Marylosides A-G, Norcycloartane Glycosides from Leaves of Cymbidium Great Flower ‘Marylaurencin’

 and
and
Abstract
:1. Introduction
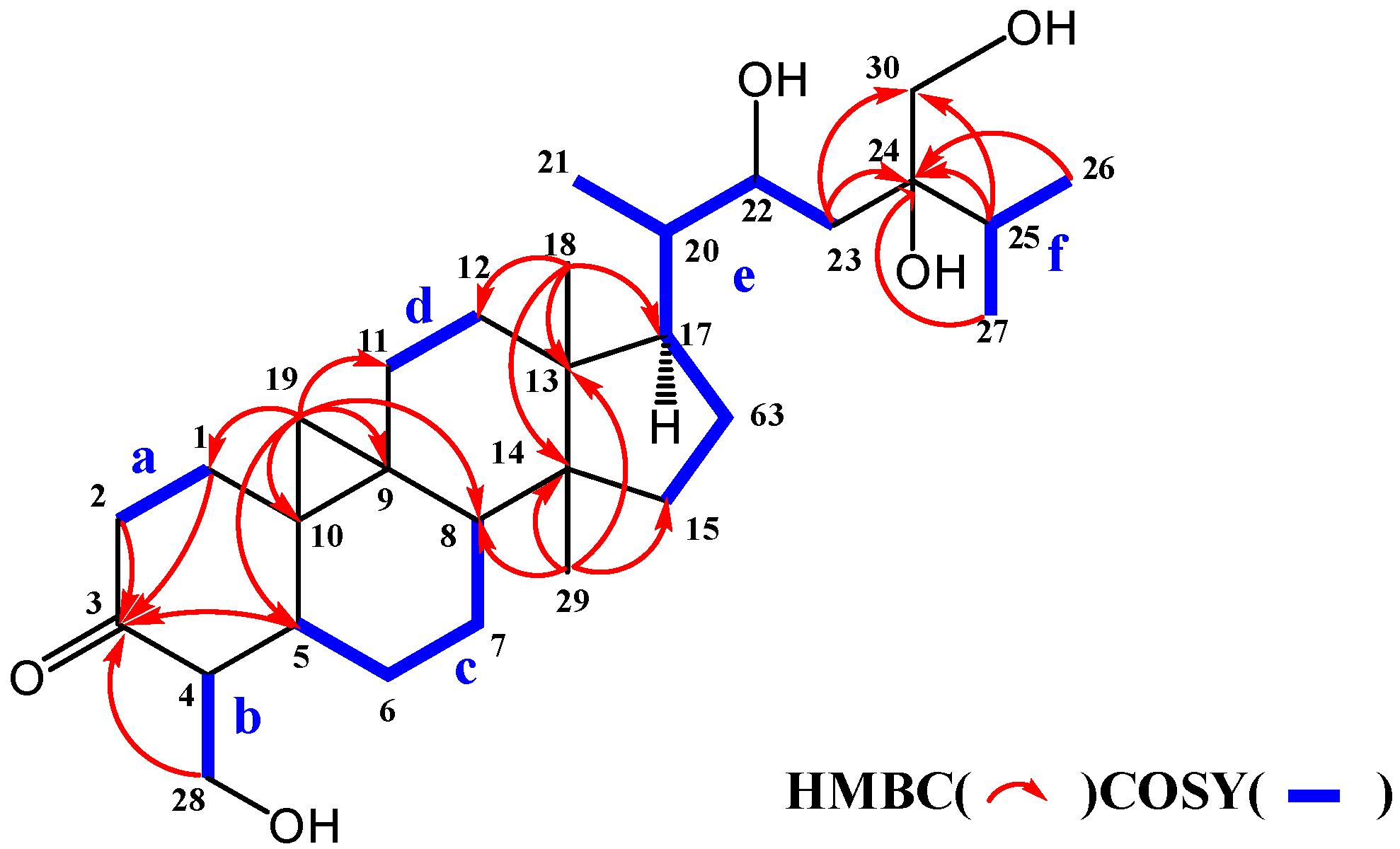
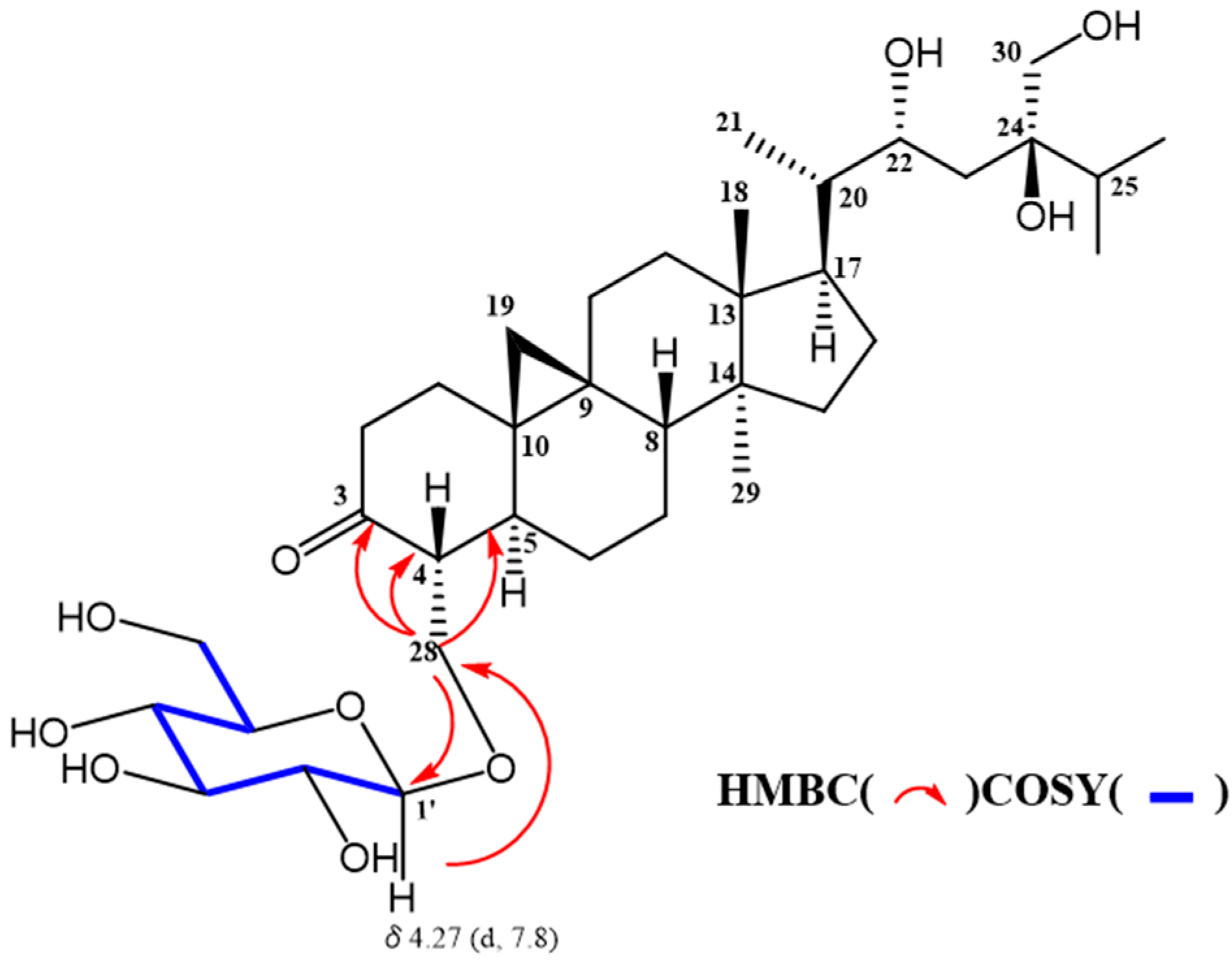
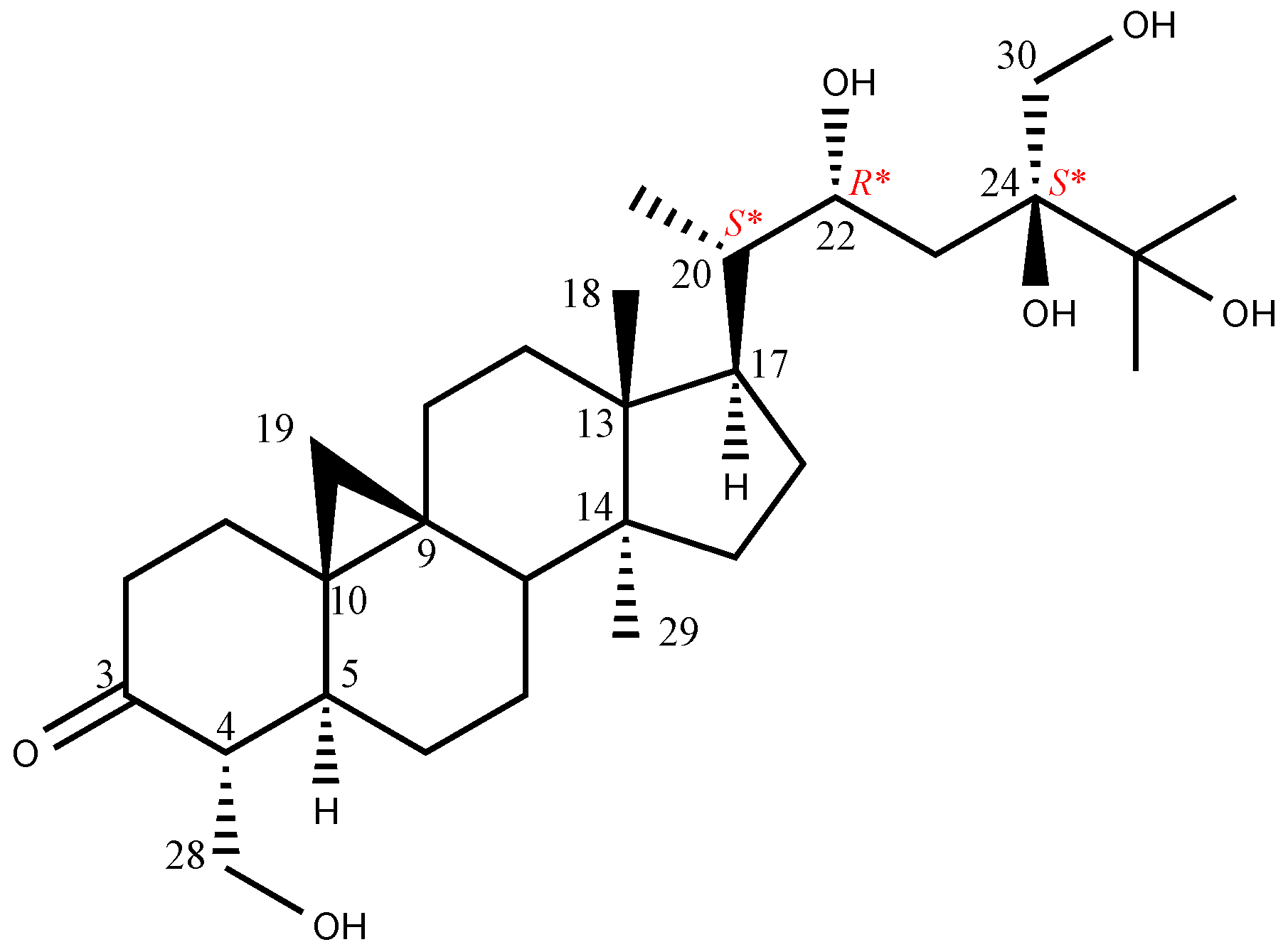
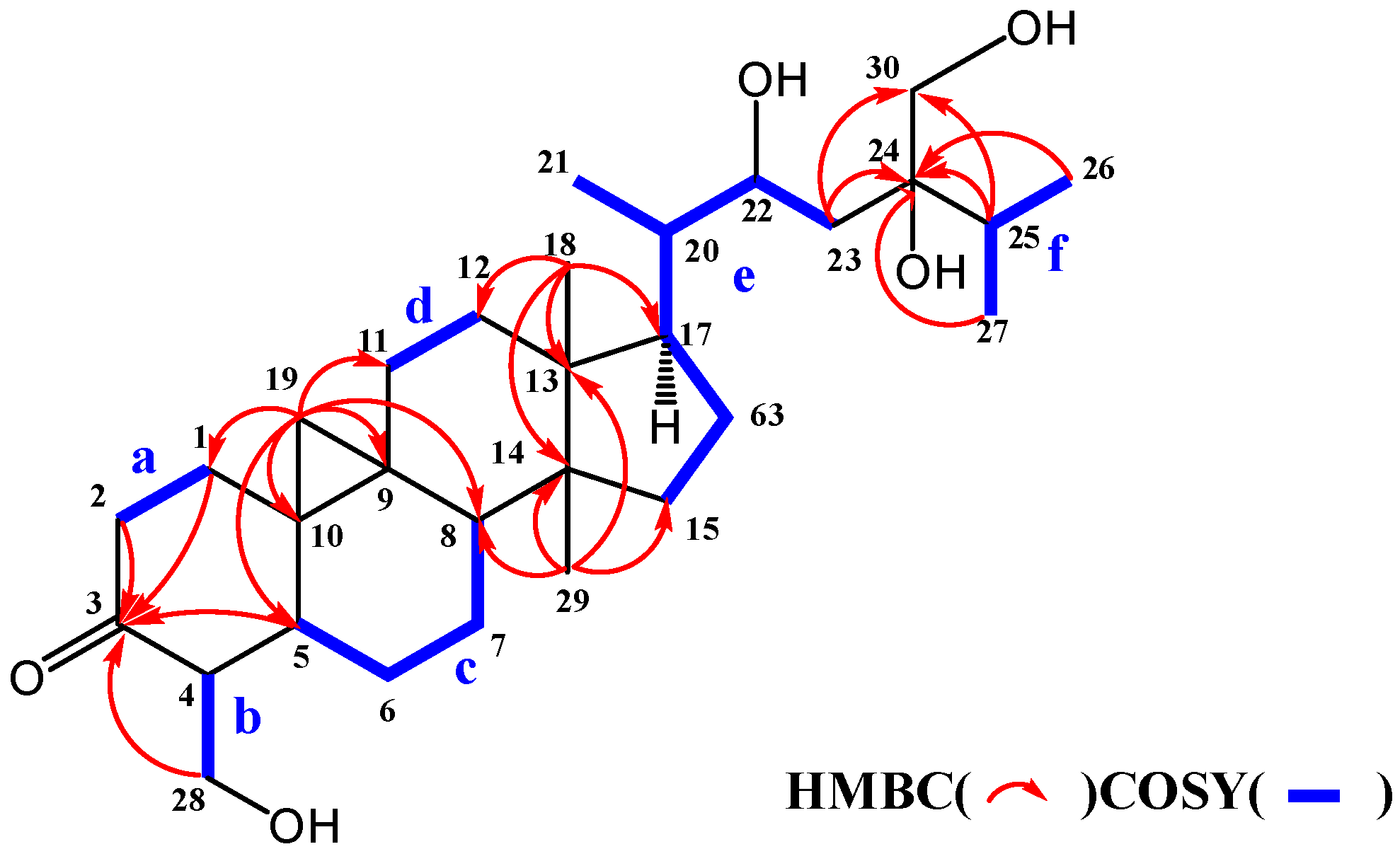
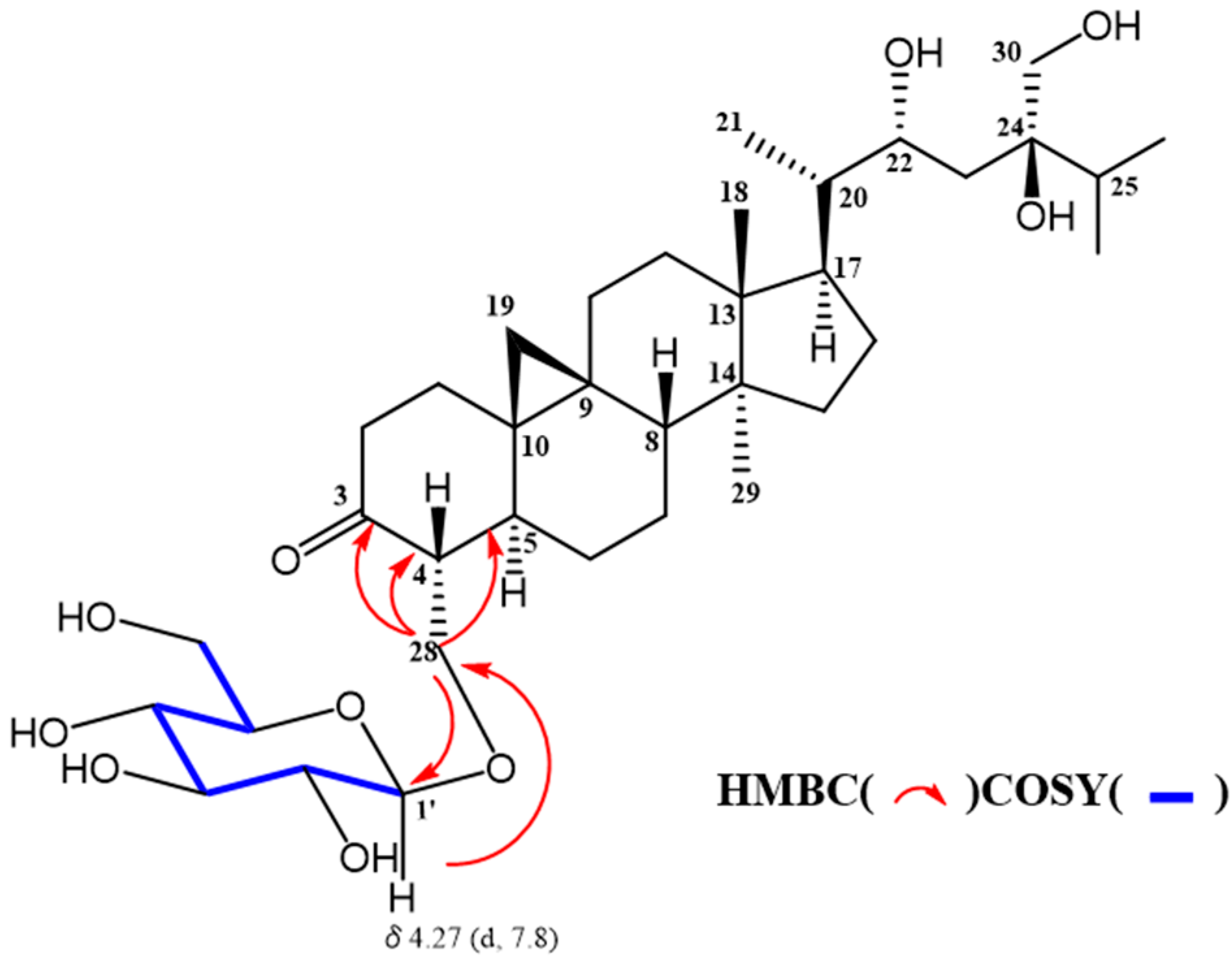
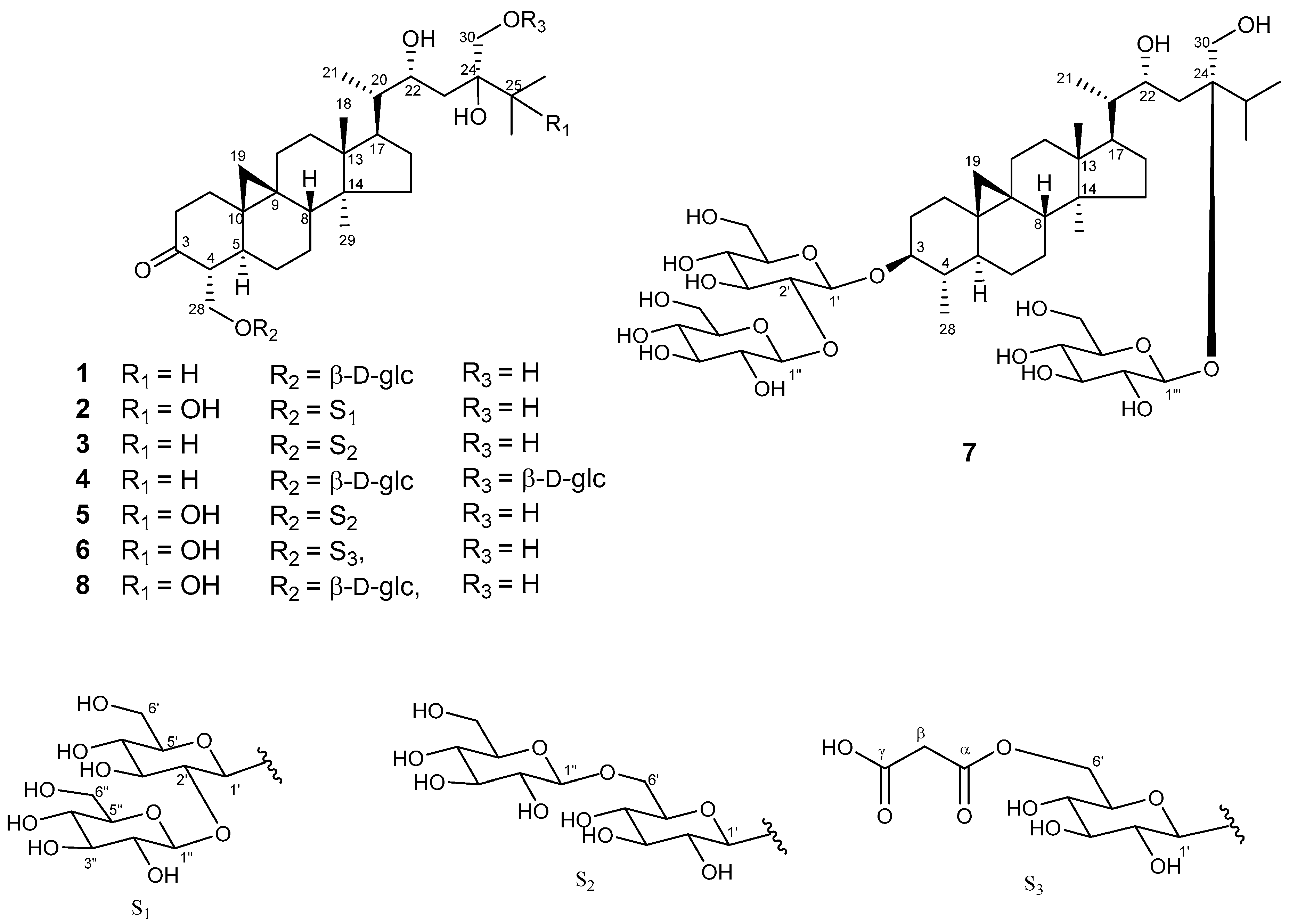
2. Results and Discussion
3. Materials and Methods
3.1. General Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Determination of Nitric Oxide (NO) Production and MTT Cell Viability Assay
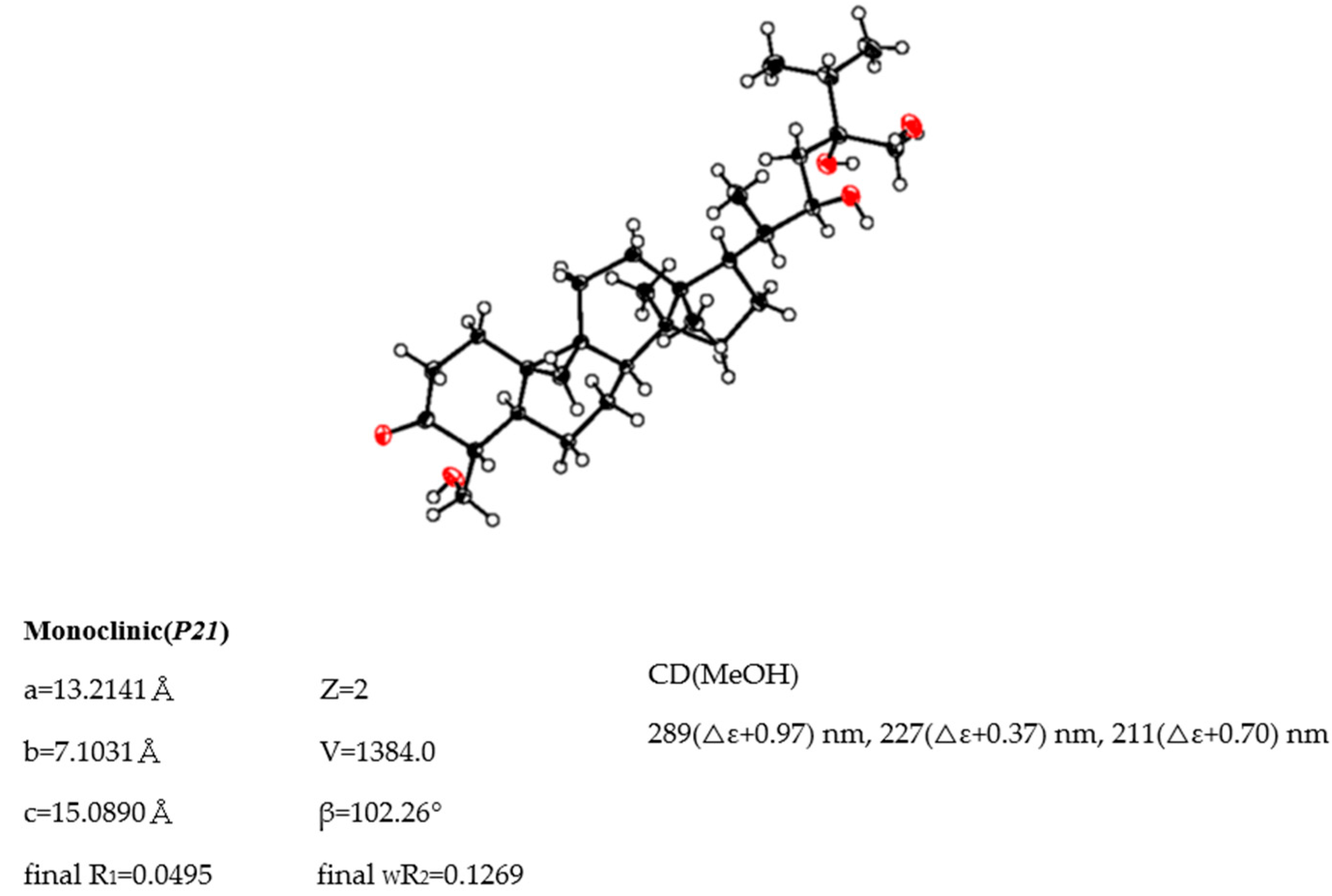
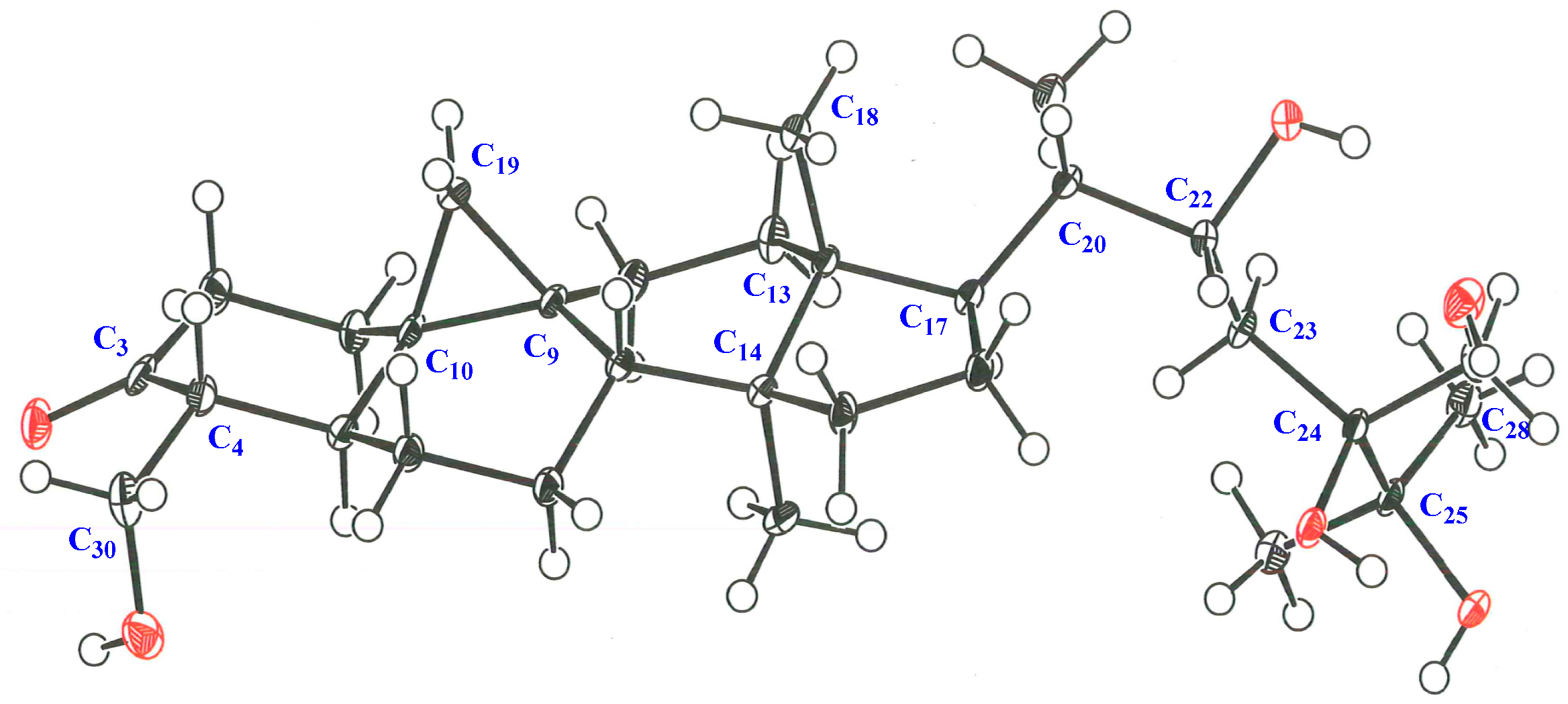
3.5. X-ray Crystallographic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yoshikawa, K.; Ito, T.; Iseki, K.; Baba, C.; Imagawa, H.; Yagi, Y.; Morita, H.; Asakawa, Y.; Kawano, S.; Hashimoto, T. Phenanthrene Derivatives from Cymbidium Great Flower Marie Laurencin and Their Biological Activities. J. Nat. Prod. 2012, 75, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, K.; Otsu, M.; Ito, T.; Asakawa, Y.; Kawano, S.; Hashimoto, T. Aromatic constituents of Cymbidium Great Flower Marie Laurencin and their antioxidative activity. J. Nat. Med. 2013, 67, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, K.; Baba, C.; Iseki, K.; Ito, T.; Asakawa, Y.; Kawano, S.; Hashimoto, T. Phenanthrene and phenylpropanoid constituents from the roots of Cymbidium Great Flower ‘Marylaurencin’ and their antimicrobial activity. J. Nat. Med. 2014, 68, 743–747. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, K.; Okahuji, M.; Iseki, K.; Ito, T.; Asakawa, Y.; Kawano, S.; Hashimoto, T. Two novel aromatic glucosides, marylaurencinosides D and E, from the fresh flowers of Cymbidium Great Flower ‘Marylaurencin’. J. Nat. Med. 2014, 68, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Dahmén, J.; Leander, K. A new triterpene glucoside from Cymbidium giganteum. Phytochemistry 1978, 17, 1978. [Google Scholar] [CrossRef]
- Tanaka, T.; Nakashima, T.; Ueda, T.; Tomii, K.; Kouno, I. Facile discrimination of aldose enantiomers by reversed-phase HPLC. Chem. Pharm. Bull. 2007, 55, 899–901. [Google Scholar] [CrossRef] [PubMed]
