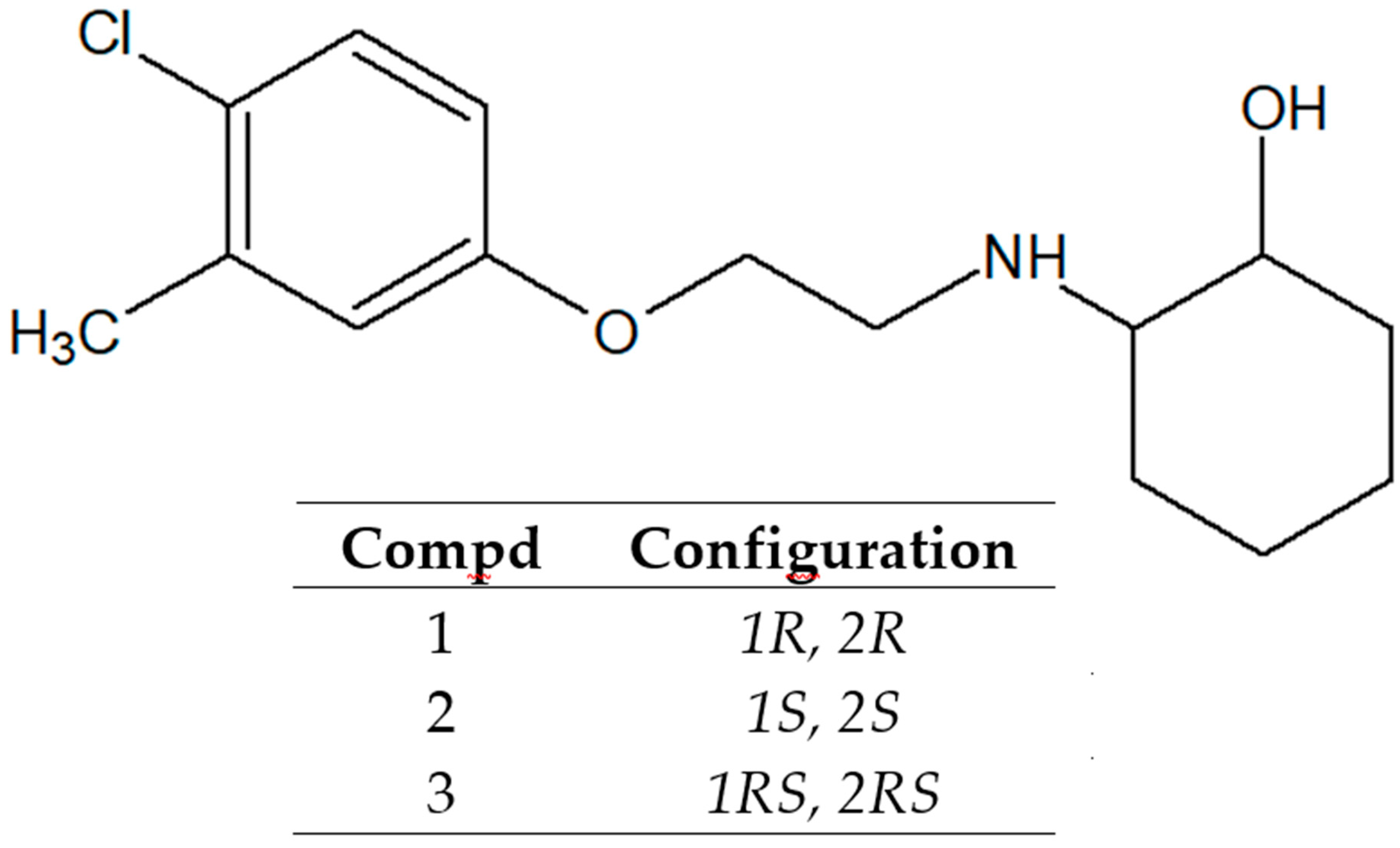
2. Results and Discussion
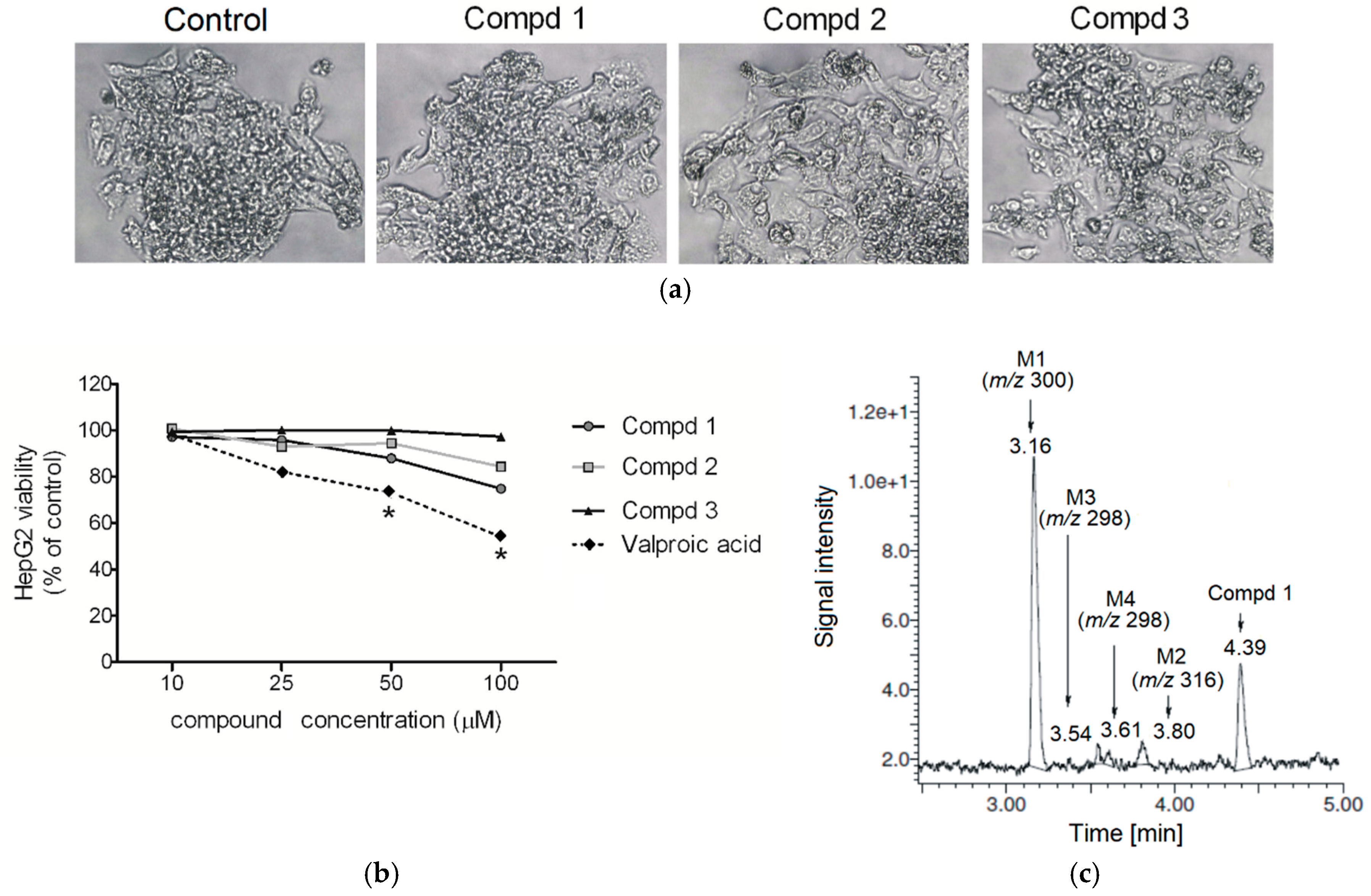
2.1. Cytotoxicity
2.2. Hepatotoxicity, Metabolic Activation and Trapping Assay
2.3. Mutagenicity and Antimutagenicity
2.4. P-gp Activity
3. Materials and Methods
3.1. Cytotoxicity Assays
3.1.1. Cell Culture
3.1.2. MTT Viability Test
3.1.3. LDH Viability Test
3.1.4. Data Analysis
3.2. Metabolic Activation and Trapping Assay
3.2.1. Chemicals and Reagents
3.2.2. Human Liver Microsomal Biotransformation and Trapping Assay
3.3. Mutagenicity and Antimutagenicity Assays
3.3.1. Chemicals and Reagents
3.3.2. Bacterial Test Systems
3.3.3. Ames Mutagenicity Testing
3.3.4. Vibrio harveyi Mutagenicity Testing
3.3.5. Ames Antimutagenicity Testing
3.3.6. Vibrio harveyi Antimutagenicity Testing
3.4. P-gp ATPase Activity Assay
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bialer, M.; White, H.S. Key factors in the discovery and development of new antiepileptic drugs. Nat. Rev. Drug Discov. 2010, 9, 68–82. [Google Scholar] [CrossRef] [PubMed]
- Froscher, W. Drug resistant epilepsy. J. Epileptol. 2012, 20, 17–23. [Google Scholar]
- Xiong, J.; Mao, D.A.; Liu, L.Q. Research progress on the role of ABC transporters in the drug resistance mechanism of intractable epilepsy. Biomed. Res. Int. 2015, 19, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Dalic, L.; Cook, M.J. Managing drug-resistant epilepsy: Challenges and solutions. Neuropsychiatr. Dis. Treat. 2016, 12, 2605–2616. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, M.; Koepp, M. ABC transporters and drug resistance in patients with epilepsy. Curr. Pharm. Des. 2016, 22, 5793–5807. [Google Scholar] [CrossRef] [PubMed]
- Muth, E.A.; Haskins, J.T.; Moyer, J.A.; Husbands, G.E.; Nielsen, S.T.; Sigg, E.B. Antidepressant biochemical profile of the novel bicyclic compound Wy-45,030, an ethyl cyclohexanol derivative. Biochem. Pharmacol. 1986, 35, 4493–4497. [Google Scholar] [CrossRef]
- Jesse, C.R.; Bortolatto, C.F.; Savegnago, L.; Rocha, J.B.; Nogueira, C.W. Involvement of L-arginine-nitric oxide-cyclic guanosine monophosphate pathway in the antidepressant-like effect of tramadol in the rat forced swimming test. Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 1838–1843. [Google Scholar] [CrossRef] [PubMed]
