Metabolites of Medicarpin and Their Distributions in Rats

Abstract
:1. Introduction
2. Results and Discussion
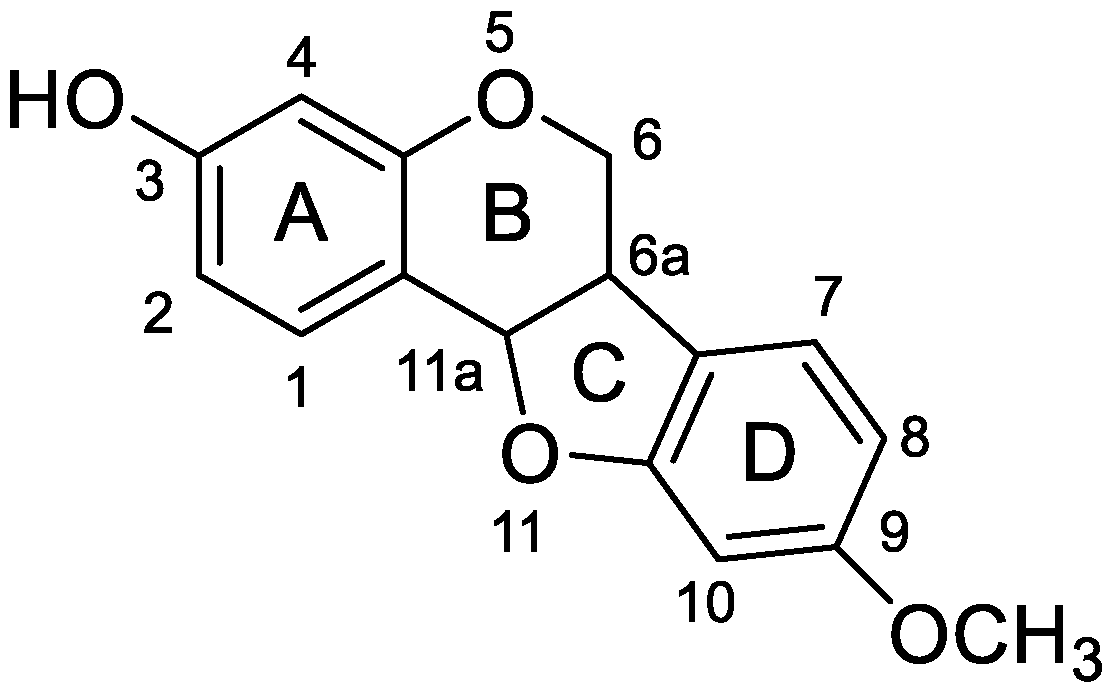
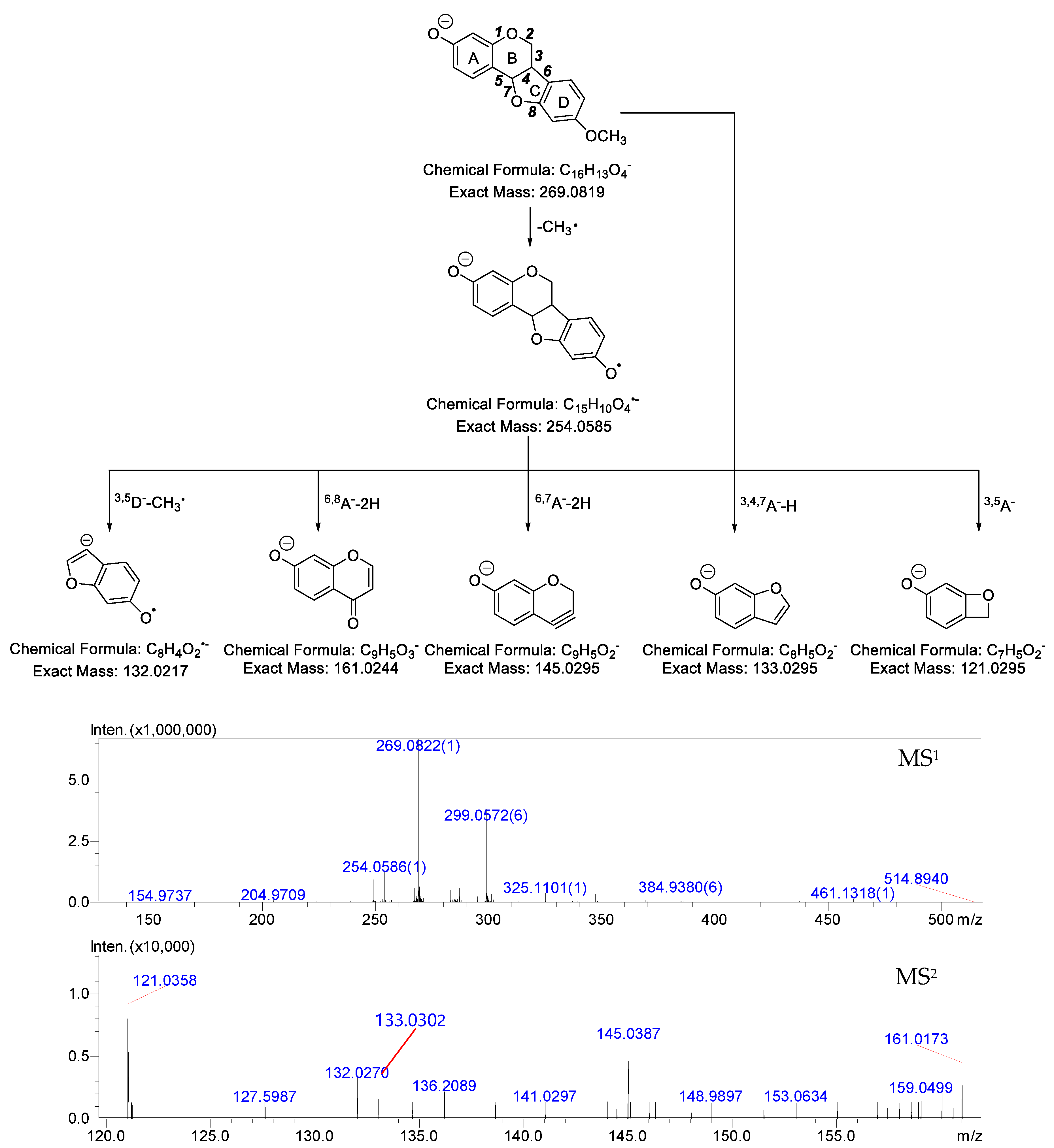
2.1. MS Fragmentation Characteristics of Medicarpin in ESI− Mode and Identification of Medicarpin in Rats
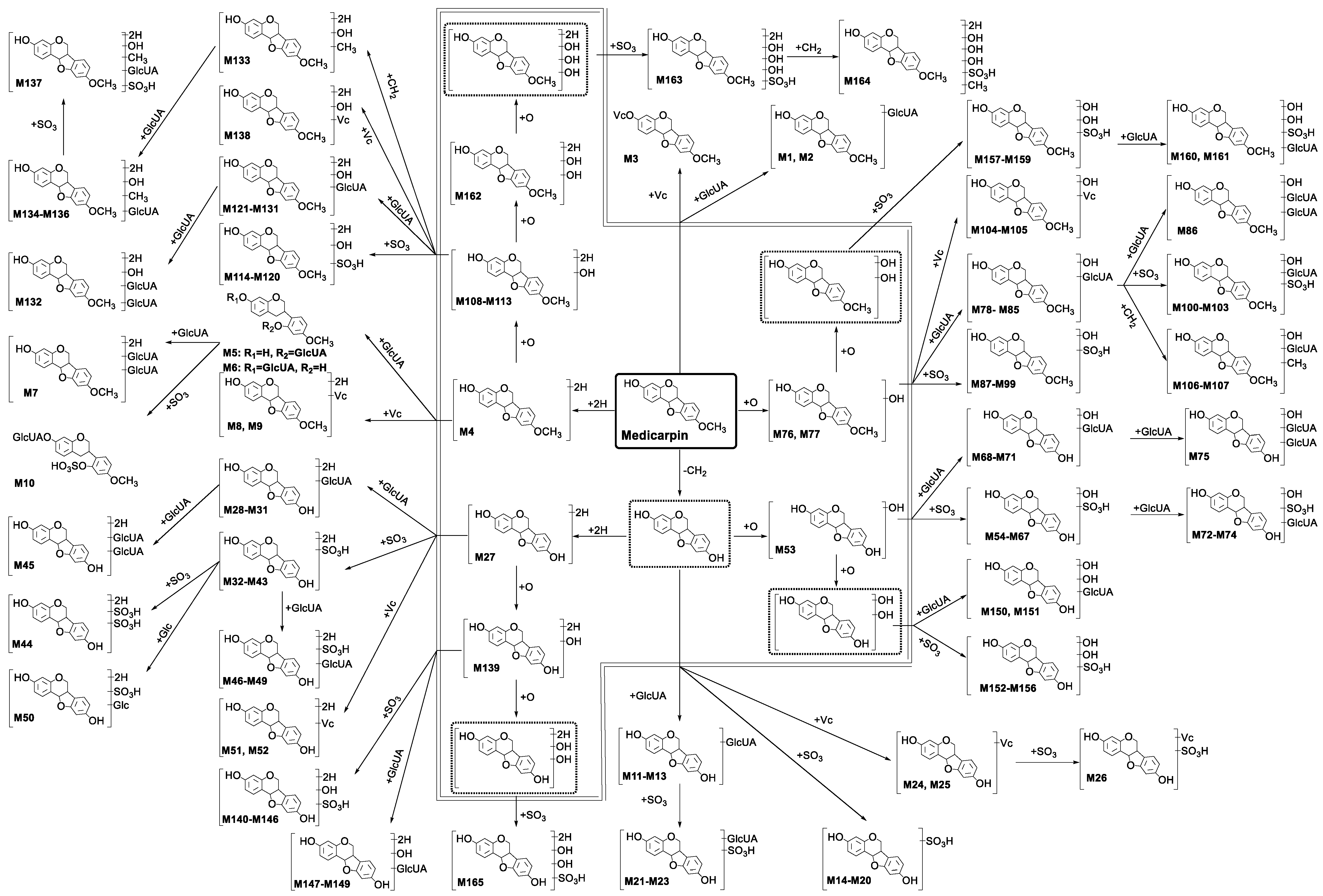
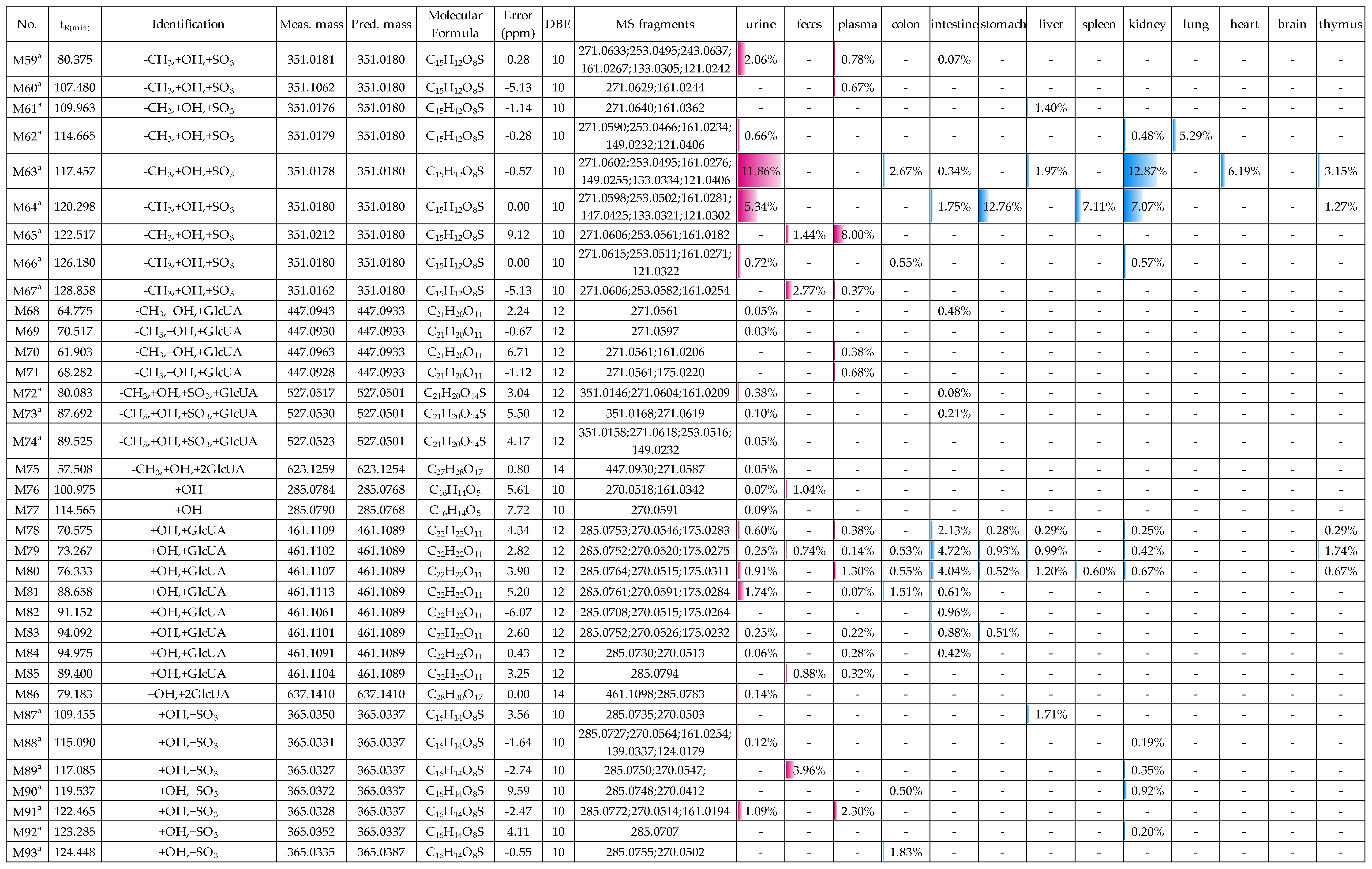
2.2. Profiling 165 Metabolites of Medicarpin in Rats
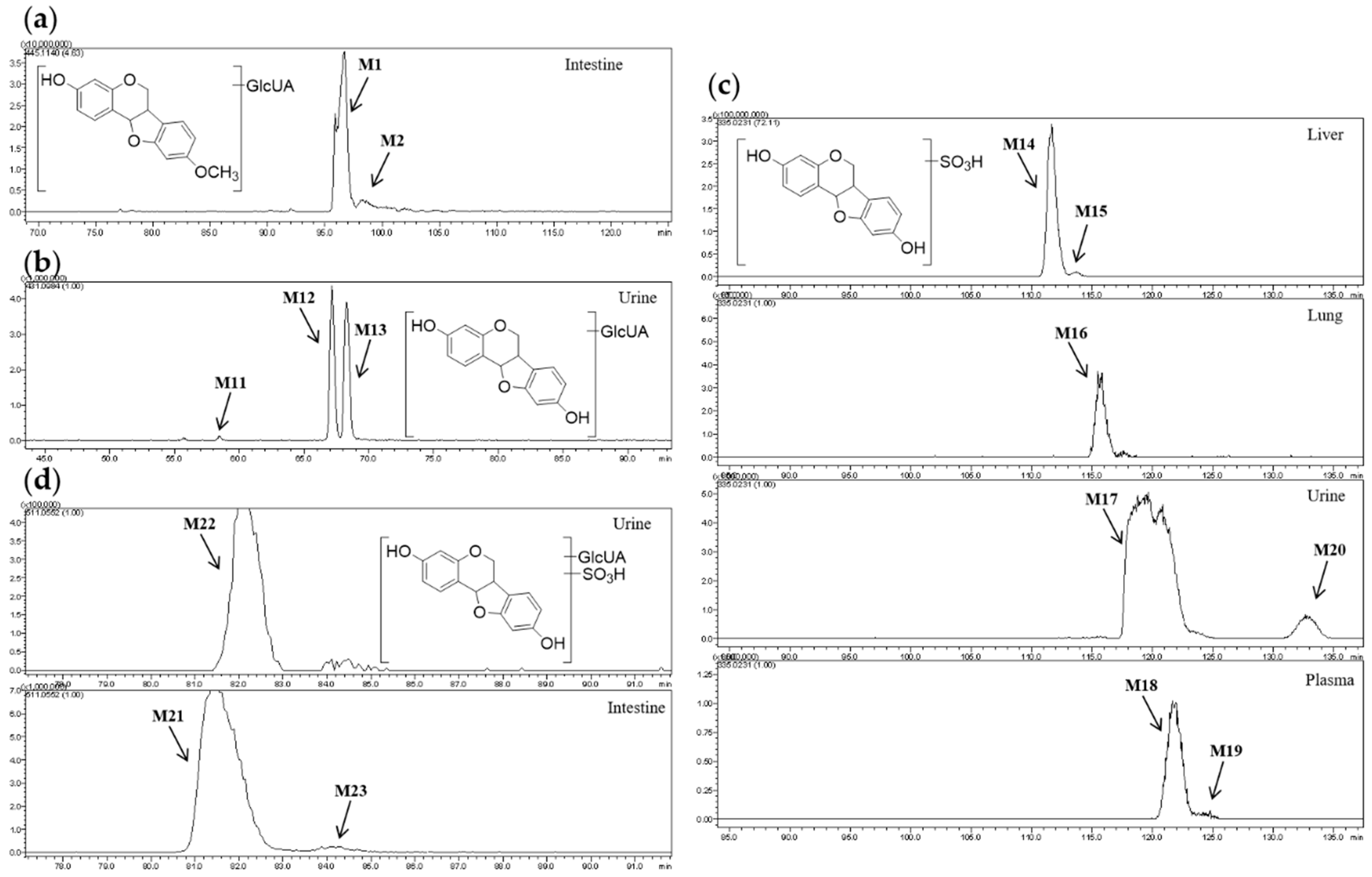
2.2.1. Analysis of Metabolites (M1–M3) Having the Skeleton of Medicarpin
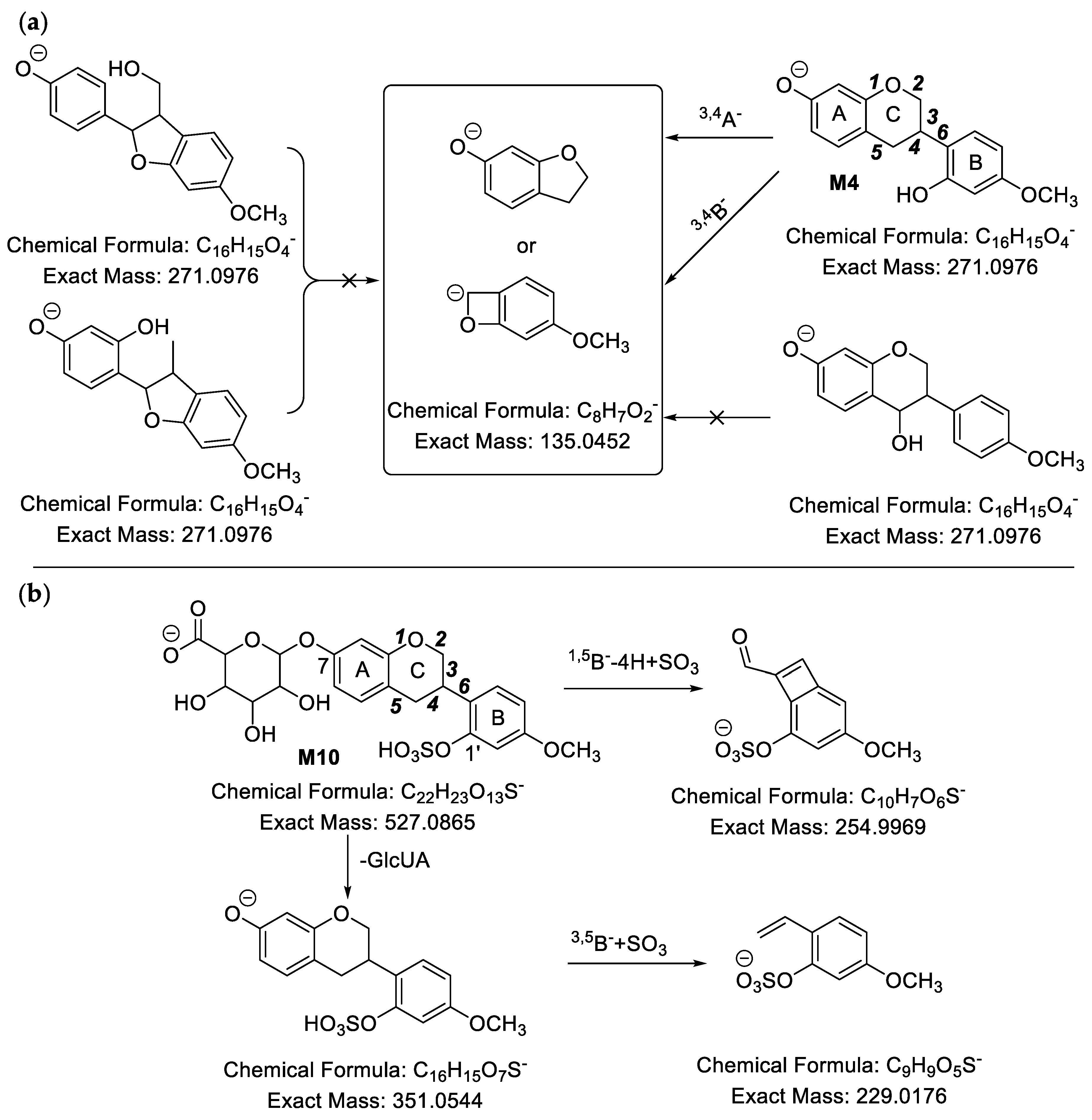
2.2.2. Analysis of Metabolites (M4–M10) Having the Skeleton of Hydrogenated Medicarpin
2.2.3. Analysis of Metabolites (M11–M26) Having the Skeleton of Demethylated Medicarpin
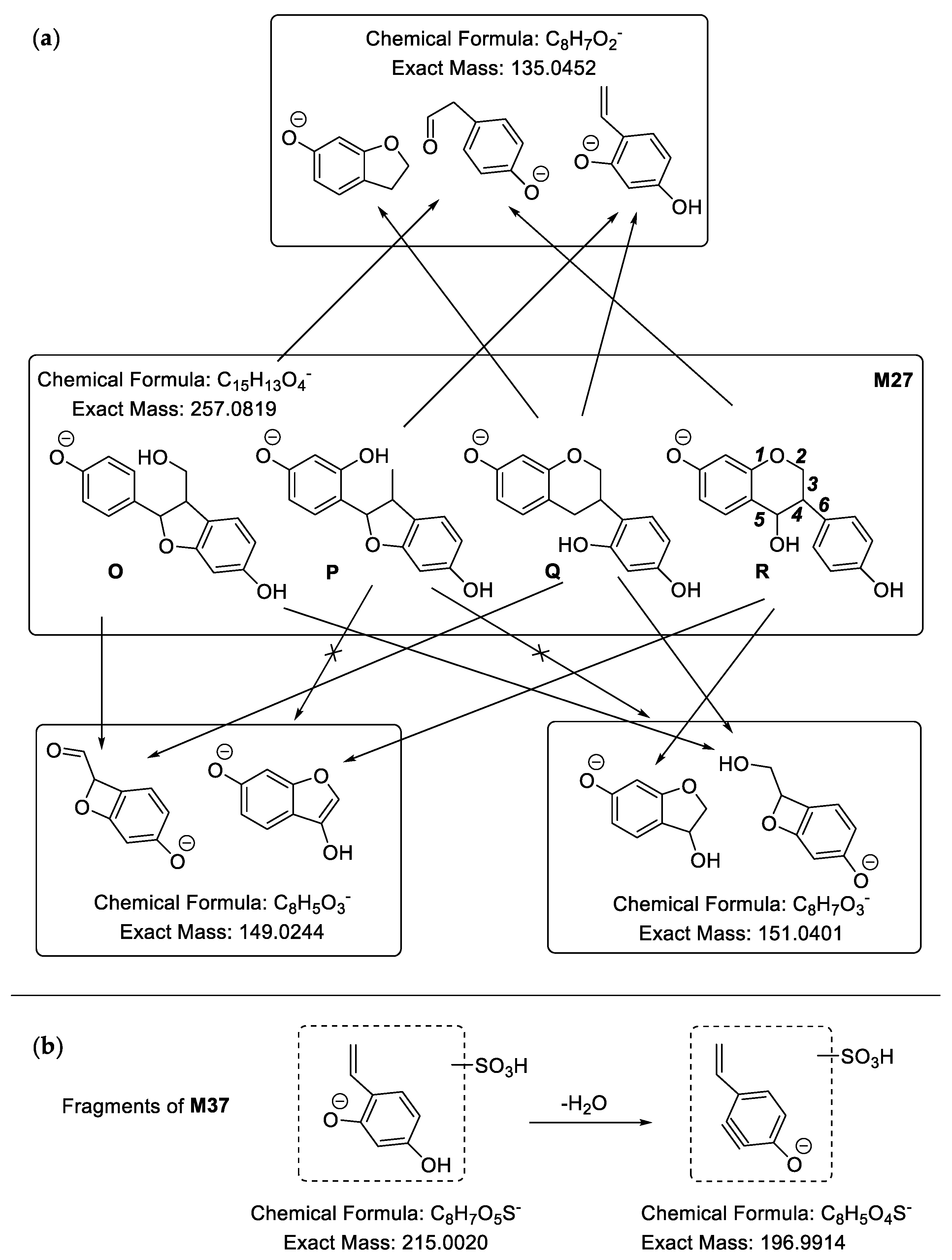
2.2.4. Analysis of Metabolites (M27–M52) Having the Skeleton of Demethylated and Hydrogenated Medicarpin
2.2.5. Analysis of Metabolites (M53–M75) Having the Skeleton of Demethylated and Hydroxylated Medicarpin