- Yahagi, T.; Yakura, N.; Matsuzaki, K.; Kitanaka, S. Inhibitory effect of chemical constituents from Artemisia scoparia Waldst. et Kit. on triglyceride accumulation in 3T3-L1 cells and nitric oxide production in RAW 264.7 cells. J. Nat. Med. 2014, 68, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Bae, I.-K.; Min, H.-Y.; Han, A.-R.; Seo, E.-K.; Lee, S.K. Suppression of lipopolysaccharide-induced expression of inducible nitric oxide synthase by brazilin in RAW 264.7 macrophage cells. Eur. J. Pharmacol. 2005, 513, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Corey, E.J.; Virgil, S.C. An Experimental Demonstration of the Stereochemistry of Enzymic Cyclization of 2,3-Oxidosqualene to the Protosterol System, Forerunner of Lanosterol and Cholesterol. J. Am. Chem. Soc. 1991, 113, 4025–4026. [Google Scholar] [CrossRef]
- Corey, E.J.; Virgil, S.C.; Sarshar, S. New Mechanistic and Stereochemical Insights on the Biosynthesis of Sterols from 2,3-Oxidosqualene. J. Am. Chem. Soc. 1991, 113, 8171–8172. [Google Scholar] [CrossRef]
- Corey, E.J.; Virgil, S.C.; David, R.L.; Sarshar, S. The Methyl Group at C (10) of 2,3-Oxidosqualene Is Crucial to the Correct Folding of This Substrate in the Cyclization-Rearrangement Step of Sterol Biosynthesis. J. Am. Chem. Soc. 1992, 114, 1524–1525. [Google Scholar] [CrossRef]
- Wang, H.; Ning, R.; Shen, Y.; Chen, Z.; Li, J.; Zhang, R.; Leng, Y.; Zhao, W. Lithocarpic Acids A-N, 3, 4-seco-Cycloartane Derivatives from the Cupules of Lithocarpus polystachyus. J. Nat. Prod. 2014, 77, 1910–1920. [Google Scholar] [CrossRef] [PubMed]
- Li, R.-T.; Han, Q.-B.; Zheng, Y.-T.; Wang, R.-R.; Yang, L.-M.; Lu, Y.; Sang, S.-Q.; Zheng, Q.-T.; Zhao, Q.S.; Sun, H.-D. Structure and anti-HIV activity of micrandilactones B and C, new nortriterpenoids possessing a unique skeleton from Schisandra micrantha. Chem. Commun. 2005, 23, 2936–2938. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 a) | 2 b) | 3 b) | 4 b) | 5 b) | 6 b) | 7 b) |
|---|---|---|---|---|---|---|---|
| 1 | 1.62 (1H, m) | 1.40 (1H, m) | 1.37 (1H, ddd, 10.8, 7.3, 2.9) | 1.38 (1H, ddd, 10.7, 4.26, 4.26) | 1.36 (1H, ddd, 13.3, 6.7, 4.9) | 1.38 (1H, ddd, 13.5, 7.2, 2.7) | 1.44 (1H, m) |