- Pękala, E.; Marona, H. Estimating the lipophilicity of a number of 2-amino-1-cyclohexanol derivatives exhibiting anticonvulsant activity. Biomed. Chromatogr. 2009, 23, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Kubowicz, P.; Marona, H.; Pękala, E. Synthesis, anticonvulsant activity and metabolism of 4-chlor-3-methylphenoxyethylamine derivatives of trans-2-aminocyclohexan-1-ol. Chirality 2015, 27, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Lippi, G.; Fernandes, C.C.; Ewell, L.A.; John, D.; Romoli, B.; Curia, G.; Taylor, S.R.; Frady, E.P.; Jensen, A.B.; Liu, J.C.; et al. MicroRNA-101 regulates multiple developmental programs to constrain excitation in adult neural networks. Neuron 2016, 92, 1337–1351. [Google Scholar] [CrossRef] [PubMed]
- Mahringer, A.; Fricker, G. ABC transporters at the blood-brain barrier. Expert Opin. Drug Metab. Toxicol. 2016, 12, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Luna-Tortós, C.; Fedrowitz, M.; Löscher, W. Several major antiepileptic drugs are substrates for human P-glycoprotein. Neuropharmacology 2008, 55, 1364–1375. [Google Scholar] [CrossRef] [PubMed]
- Luna-Tortós, C.; Rambeck, B.; Jürgens, U.H.; Löscher, W. The antiepileptic drug topiramate is a substrate for human P-glycoprotein but not multidrug resistance proteins. Pharm. Res. 2009, 26, 2464–2470. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Kwan, P.; Zu, Z.; Baum, L. The transport of antiepileptic drugs by P-glycoprotein. Adv. Drug Deliv. Rev. 2012, 64, 930–942. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.L. P-glycoprotein Inhibition for Optimal Drug Delivery. Drug Target Insights 2013, 7, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J.; Scorza, F.A. Effects of antiepileptic drugs on mitochondrial functions, morphology, kinetics, iogenesis, and survival. Epilepsy Res. 2017, 136, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Na, L.; Wartenberg, M.; Nau, H.; Hescheler, J.; Sauer, H. Anticonvulsant valproic acid inhibits cardiomyocyte differentiation of embryonic stem cells by increasing intracellular levels of reactive oxygen species. Birth Defects Res. A Clin. Mol. Teratol. 2003, 67, 174–180. [Google Scholar] [CrossRef]
- Ahir, B.K.; Pratten, M.K. Developmental cardiotoxicity effects of four commonly used antiepileptic drugs in embryonic chick heart micromass culture and embryonic stem cell culture systems. Toxicol. In Vitro 2014, 28, 948–960. [Google Scholar] [CrossRef]
- Kudin, A.P.; Mawasi, H.; Eisenkraft, A.; Elger, C.E.; Bialer, M.; Kunz, W.S. Mitochondrial Liver Toxicity of Valproic Acid and Its Acid Derivatives Is Related to Inhibition of α-Lipoamide Dehydrogenase. Int. J. Mol. Sci. 2017, 18, 1912. [Google Scholar] [CrossRef] [PubMed]