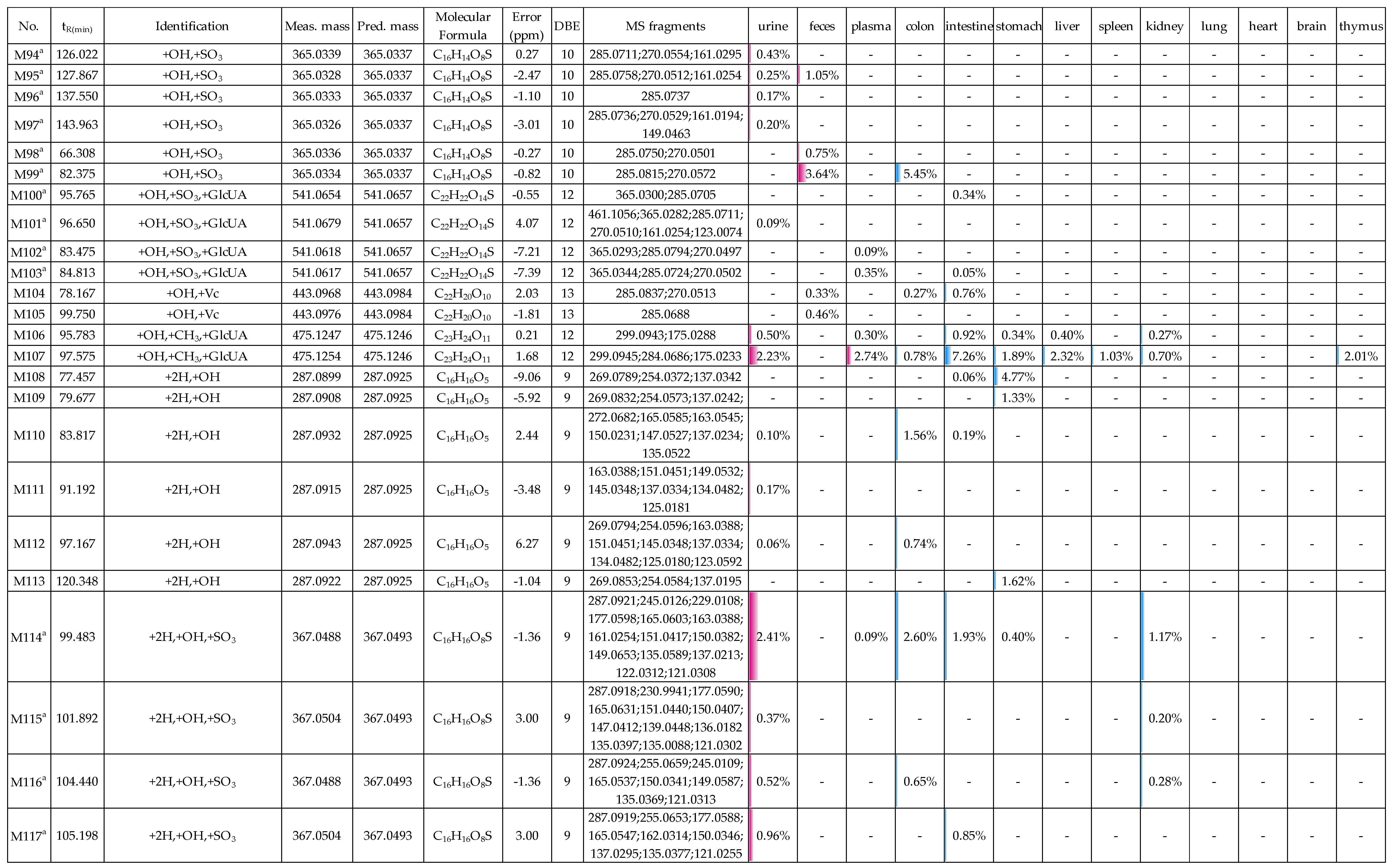
2.2.6. Analysis of Metabolites (M76–M107) Having the Skeleton of Hydroxylated Medicarpin
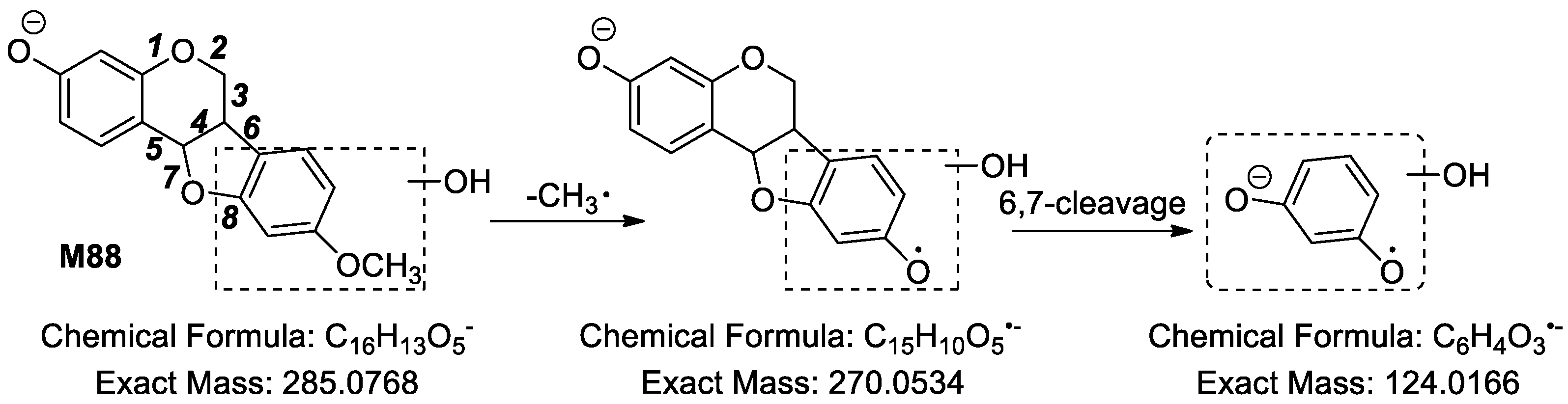
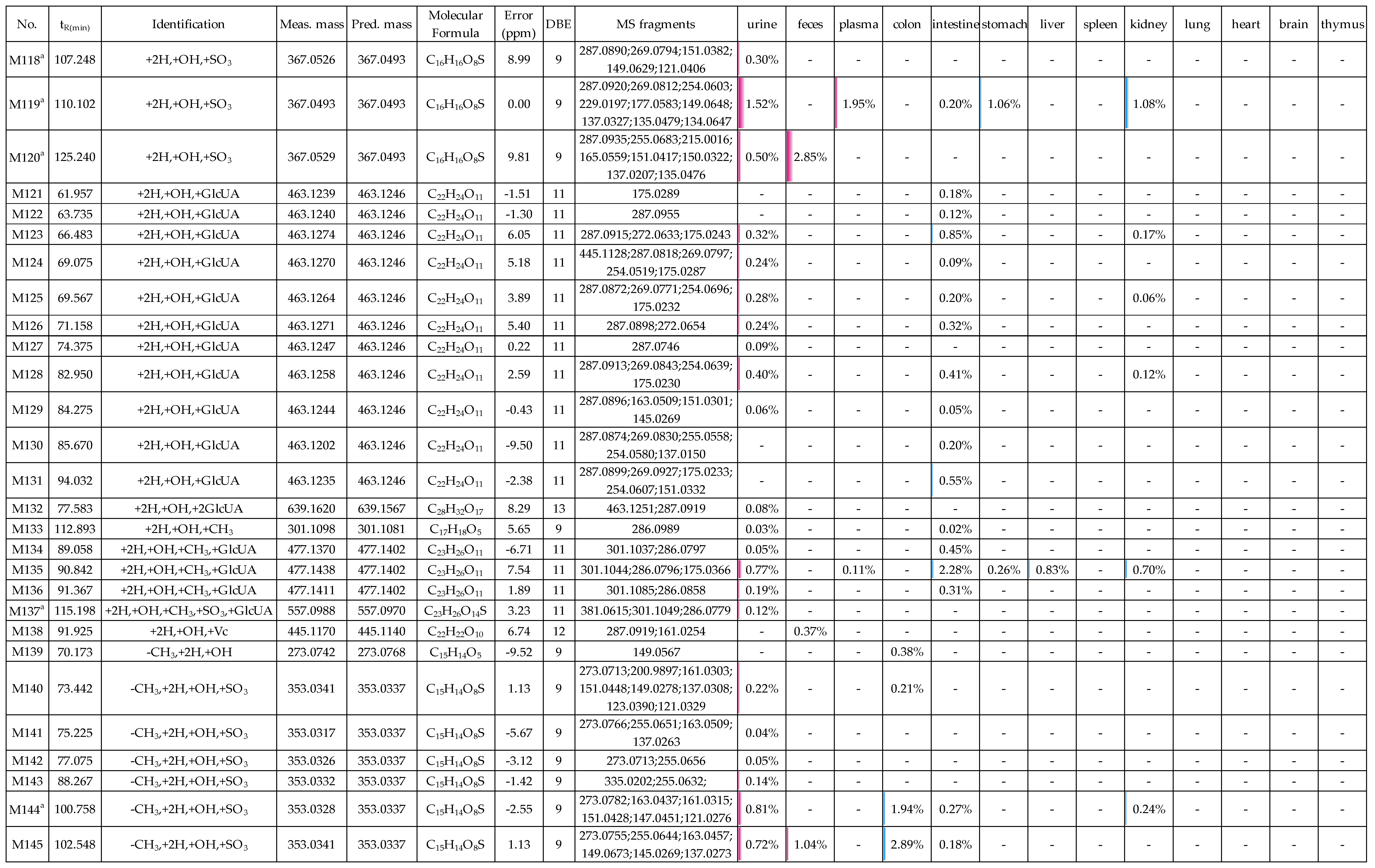
2.2.7. Analysis of Metabolites (M108–M138) Having the Skeleton of Hydrogenated and Hydroxylated Medicarpin
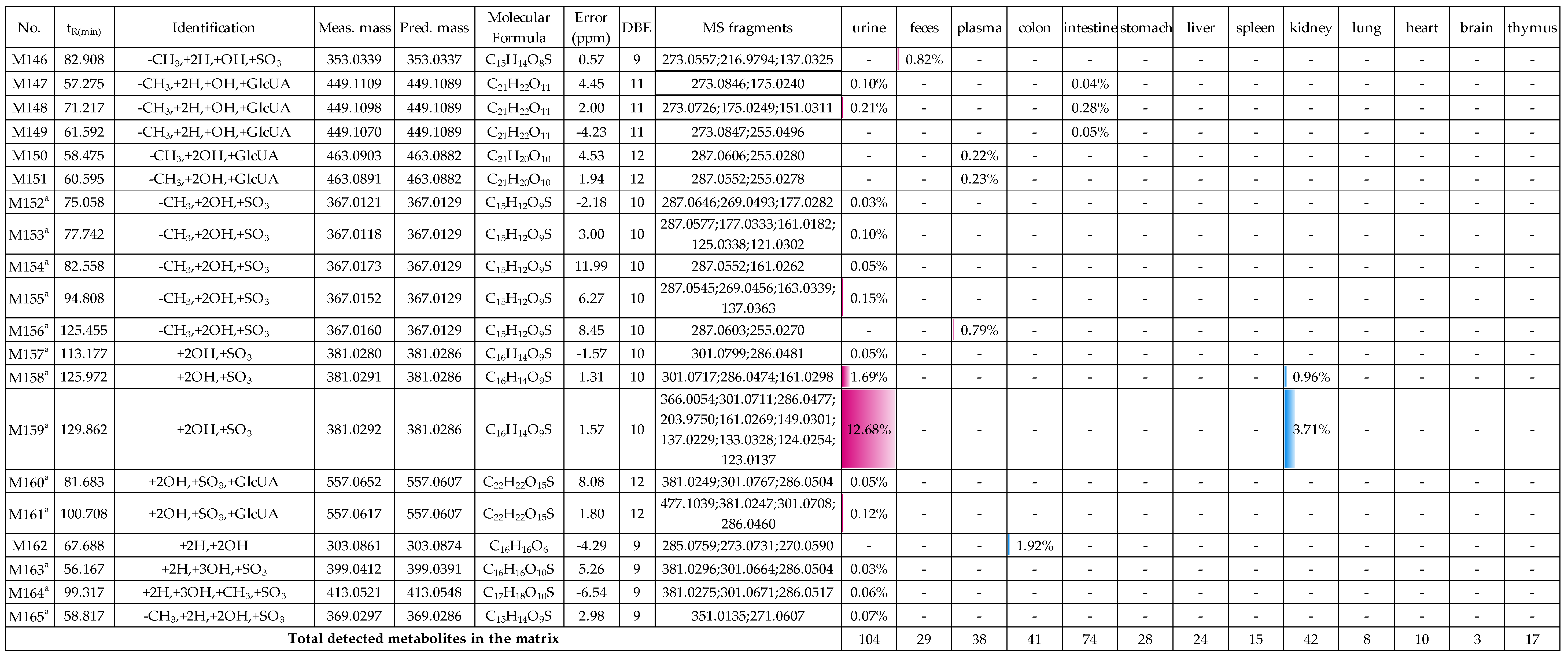
2.2.8. Analysis of Metabolites (M139–M149) Having the Skeleton of Demethylated, Hydrogenated and Hydroxylated Medicarpin
2.2.9. Analysis of Metabolites (M150–M156) Having the Skeleton of Demethylated and Dihydroxylated Medicarpin
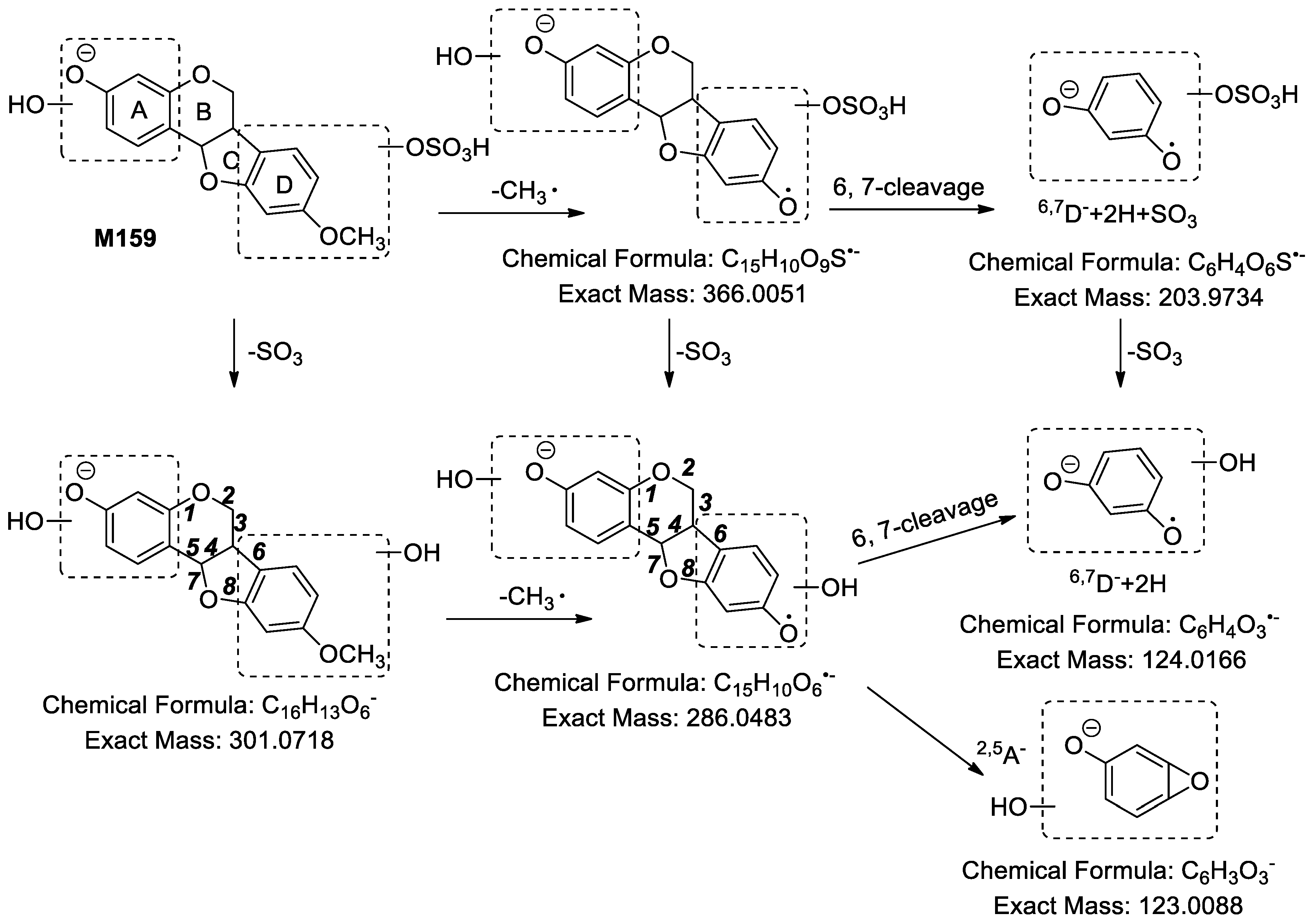
2.2.10. Analysis of Metabolites (M157–M161) Having the Skeleton of Dihydroxylated Medicarpin
2.2.11. Analysis of Metabolites M162 Having the Skeleton of Hydrogenated and Dihydroxylated Medicarpin
2.2.12. Analysis of Metabolites M163–M164 Having the Skeleton of Hydrogenated and Trihydroxylated Medicarpin
2.2.13. Analysis of Metabolite M165 Having the Skeleton of Demethylated, Hydrogenated and Dihydroxylated Medicarpin