| 1.90 (1H, m) | 1.95 (1H, ddd, 12.9, 12.9, 4.9) | 1.68 (1H, m) | 1.69 (1H, m) | 1.64 (1H, m) | 1.69 (1H, m) | 1.16 (1H, m) | |
| 2 | 2.41 (1H, m) | 2.40 (1H, m) | 2.33 (1H, m) | 2.34 (1H, m) | 2.33 (1H, m) | 2.32 (1H, m) | 1.75 (1H, m) |
| 2.46 (1H, m) | 2.57 (1H, ddd, 11.7, 4.9, 4.9) | 2.49 (1H, ddd, 11.8, 4.8, 2.9) | 2.50 (1H, m) | 2.48 (1H, ddd, 14.4, 4.9, 2.7) | 2.48 (1H, ddd, 14.6, 4.8, 2.7) | 2.54 (1H, m) | |
| 3 | 3.45 (1H, ddd, 10.6, 10.6, 4.6) | ||||||
| 4 | 2.37 (1H, dt, 11.8, 4.2) | 2.40 (1H, m) | 2.25 (1H, dt, 11.8, 3.8) | 2.33 (1H, m) | 2.24 (1H, dt, 11.9, 3.8) | 2.25 (1H, dt, 11.8, 3.2) | 1.59 (1H, m) |
| 5 | 2.06 (1H, ddd, 11.8, 11.8, 5.1) | 2.198 (1H, m) | 2.15 (1H, m) | 2.09 (1H, ddd, 12.4, 12.4, 4.8) | 2.11 (1H, m) | 2.10 (1H, m) | 1.17 (1H, m) |
| 6 | 0.83 (1H, dddd, 12.4, 12.4, 12.4, 1.8) | 0.75 (1H, m) | 0.74 (1H, m) | 0.72 (1H, m) | 0.73 (1H, m) | 0.71 (1H, m) | 0.50 (1H, m) |
| 1.88 (1H, m) | 1.92 (1H, m) | 2.03 (1H, m) | 1.95 (1H, m) | 2.02 (1H, m) | 2.00(1H, m) | 1.63 (1H, m) | |
| 7 | 1.17 (1H, dddd, 12.4, 12.4, 12.4, 2.4) | 1.07 (1H, m) | 0.93 (1H, m) | 0.93 (1H, m) | 0.91 (1H, m) | 0.90 (1H, m) | 0.96 (1H, m) |
| 1.34 (1H, m) | 1.21 (1H, m) | 1.22 (1H, m) | 1.11 (1H, m) | 1.17 (1H, m) | 1.14 (1H, m) | 1.18 (m) | |
| 8 | 1.68 (1H, m) | 1.57 (1H, m) | 1.49 (1H, dd, 2.5, 2.5) | 1.49 (1H, m) | 1.48 (1H, m) | 1.48 (1H, m) | 1.52 (m) |
| 11 | 2.13 (2H, m) | 1.16 (1H, ddd, 11.1, 7.0, 7.0) | 1.94 (1H, m) | 1.13 (1H, m) | 1.12 (1H, m) | 1.12 (1H, m) | 1.16 (m) |
| 2.05 (1H, m) | 1.13 (1H, ddd, 13.3, 9.7, 4.8) | 1.94 (1H, m) | 1.92 (1H, m) | 1.92 (1H, m) | 1.89 (m) | ||
| 12 | 1.54 (1H, m) | 1.64 (2H, ddd, 11.1, 7.0, 7.0) | 1.62 (2H, m) | 1.61 (2H, m) | 1.58 (2H, m) | 1.62 (2H, m) | 1.60 (2H, m) |
| 1.57 (1H, m) | |||||||
| 15 | 1.35 (2H, m) | 1.28 (2H, m) | 1.22 (2H, m) | 1.25 (2H, m) | 1.24 (2H, m) | 1.22 (2H, m) | 1.18 (2H, m) |
| 16 | 1.39 (1H, m) | 1.45 (1H, m) | 1.47 (1H, m) | 1.48 (1H, m) | 1.46 (1H, m) | 1.46 (1H, m) | 1.37 (m) |
| 1.95 (1H, m) | 2.32 (1H, m) | 2.12 (1H, m) | 2.18 (1H, m) | 2.30 (1H, m) | 2.0(1H, m) | 2.12 (m) | |