- Hebert, S.A.; Bohan, T.P.; Erikson, C.L.; Swinford, R.D. Thrombotic microangiopathy associated with Valproic acid toxicity. BMC Nephrol. 2017, 18, 262. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed]
- Gualtieri, F.; Curia, G.; Marinelli, C.; Biagini, G. Increased perivascular laminin predicts damage to astrocytes in CA3 and piriform cortex following chemoconvulsive treatments. Neuroscience 2012, 218, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Curia, G.; Lucchi, C.; Vinet, J.; Gualtieri, F.; Marinelli, C.; Torsello, A.; Constantino, L.; Biagini, G. Pathophysiogenesis of mesial temporal lobe epilepsy: Is prevention of damage antiepileptogenic? Curr. Med. Chem. 2014, 21, 663–688. [Google Scholar] [CrossRef] [PubMed]
- Rapacz, A.; Waszkielewicz, A.M.; Pańczyk, K.; Pytka, K.; Koczurkiewicz, P.; Piska, K.; Pękala, E.; Budziszewska, B.; Starek-Śmiechowicz, B.; Marona, H. Design, synthesis and anticonvulsant-analgesic activity of new N-[(phenoxy)alkyl]- and N-[(phenoxy)ethoxyethyl]aminoalkanols. MedChemComm 2016, 8, 220–238. [Google Scholar] [CrossRef] [PubMed]
- Coccini, T.; Caloni, F.; Ramírez Cando, L.J.; De Simone, U. Cytotoxicity and proliferative capacity impairment induced on human brain cell cultures after short- and long-term exposure to magnetite nanoparticles. J. Appl. Toxicol. 2017, 37, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Lian, D.; Chonghua, Z.; Wen, G.; Hongwei, Z.; Xuetau, B. Label-free and dynamic monitoring of cytotoxicity to the blood-brain barrier cells treated with nanometre copper oxide. IET Nanobiotechnol. 2017, 11, 948–956. [Google Scholar] [CrossRef] [PubMed]
- Pańczyk, K.; Żelaszczyk, D.; Koczurkiewicz, P.; Słoczyńska, K.; Pękala, E.; Żesławska, E.; Nitek, W.; Żmudzki, P.; Marona, H.; Waszkielewicz, A. Synthesis and anticonvulsant activity of phenoxyacetyl derivatives of amines, including aminoalkanols and amino acids. MedChemComm 2018, 9, 1933–1948. [Google Scholar] [CrossRef]
- Matysiak, S.; Chmiel, A.; Skolimowski, J.; Kula, J.; Pasternak, B.; Blaszczyk, A. Synthesis and cytotoxicity of (R)- and (S)-ricinoleic acid amides and their acetates. Chirality 2017, 29, 616–622. [Google Scholar] [CrossRef]
- Sun, Q.; Mao, R.; Wang, D.; Hu, C.; Zheng, Y.; Sun, D. The cytotoxicity study of praziquantel enantiomers. Drug Des. Devel. Ther. 2016, 10, 2061–2068. [Google Scholar] [CrossRef]