2.3. Distribution of 165 Metabolites in Rats
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Animals and Drug Administration
3.3. Sample Collection and Preparation
3.4. Instrumentation and Analytical Conditions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Harper, S.H.; Kemp, A.D.; Underwood, W.G.E. Heartwood constituents of Stoarfzia madagascariensis. Chem. Comm. 1965, 14, 309–310. [Google Scholar]
- Belofsky, G.; Kolaczkowski, M.; Adams, E.; Schreiber, J.; Eisenberg, V.; Coleman, C.M.; Zou, Y.; Ferreira, D. Fungal ABC tansporter-associated activity of isoflavonoids from the root extract of Dalea formosa. J. Nat. Prod. 2013, 76, 915–925. [Google Scholar] [CrossRef]
- Rukachaisirikul, T.; Innok, P.; Aroonrerk, N.; Boonamnuaylap, W.; Limrangsun, S.; Boonyon, C.; Woonjina, U.; Suksamrarn, A. Antibacterial pterocarpans from Erythrina subumbrans. J. Ethnopharmacol. 2007, 110, 171–175. [Google Scholar] [CrossRef]
- Weng, J.; Tsao, L.; Yen, M.; Wang, J.; Lin, C. Anti-inflammatory constituents and new pterocarpanoid of Crotalaria pallida. J. Nat. Prod. 2003, 66, 404–407. [Google Scholar] [CrossRef]
- Zhou, H.; Lutterodt, H.; Cheng, Z.; Yu, L.L. Anti-inflammatory and antiproliferative activities of trifolirhizin, a flavonoid from Sophora flavescens Roots. J. Agric. Food Chem. 2009, 57, 4580–4585. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, H.; Hsiao, P.; Kuo, L.Y.; Lee, I.; Wu, T.; Chiou, W.; Kuo, Y. New isoflavonoid glycosides and related constituents from Astragali Radix (Astragalus membranaceus) and their inhibitory activity on nitric oxide production. J. Agric. Food Chem. 2011, 59, 1131–1137. [Google Scholar] [CrossRef]
- Kuete, V.; Sandjo, L.P.; Djeussi, D.E.; Zeino, M.; Kwamou, G.M.N.; Ngadjui, B.; Efferth, T. Cytotoxic flavonoids and isoflavonoids from Erythrina sigmoidea towards multi-factorial drug resistant cancer cells. Invest. New Drugs 2014, 32, 1053–1062. [Google Scholar] [CrossRef]