| 17 | 1.63 (1H, m) | 1.88 (1H, m) | 1.78 (1H, d, 9.3) | 1.80 (1H, dd, 10.7, 10.7) | 1.85 (1H, m) | 1.86 (1H, m) | 1.80 (m) |
| 18 | 1.09 (3H, s) | 1.03 (3H, s) | 1.00 (3H, s) | 1.02 (3H, s) | 1.00 (3H, s) | 1.00 (3H, s) | 0.99 (3H, s) |
| 19 | 0.46 (1H, d, 4.1) | 0.30 (1H, d, 4.0) | 0.26 (1H, d, 4.0) | 0.27 (1H, d, 4.0) | 0.25 (1H, d, 3.8) | 0.27 (1H, d, 4.0) | 0.10 (d, 3.7) |
| 0.67 (1H, d, 4.1) | 0.55 (1H, d, 4.0) | 0.43 (1H, d, 4.0) | 0.46 (1H, d, 4.0) | 0.43 (1H, d, 3.8) | 0.45 (1H, d, 4.0) | 0.33 (d, 3.7) | |
| 20 | 1.68 (1H, m) | 2.08 (1H, m) | 2.01 (1H, m) | 2.02 (1H, m) | 2.06 (1H, m) | 2.06 (1H, ddd, 7.0, 3.4, 3.3) | 2.08 (m) |
| 21 | 0.92 (3H, d, 6.5) | 1.22 (3H, d, 6.6) | 1.19 (3H, d, 6.9) | 1.19 (3H, d, 6.6) | 1.22 (3H, d, 6.7) | 1.22 (3H, d, 7.0) | 1.22 (3H, d, 6.7) |
| 22 | 4.07 (1H, dd, 10.2, 4.2) | 4.68 (1H, dd, 9.7, 2.7) | 4.59 (1H, dd, 10.4, 2.3) | 4.56 (1H, dd, 5.1, 2.6) | 4.70 (1H, dd, 9.9, 2.9) | 4.70 (1H, dd, 9.9, 3.3) | 4.56 (m) |
| 23 | 1.70 (1H, m) | 2.197 (1H, m) | 1.99 (1H, m) | 1.99 (1H, m) | 2.36 (1H, m) | 2.15 (1H, m) | 2.02 (m) |
| 2.34 (1H, m) | 2.11 (1H, m) | 1.61 (1H, m) | 2.19 (1H, dd, 14.7, 9.9) | 2.32 (1H, m) | 2.08 (m) | ||
| 25 | 1.93 (1H, m) | 2.35 (1H, sep, 7.0) | 2.46 (1H, sep, 9.2) | 2.48 (sext, 6.9) | |||
| 26 | 0.95 (3H, d, 7.1) | 1.69 (3H, s) | 1.25 (3H, d, 7.0) | 1.18 (3H, d, 9.2) | 1.694 (3H, s) | 1.69(3H, s) | 1.27 (3H, d, 6.9) |
| 27 | 0.964 (3H, d, 7.3) | 1.68 (3H, s) | 1.22 (3H, d, 7.0) | 1.17 (3H, d, 9.2) | 1.687 (3H, s) | 1.68(3H, s) | 1.13 (3H, d, 6.9) |
| 28 | 3.87 (1H, dd, 10.2, 2.7) | 4.18 (1H, m) | 4.22 (1H, dd, 10.2, 3.8) | 4.19 (1H, dd, 10.4, 3.0) | 4.21 (1H, m) | 4.27 (1H, dd,10.3,3.2) | 1.41 (3H, d, 6.5) |
| 4.07 (1H, dd, 10.2, 4.2) | 4.42 (1H, m) | 4.45 (1H, dd, 10.2, 3.8) | 4.44 (1H, dd, 10.4, 4.0) | 4.45 (1H, dd, 10.2, 3.8) | 4.40 (1H, m) | ||
| 29 | 0.957 (3H, s) | 0.85 (3H, s) | 0.81 (3H, s) | 0.83 (3H, s) | 0.79 (3H, s) | 0.79 (3H, s) | 0.85 (3H, s) |
| 30 | 3.49 (1H, d, 11.4) | 4.12 (1H, d, 10.7) | 4.04 (1H, d, 11.1) | 4.17 (1H, d, 11.1) | 4.37 (1H, d, 11.2) | 4.36 (1H, d, 11.3) | 4.06 (d, 12.4) |