- Tokunaga, E.; Akiyama, H.; Soloshonok, V.A.; Inoue, Y.; Hara, H.; Shibata, N. Biological evaluation of both enantiomers of fluoro-thalidomide using human myeloma cell line H929 and others. PLoS ONE 2017, 12, e0182152. [Google Scholar] [CrossRef]
- Tarasenko, N.; Cutts, S.M.; Phillips, D.R.; Berkovitch-Luria, G.; Bardugo-Nissim, E.; Weitman, M.; Nudelman, A.; Rephaeli, A. A novel valproic acid prodrug as an anticancer agent that enhances doxorubicin anticancer activity and protects normal cells against its toxicity in vitro and in vivo. Biochem. Pharmacol. 2014, 15, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Kwiecińska, P.; Taubøll, E.; Grzyb, E.; Fiedor, E.; Ptak, A.; Gregoraszczuk, E.L. Valproic Acid as a Promising Co-Treatment with Paclitaxel and Doxorubicin in Different Ovarian Carcinoma Cell Lines. Int. J. Gynecol. Cancer 2016, 26, 1546–1556. [Google Scholar] [CrossRef] [PubMed]
- Leung, L.; Kalgutkar, A.S.; Obach, R.S. Metabolic activation in drug induced liver injury. Drug Metab. Rev. 2012, 44, 18–33. [Google Scholar] [CrossRef] [PubMed]
- Pessayre, D.; Fromenty, B.; Berson, A.; Robin, M.A.; Letteron, P.; Moreau, R.; Mansouri, A. Central role of mitochondria in drug-induced liver injury. Drug Metab. Rev. 2012, 44, 34–87. [Google Scholar] [CrossRef] [PubMed]
- Sadeque, A.J.; Fisher, M.B.; Korzekwa, K.R.; Gonzalez, F.J.; Rettie, A.E. Human CYP2C9 and CYP2A6 mediate formation of the hepatotoxin 4-ene-valproic acid. J. Pharmacol. Exp. Ther. 1997, 282, 698–703. [Google Scholar]
- Dalvie, D.; Kalgutkar, A.S.; Chen, W. Practical approaches to resolving reactive metabolite liabilities in early discovery. Drug Metab. Rev. 2015, 47, 56–70. [Google Scholar] [CrossRef] [PubMed]
- Lassila, T.; Mattila, S.; Turpeinen, M.; Tolonen, A. Glutathione trapping of reactive drug metabolites produced by biomimetic metalloporphyrin catalysts. Rapid Commun. Mass Spectrom. 2015, 30, 521–532. [Google Scholar] [CrossRef]
- Gómez-Lechón, M.J.; Tolosa, L.; Donato, M.T. Metabolic activation and drug-induced liver injury: In vitro approaches for the safety risk assessment of new drugs. J. Appl. Toxicol. 2016, 36, 752–768. [Google Scholar] [CrossRef]
- Migliore, L.; Coppedč, F. Genetic and environmental factors in cancer and neurodegenerative diseases. Mutat. Res. 2002, 512, 135–153. [Google Scholar] [CrossRef]
- Smith, S.W. Chiral toxicology: it’s the same thing…only different. Toxicol. Sci. 2009, 110, 4–30. [Google Scholar] [CrossRef]