- Mahajan, P.; Gnana, O.R.; Jachak, S.M.; Bharate, S.B.; Chaudhuri, B. Antioxidant and antiproliferative activity of indigocarpan, a pterocarpan from Indigofera aspalathoides. J. Pharm. Pharmacol. 2016, 68, 1331–1339. [Google Scholar] [CrossRef]
- Nguyen, P.H.; Le, T.V.T.; Thuong, P.T.; Dao, T.T.; Ndinteh, D.T.; Mbafor, J.T.; Kang, K.W.; Oh, W.K. Cytotoxic and PTP1B inhibitory activities from Erythrina abyssinica. Bioorg. Med. Chem. Lett. 2009, 19, 6745–6749. [Google Scholar] [CrossRef]
- Bhargavan, B.; Singh, D.; Gautam, A.K.; Mishra, J.S.; Kumar, A.; Goel, A.; Dixit, M.; Pandey, R.; Manickavasagam, L.; Dwivedi, S.D.; et al. Medicarpin, a legume phytoalexin, stimulates osteoblast differentiation and promotes peak bone mass achievement in rats: Evidence for estrogen receptor β-mediated osteogenic action of medicarpin. J. Nutr. Biochem. 2012, 23, 27–38. [Google Scholar] [CrossRef]
- Kureel, J.; Dixit, M.; Tyagi, A.M.; Mansoori, M.N.; Srivastava, K.; Raghuvanshi, A.; Maurya, R.; Trivedi, R.; Goel, A.; Singh, D. miR-542-3p suppresses osteoblast cell proliferation and differentiation, targets BMP-7 signaling and inhibits bone formation. Cell Death Dis. 2014, 5, e1050. [Google Scholar] [CrossRef]
- Tyagi, A.M.; Gautam, A.K.; Kumar, A.; Srivastava, K.; Bhargavan, B.; Trivedi, R.; Saravanan, S.; Yadav, D.K.; Singh, N.; Pollet, C.; et al. Medicarpin inhibits osteoclastogenesis and has nonestrogenic bone conserving effect in ovariectomized mice. Mol. Cell Endocrinol. 2010, 325, 101–109. [Google Scholar] [CrossRef]
- Vieira, N.C.; Espíndola, L.S.; Santana, J.M.; Veras, M.L.; Pessoa, O.D.; Pinheiro, S.M.; de Araujo, R.M.; Lima, M.A.; Silveira, E.R. Trypanocidal activity of a new pterocarpan and other secondary metabolites of plants from Northeastern Brazil flora. Bioorg. Med. Chem. 2008, 16, 1676–1682. [Google Scholar] [CrossRef]
- Rosa, A.; Deiana, M.; Corona, G.; Atzeri, A.; Incani, A.; Appendino, G.; Dessì, M.A. Antioxidant properties of extracts and compounds from Psoralea morisiana. Eur. J. Lipid Sci. Technol. 2005, 107, 521–529. [Google Scholar] [CrossRef]