| 3.52 (1H, d, 11.4) | 4.39 (1H, d, 10.7) | 4.06 (1H, d, 11.1) | 4.49 (1H, d, 11.1) | 4.41 (1H, d, 11.2) | 4.40 (1H, d, 11.3) | 4.26 (d, 12.4) | |
| glucoside at C-28 or C-3 | |||||||
| 1’ | 4.27 (1H, d, 7.8) | 4.97 (d, 7.7) | 4.87 (1H, d, 7.8) | 4.97 (1H, d, 8.8) | 4.88 (1H, d, 8.0) | 4.90 (1H, d, 7.8) | 4.98 (1H, d, 7.7) |
| 2’ | 3.13 (1H, dd, 9.1, 7.8) | 4.17 (1H, m) | 3.97 (1H, dd, 8.8, 7.8) | 4.03 (1H, dd, 8.8, 8.8) | 3.97 (1H, dd, 8.8, 8.0) | 4.00 (1H, dd, 8.8, 7.8) | 4.26 (1H, m) |
| 3’ | 3.35 (1H, dd, 9.1, 9.1) | 4.209 (1H, m) | 4.17 (1H, dd, 9.0, 8.8) | 4.24 (1H, m) | 4.17 (1H, dd, 8.8, 8.8) | 4.19 (1H, dd, 8.8, 8.8) | 4.34 (dd, 11.5, 11.5) |
| 4’ | 3.28 (1H, dd, 9.6, 9.6) | 4.207 (1H, m) | 4.12 (1H, dd, 9.0) | 4.25 (1H, m) | 4.12 (1H, dd, 9.1, 8.8) | 4.08 (1H, dd, 9.1, 8.8) | 4.21 (dd, 11.5, 11.5) |
| 5’ | 3.25 (1H, ddd, 9.6, 9.6, 2.3) | 3.94 (1H, m) | 4.06 (1H, m) | 3.94 (1H, ddd, 5.5, 2.7, 2.7) | 4.06 (1H, m) | 4.05 (1H, m) | 3.89 (ddd, 11.5, 5.4, 2.6) |
| 6’ | 3.66 (1H, dd, 11.8, 9.6) | 4.40 (1H, dd, 11.7, 5.2) | 4.38 (1H, dd, 11.5, 5.2) | 4.38 (1H, dd, 11.4, 5.5) | 4.35 (1H, dd, 11.5, 6.0) | 4.89 (1H, dd, 11.6, 5.9) | 4.35 (dd, 11.5, 11.5) |
| 3.85 (1H, dd, 11.8, 2.3) | 4.55 (1H, dd, 11.7, 2.7) | 4.83 (1H, dd, 11.5, 1.7) | 4.54 (1H, dd, 11.4, 2.7) | 4.83 (1H, dd, 11.5, 1.7) | 5.06 (1H, dd, 11.6, 1.6) | 4.49 (dd, 11.5, 3.0) | |
| 1’’ | 5.37 (1H, d, 7.7) | 5.15 (1H, d, 7.8) | 5.15 (1H, d, 7.8) | 5.43 (d, 9.1) | |||
| 2’’ | 4.04 (1H, dd, 9.1, 7.7) | 4.05 (1H, m) | 4.05 (1H, dd, 7.8, 6.5) | 4.15 (dd, 9.1, 9.1) | |||
| 3’’ | 4.28 (1H, dd, 9.1, 9.1) | 4.24 (1H, m) | 4.23 (1H, dd, 6.5, 6.5) | 4.24(m) | |||
| 4’’ | 4.197 (1H, m) | 4.24 (1H, m) | 4.24 (1H, dd, 6.5, 6.0) | 4.28 (dd, 9.5, 1.9) | |||
| 5’’ | 3.86 (1H, ddd, 7.7, 5.4, 2.6) | 3.94 (1H, ddd, 8.8, 6.3, 3.4) | 3.94 (1H, ddd, 3.2, 6.0, 8.9) | 3.94(ddd,9.5,4.7,3.3) | |||
| 6’’ | 4.37 (1H, m) | 4.35 (1H, dd, 11.8, 6.3) | 4.36 (1H, m) | 4.41 (dd, 11.9, 4.7) | |||
| 4.49 (1H, dd, 11.8, 2.6) | 4.52 (1H, dd, 11.8, 3.4) | 4.53 (1H, dd, 11.7, 3.2) | 4.52 (dd, 11.9, 3.3) | ||||
| malonyl moiety β | 3.78 (2H, d, 4.8) | ||||||
| glucoside at C-28 or C-3 | |||||||