- Słoczyńska, K.; Powroźnik, B.; Pękala, E.; Waszkielewicz, A.M. Antimutagenic compounds and their possible mechanisms of action. J. Appl. Genet. 2014, 55, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Fronza, G.; Campomenosi, P.; Iannone, R.; Abbondandol, A. The 4-nitroquinoline 1 oxide mutational spectrum in single stranded DNA is characterized by guanine to pyrimidine transversions. Nucleic Acids Res. 1992, 20, 1283–1287. [Google Scholar] [CrossRef] [PubMed]
- Ajith, T.A.; Soja, M.A. Comparative study on the antimutagenicity of atorvastatin and lovastatin against directly acting mutagens. Cell Biol. Toxicol. 2006, 22, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Ambudkar, S.V.; Dey, S.; Hrycyna, C.A.; Ramachandra, M.; Pastan, I.; Gottesman, M.M. Biochemical, cellular and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 361–398. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.H.; Jonker, J.W. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: An overview. Adv. Drug Deliv. Rev. 2003, 55, 3–29. [Google Scholar] [CrossRef]
- Nobili, S.; Landini, I.; Giglioni, B.; Mini, E. Pharmacological strategies for overcoming multidrug resistance. Curr. Drug Targets 2006, 1, 861–879. [Google Scholar] [CrossRef]
- Aller, S.G.; Yu, J.; Ward, A.; Weng, Y.; Chittaboina, S.; Zhuo, R.; Harrell, P.M.; Trinh, Y.T.; Zhang, Q.; Urbatsch, I.L.; et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009, 27, 1718–1722. [Google Scholar] [CrossRef]
- Zhou, Q.; Yu, L.S.; Zeng, S. Stereoselectivity of chiral drug transport: A focus on enantiomer- transporter interaction. Drug Metab. Rev. 2014, 46, 283–290. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, M.; Wang, D.; Zhao, Y.; Yu, Z. Comparison of intestinal permeability and p-glycoprotein effects on the intestinal absorption of enantiomers of 2-(2-hydroxypropanamido) benzoic acid in rats. Chirality 2017, 29, 26–32. [Google Scholar] [CrossRef]
- Lopes, A.; Martins, E.; Silva, R.; Pinto, M.M.M.; Remião, F.; Sousa, E.; Fernandes, C. Chiral Thioxanthones as Modulators of P- glycoprotein: Synthesis and Enantioselectivity Studies. Molecules 2018, 23, 626. [Google Scholar] [CrossRef]
- Pham, Y.T.; Régina, A.; Farinotti, R.; Couraud, P.O.; Wainerm, I.W.; Roux, F.; Gimenez, F. Interactions of racemic mefloquine and its enantiomers with P-glycoprotein in an immortalised rat braincapillary endothelial cell line, GPNT. Biochim. Biophys. Acta 2000, 1524, 212–219. [Google Scholar] [CrossRef]