- Fotso, G.W.; Maher, F.A.; Ngnintedo, D.; Ango, P.Y.; Kapche, D.G.F.W.; Ngameni, B.; Ngwenya, B.; Yeboah, S.O.; Ngadjui, B.T.; Andrae-Marobela, K. Three new isoflavonoids with antioxidant properties from Ptycholobium contortum (N.E.Br.) Brummitt (Leguminosae). Phytochem. Lett. 2015, 14, 254–259. [Google Scholar] [CrossRef]
- Selvam, C.; Jachak, S.M.; Gnana Oli, R.; Thilagavathi, R.; Chakraborti, A.K.; Bhutani, K.K. A new cyclooxygenase (COX) inhibitory pterocarpan from Indigofera aspalathoides: Structure elucidation and determination of binding orientations in the active sites of the enzyme by molecular docking. Tetrahedron Lett. 2004, 45, 4311–4314. [Google Scholar] [CrossRef]
- Yuk, H.J.; Curtis-Long, M.J.; Ryu, H.W.; Jang, K.C.; Seo, W.D.; Kim, J.Y.; Kang, K.Y.; Park, K.H. Pterocarpan profiles for soybean leaves at different growth stages and investigation of their glycosidase inhibitions. J. Agric. Food Chem. 2011, 59, 12683–12690. [Google Scholar] [CrossRef]