| 1’’’ | 5.00 (d, 7.8) | 5.40 (d, 8.0) | |||||
| 2’’’ | 4.07 (dd, 7.8, 4.1) | 4.12 (dd, 9.1, 8.0) | |||||
| 3’’’ | 4.20 (m) | 4.26 (dd, 9.1, 3.1) | |||||
| 4’’’ | 4.19 (m) | 4.13 (m) | |||||
| 5’’’ | 3.96 (ddd, 9.2, 6.3, 3.3) | 4.02 (ddd, 9.7, 6.7, 3.0) | |||||
| 6’’’ | 4.38 (dd, 11.4, 6.3) | 4.25 (m) | |||||
| 4.54 (dd, 11.4, 3.3) | 4.54 (m) |
| Position | 1 a) | 1a b) | 2 b) | 3 b) | 4 b) | 5 b) | 6 b) | 7 b) | 8a a) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 33.1 | 32.1 | 32.2 | 31.9 | 32.0 | 31.9 | 31.8 | 30.9 | 33.0 |
| 2 | 41.7 | 41.3 | 41.2 | 41.0 | 41.1 | 41.0 | 41.0 | 34.0 | 41.6 |
| 3 | 213.8 | 211.9 | 210.8 | 210.1 | 210.2 | 210.1 | 210.1 | 86.5 | 213.8 |
| 4 | 56.7 | 57.6 | 56.2 | 55.9 | 56.0 | 55.9 | 55.9 | 43.9 | 58.1 |
| 5 | 42.1 | 40.4 | 41.3 | 40.8 | 41.2 | 40.8 | 40.8 | 43.8 | 41.8 |
| 6 | 26.6 | 25.8 | 26.0 | 25.8 | 25.8 | 25.8 | 25.7 | 24.9 | 26.5 |
| 7 | 26.3 | 25.3 | 25.5 | 25.3 | 25.4 | 25.3 | 25.3 | 25.3 | 26.1 |
| 8 | 48.6 | 47.4 | 47.7 | 47.5 | 47.5 | 47.5 | 47.5 | 46.8 | 48.6 |
| 9 | 26.1 | 24.7 | 24.8 | 24.7 | 24.8 | 24.7 | 24.7 | 23.6 | 25.9 |
| 10 | 30.0 | 28.8 | 28.9 | 28.8 | 28.9 | 28.8 | 28.8 | 30.0 | 29.7 |
| 11 | 28.1 | 27.1 | 27.2 | 27.1 | 27.1 | 26.8 | 27.1 | 27.1 | 28.0 |
| 12 | 34.1 | 33.0 | 33.0 | 33.0 | 33.0 | 33.0 | 33.0 | 33.1 | 34.2 |
| 13 | 46.8 | 45.9 | 45.9 | 45.9 | 45.9 | 45.8 | 45.8 | 45.9 | 46.7 |
| 14 | 49.6 | 48.6 | 48.6 | 48.5 | 48.6 | 48.6 | 48.6 | 48.7 | 49.5 |
| 15 | 36.8 | 35.8 | 35.8 | 35.8 | 35.9 | 35.8 | 35.8 | 35.6 | 36.6 |
| 16 | 28.4 | 27.7 | 27.7 | 27.8 | 27.8 | 27.7 | 27.7 | 27.6 | 28.3 |
| 17 | 50.2 | 49.4 | 49.6 | 49.4 | 49.4 | 49.6 | 49.6 | 49.4 | 50.2 |
| 18 | 18.5 | 18.1 | 18.4 | 18.2 | 18.2 | 18.2 | 18.3 | 17.9 | 18.6 |
| 19 | 27.7 | 26.8 | 27.2 | 26.8 | 26.8 | 27.1 | 26.8 | 26.9 | 27.5 |
| 20 | 44.2 | 43.6 | 43.8 | 43.6 | 43.9 | 43.9 | 43.9 | 43.6 | 44.3 |
| 21 | 12.5 | 12.6 | 12.6 | 12.6 | 12.7 | 12.6 | 12.6 | 12.7 | 12.5 |
| 22 | 70.3 | 69.3 | 69.5 | 69.4 | 69.3 | 69.4 | 69.4 | 68.1 | 70.2 |
| 23 | 33.9 | 34.1 | 34.2 | 34.1 | 33.2 | 34.2 | 34.2 | 32.2 | 33.8 |
| 24 | 76.9 | 76.1 | 77.6 | 76.1 | 76.2 | 77.6 | 77.6 | 83.4 | 77.9 |