- Shen, S.; He, Y.; Zeng, S. Stereoselective regulation of mdr1 expression in caco-2 cells by cetirizine enantiomers. Chirality 2007, 19, 485–490. [Google Scholar] [CrossRef]
- Hu, Z.; Zhou, Z.; Hu, Y.; Wu, J.; Li, Y.; Huang, W. HZ08 reverse P-glycoprotein mediated multidrug resistance in vitro and in vivo. PLoS ONE 2015, 10, 68–86. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Luna-Tortós, C.; Römermann, K.; Fedrowitz, M. Do ATP-binding cassette transporters cause pharmacoresistance in epilepsy? Problems and approaches in determining which antiepileptic drugs are affected. Curr. Pharm. Des. 2011, 17, 2808–2828. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Shen, J.; Shi, H.; Hornicek, F.J.; Kan, Q.; Duan, Z. Novel mechanisms and approaches to overcome multidrug resistance in the treatment of ovarian cancer. Biochim. Biophys. Acta 2016, 1866, 266–275. [Google Scholar] [CrossRef]
- Dewanjee, S.; Dua, T.K.; Bhattacharjee, N.; Das, A.; Gangopadhyay, M.; Khanra, R.; Joardar, S.; Riaz, M.; Feo, V.; Zia-Ul-Haq, M. Natural products as alternative choices for P-glycoprotein (P-gp) inhibition. Molecules 2017, 22, 871. [Google Scholar] [CrossRef]
- Joshi, P.; Vishwakarma, R.A.; Bharate, S.B. Natural alkaloids as P gp inhibitors for multidrug resistance reversal in cancer. Eur. J. Med. Chem. 2017, 138, 273–292. [Google Scholar] [CrossRef]
- Kambli, L.; Bhatt, L.K.; Oza, M.; Prabhavalkar, K. Novel therapeutic targets for epilepsy intervention. Seizure 2017, 51, 27–34. [Google Scholar] [CrossRef]
- Yu, J.; Mathisen, D.E.; Burdette, D.; Brown, D.G.; Becker, C.; Aharony, D. Identification of multiple glutathione conjugates of 8-amino-2-methyl-4-phenyl-1,2,3,4-tetrahydroisoquinoline maleate (nomifensine) in liver microsomes and hepatocyte preparations: Evidence of the bioactivation of nomifensine. Drug Metab. Dispos. 2010, 38, 46–60. [Google Scholar] [CrossRef]
- Maron, D.M.; Ames, B.N. Revised methods for the Salmonella mutagenicity test. Mutat. Res. 1983, 4, 173–215. [Google Scholar] [CrossRef]
- Mortelmans, K.; Zeiger, E. The Ames Salmonella/microsome mutagenicity assay. Mutat. Res. 2000, 455, 29–60. [Google Scholar] [CrossRef]
- Gulluce, M.; Agar, G.; Baris, O.; Karadayi, M.; Orhan, F.; Sahin, F. Mutagenic and antimutagenic effects of hexane extract of some Astragalus species grown in the eastern Anatolia region of Turkey. Phytother. Res. 2010, 24, 1014–1018. [Google Scholar] [PubMed]
- Czyż, A.; Jasiecki, J.; Bogdan, A.; Szpilewska, H.; Węgrzyn, G. Genetically modified Vibrio harveyi strains as potential bioindicators of mutagenic pollution of marine environments. Appl. Environ. Microbiol. 2000, 66, 599–605. [Google Scholar] [CrossRef]
- Podgórska, B.; Chęć, E.; Ulanowska, K.; Węgrzyn, G. Optimisation of the microbiological mutagenicity assay based on genetically modified Vibrio harveyi strains. J. Appl. Genet. 2005, 46, 241–246. [Google Scholar] [PubMed]
- Słoczyńska, K.; Waszkielewicz, A.M.; Marona, H. Preliminary assessment of mutagenic and anti-mutagenic potential of some aminoalkanolic derivatives of xanthone by use of the Vibrio harveyi assay. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2014, 21, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Evandri, M.G.; Battinelli, L.; Daniele, C.; Mastrangelo, S.; Bolle, P.; Mazzanti, G. The antimutagenic activity of Lavandulaangustifolia (lavender) essential oil in the bacterial reverse mutation assay. Food Chem. Toxicol. 2005, 43, 1381–1387. [Google Scholar] [CrossRef] [PubMed]
- Resende, F.A.; Barbosa, L.C.; Tavares, D.C.; de Camargo, M.S.; de Souza Rezende, K.C.; E Silva, M.L.; Varanda, E.A. Mutagenicity and antimutagenicity of (-)-hinokinin a trypanosomicidal compound measured by Salmonella microsome and comet assays. BMC Complement. Altern. Med. 2012, 12, 203. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1–3 are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Dose (mg/kg) | TD50 a | ED50(MES) b | PI(MES) |
|---|---|---|---|---|
| 1 | 30 | 66.03 # | 38.34 # | 1.722 # |
| 100 | 66.03 #scMet | >90.00 #scMet | <1.722 #scMet | |
| 2 | 30 | n.d. | n.d. | n.d. |
| 100 | n.d. | n.d. | n.d. | |
| 3 | 30 | 71.04 # | 25.05 # | 2.839 # |
| 100 | 71.04 #scMet | >150 #scMet | <0.474 #scMet | |
| 300 | >250 ^ | 99.25 ^ | 2.519 ^ | |
| PHT | 34.45 # | 6.48 # | 6.60 # | |
| >500 ^ | 32.2 ^ | >22 ^ | ||
| CBZ | 47.8 # | 9.85 # | 4.90 # | |
| 361 ^ | 3.57 ^ | 101 ^ | ||
| VPA | 483 # | 287 # | 1.70 # | |
| 859 ^ | 395 ^ | 2.2 ^ |
| Compound | Ames Test | Vibrio harveyi Test | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| TA100 a | BB7 a | BB7X a | BB7M a | BB7XM a | ||||||
| Mean ± SD | MI b | Mean ± SD | MI b | Mean ± SD | MI b | Mean ± SD | MI b | Mean ± SD | MI b | |
| H2O | 8 ± 3 | 19 ± 4 | 17 ± 6 | 22 ± 3 | 9 ± 4 | |||||
| DMSO | 16 ± 5 | 16 ± 3 | 13 ± 4 | 16 ± 2 | 11 ± 4 | |||||
| NQNO | 33 ± 4 | 2.1 | 32 ± 4 | 2.0 | 26 ± 2 | 2.0 | 32 ± 2 | 2.0 | 24 ± 3 | 2.2 |