| 25 | 36.1 | 35.7 | 75.4 | 35.7 | 34.9 | 75.4 | 75.4 | 33.9 | 76.2 |
| 26 | 17.9 | 18.0 | 26.1 | 18.0 | 17.9 | 26.1 | 26.1 | 18.1 | 25.6 |
| 27 | 17.4 | 17.6 | 26.1 | 17.6 | 17.7 | 26.1 | 26.1 | 17.7 | 25.6 |
| 28 | 65.9 | 57.7 | 66.2 | 66.9 | 65.6 | 65.4 | 65.4 | 15.2 | 58.1 |
| 29 | 19.8 | 19.4 | 19.6 | 19.5 | 19.5 | 19.5 | 19.5 | 19.3 | 19.8 |
| 30 | 66.8 | 66.8 | 65.7 | 65.4 | 75.9 | 66.2 | 66.2 | 65.3 | 66.0 |
| glucoside at C-28 or C-3 | |||||||||
| 1’ | 104.9 | 103.3 | 105.1 | 105.3 | 105.1 | 105.1 | 104.1 | ||
| 2’ | 75.1 | 82.7 | 75.2 | 75.4 | 75.2 | 75.2 | 83.6 | ||
| 3’ | 77.9 | 78.1 | 78.3 | 78.5 | 78.3 | 78.3 | 78.3 | ||
| 4’ | 71.6 | 71.9 | 71.7 | 71.6 | 71.7 | 71.5 | 71.5 | ||
| 5’ | 78.0 | 78.5 | 77.4 | 78.6 | 77.4 | 75.2 | 78.0 | ||
| 6’ | 62.8 | 63.1 | 70.1 | 63.0 | 70.1 | 65.5 | 63.0 | ||
| 1’’ | 105.6 | 105.5 | 105.5 | 106.0 | |||||
| 2’’ | 76.6 | 75.3 | 75.3 | 76.9 | |||||
| 3’’ | 78.3 | 78.4 | 78.4 | 78.0 | |||||
| 4’’ | 71.4 | 71.7 | 71.7 | 71.8 | |||||
| 5’’ | 78.5 | 78.6 | 78.5 | 78.4 | |||||
| 6’’ | 62.6 | 62.8 | 62.8 | 62.9 | |||||
| malonyl moiety | |||||||||
| α | 168.1 | ||||||||
| β | 42.8 | ||||||||
| γ | 169.6 | ||||||||
| glucoside at C-30 or C-24 | |||||||||
| C-1‴ | 106.1 | 97.6 | |||||||
| C-2‴ | 75.4 | 75.8 | |||||||
| C-3‴ | 78.5 | 79.2 | |||||||
| C-4‴ | 71.6 | 72.2 | |||||||
| C-5‴ | 78.6 | 78.4 | |||||||
| C-6‴ | 63.0 | 63.0 |
| Position | 1a b) | 8a a) |
|---|---|---|
| 1 | 1.45 (1H, ddd, 13.2, 6.9, 2.8) | 1.53 (1H, ddd, 13.5, 6.6, 3.3) |
| 1.83 (1H, m) | 1.76 (1H, m) | |
| 2 | 2.41 (1H, ddd, 14.1, 6.9, 6.9) | 1.98 (1H, ddd, 11.9, 11.9, 6.6) |
| 2.53 (1H, ddd, 14.1, 2.8, 2.8) | 2.37 (1H, m) | |
| 4 | 2.251 (1H, m) | 2.18 (1H, ddd, 11.9, 4.7, 2.6) |
| 5 | 2.245 (1H, m) | 2.37 (1H, m) |
| 6 | 0.76 (1H, m) | 0.75 (1H, dddd, 12.9, 12.9, 12.9, 2.7) |
| 1.95 (1H, m) | 1.72 (1H, m) | |
| 7 | 1.07 (1H, dddd, 12.6, 12.6, 12.6, 2.9) | 1.08 (1H, dddd, 12.9, 12.9, 12.9, 2.7) |
| 1.25 (1H, m) | 1.72 (1H, m) | |
| 8 | 1.58 (1H, dd, 12.6, 4.9) | 1.61 (1H, m) |
| 11 | 2.02 (1H, m) | 1.22 (2H, m) |
| 1.19 (1H, ddd, 14.0, 9.7, 4.8) | ||
| 12 | 1.65 (2H, m) | 1.81 (2H, m) |
| 15 | 1.27 (2H, m) | 1.30 (2H, m) |
| 16 | 1.49 (1H, m) | 1.35 (1H, m) |
| 2.15 (1H, m) | 2.04 (1H, m) | |
| 17 | 1.80 (1H, dd, 11.0, 9.1) | 1.65 (1H, m) |
| 18 | 1.04 (3H, s) | 1.01 (3H, s) |
| 19 | 0.33 (1H, d, 4.1) | 0.38 (1H, d, 4.1) |
| 0.56 (1H, d, 4.1) | 0.58 (1H, d, 4.1) | |
| 20 | 2.05 (1H, m) | 1.72 (1H, m) |
| 21 | 1.21 (3H, d, 6.0) | 0.93 (3H, d, 6.6) |
| 22 | 4.61 (1H, d, 9.9) | 4.14 (1H, dd, 9.7, 2.7) |
| 23 | 2.12 (1H, d, 10.6) | 1.62 (1H, m) |
| 2.00 (1H, m) | 1.85 (1H, d, 14.8) | |
| 25 | 2.37 (1H, sep, 7.0) | |
| 26 | 1.22 (3H, d, 7.0) | 1.29 (3H, s) |
| 27 | 1.25 (3H, d, 7.0) | 1.27 (3H, s) |
| 28 | 3.96 (1H, d, 11.0) | 3.66 (1H, dd, 11.3, 4.7) |
| 4.39 (1H, d, 11.0) | 3.98 (1H, dd, 11.3, 2.6) | |
| 29 | 0.86 (3H, s) | 0.87 (3H, s) |
| 30 | 4.05 (1H, d, 11.9) | 3.82 (1H, d, 11.3) |
| 4.06 (1H, d, 11.9) | 3.97 (1H, d, 11.3) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoneyama, T.; Iseki, K.; Noji, M.; Imagawa, H.; Hashimoto, T.; Kawano, S.; Baba, M.; Kashiwada, Y.; Yahagi, T.; Matsuzaki, K.; et al. Marylosides A-G, Norcycloartane Glycosides from Leaves of Cymbidium Great Flower ‘Marylaurencin’. Molecules 2019, 24, 2504. https://doi.org/10.3390/molecules24132504
Yoneyama T, Iseki K, Noji M, Imagawa H, Hashimoto T, Kawano S, Baba M, Kashiwada Y, Yahagi T, Matsuzaki K, et al. Marylosides A-G, Norcycloartane Glycosides from Leaves of Cymbidium Great Flower ‘Marylaurencin’. Molecules. 2019; 24(13):2504. https://doi.org/10.3390/molecules24132504
Chicago/Turabian StyleYoneyama, Tatsuro, Kanako Iseki, Masaaki Noji, Hiroshi Imagawa, Toshihiro Hashimoto, Sachiko Kawano, Masaki Baba, Yoshiki Kashiwada, Tadahiro Yahagi, Keiichi Matsuzaki, and et al. 2019. "Marylosides A-G, Norcycloartane Glycosides from Leaves of Cymbidium Great Flower ‘Marylaurencin’" Molecules 24, no. 13: 2504. https://doi.org/10.3390/molecules24132504
APA StyleYoneyama, T., Iseki, K., Noji, M., Imagawa, H., Hashimoto, T., Kawano, S., Baba, M., Kashiwada, Y., Yahagi, T., Matsuzaki, K., & Umeyama, A. (2019). Marylosides A-G, Norcycloartane Glycosides from Leaves of Cymbidium Great Flower ‘Marylaurencin’. Molecules, 24(13), 2504. https://doi.org/10.3390/molecules24132504