| 1 | 9 ± 4 | 0.6 | 21 ± 3 | 1.1 | 16 ± 5 | 0.9 | 22 ± 3 | 1.0 | 18 ± 2 | 1.6 |
| 2 | 3 ± 1 | 0.2 | 23 ± 5 | 1.2 | 10 ± 6 | 0.6 | 23 ± 3 | 1.1 | 10 ± 3 | 0.9 |
| 3 | 6 ± 3 | 0.4 | 23 ± 3 | 1.4 | 13 ± 4 | 1.0 | 27 ± 4 | 1.7 | 15 ± 4 | 1.4 |
| Compound | Ames Test | Vibrio harveyi Test | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| TA100 a | BB7 a | BB7X a | BB7M a | BB7XM a | ||||||
| Mean ± SD | Inhib. (%) b | Mean ± SD | Inhib. (%) b | Mean ± SD | Inhib. (%) b | Mean ± SD | Inhib. (%) b | Mean ± SD | Inhib. (%) b | |
| H2O | 5 ± 2 | 12 ± 2 | 11 ± 4 | 16 ± 3 | 13 ± 4 | |||||
| DMSO | 6 ± 2 | 14 ± 3 | 13 ± 3 | 10 ± 2 | 15 ± 3 | |||||
| NQNO | 22 ± 4 | 38 ± 5 | 29 ± 4 | 35 ± 3 | 24 ± 4 | |||||
| 1 | 16 ± 2 | (27) | 26 ± 3 | (32) | 20 ± 4 | (31) | 22 ± 2 | (37) | 16 ± 2 | (35) |
| 2 | 14 ± 3 | (38) | 23 ± 4 | (38) | 21 ± 3 | (30) | 20 ± 4 | (41) | 14 ± 4 | (40) |
| 3 | 13 ± 3 | (41) | 19 ± 3 | (51) | 21 ± 4 | (29) | 22 ± 5 | (36) | 10 ± 4 | (57) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Słoczyńska, K.; Koczurkiewicz, P.; Piska, K.; Powroźnik, B.; Wójcik-Pszczoła, K.; Klaś, K.; Wyszkowska-Kolatko, M.; Pękala, E. Similar Safety Profile of the Enantiomeric N-Aminoalkyl Derivatives of Trans-2-Aminocyclohexan-1-ol Demonstrating Anticonvulsant Activity. Molecules 2019, 24, 2505. https://doi.org/10.3390/molecules24132505
Słoczyńska K, Koczurkiewicz P, Piska K, Powroźnik B, Wójcik-Pszczoła K, Klaś K, Wyszkowska-Kolatko M, Pękala E. Similar Safety Profile of the Enantiomeric N-Aminoalkyl Derivatives of Trans-2-Aminocyclohexan-1-ol Demonstrating Anticonvulsant Activity. Molecules. 2019; 24(13):2505. https://doi.org/10.3390/molecules24132505
Chicago/Turabian StyleSłoczyńska, Karolina, Paulina Koczurkiewicz, Kamil Piska, Beata Powroźnik, Katarzyna Wójcik-Pszczoła, Katarzyna Klaś, Magdalena Wyszkowska-Kolatko, and Elżbieta Pękala. 2019. "Similar Safety Profile of the Enantiomeric N-Aminoalkyl Derivatives of Trans-2-Aminocyclohexan-1-ol Demonstrating Anticonvulsant Activity" Molecules 24, no. 13: 2505. https://doi.org/10.3390/molecules24132505
APA StyleSłoczyńska, K., Koczurkiewicz, P., Piska, K., Powroźnik, B., Wójcik-Pszczoła, K., Klaś, K., Wyszkowska-Kolatko, M., & Pękala, E. (2019). Similar Safety Profile of the Enantiomeric N-Aminoalkyl Derivatives of Trans-2-Aminocyclohexan-1-ol Demonstrating Anticonvulsant Activity. Molecules, 24(13), 2505. https://doi.org/10.3390/molecules24132505