- Ryu, Y.B.; Curtis-Long, M.J.; Kim, J.H.; Jeong, S.H.; Yang, M.S.; Lee, K.W.; Lee, W.S.; Park, K.H. Pterocarpans and flavanones from Sophora flavescens displaying potent neuraminidase inhibition. Bioorg. Med. Chem. Lett. 2008, 18, 6046–6049. [Google Scholar] [CrossRef]
- Woo, H.S.; Kim, D.W.; Curtis-Long, M.J.; Lee, B.W.; Lee, J.H.; Kim, J.Y.; Kang, J.E.; Park, K.H. Potent inhibition of bacterial neuraminidase activity by pterocarpans isolated from the roots of Lespedeza bicolor. Bioorg. Med. Chem. Lett. 2011, 21, 6100–6103. [Google Scholar] [CrossRef]
- Mori-Hongo, M.; Takimoto, H.; Katagiri, T.; Kimura, M.; Ikeda, Y.; Miyase, T. Melanin synthesis inhibitors from Lespedeza floribunda. J. Nat. Prod. 2009, 72, 194–203. [Google Scholar] [CrossRef]
- Boué, S.M.; Burow, M.E.; Wiese, T.E.; Shih, B.Y.; Elliott, S.; Carter-Wientjes, C.H.; McLachlan, J.A.; Bhatnagar, D. Estrogenic and antiestrogenic activities of phytoalexins from red kidney bean (Phaseolus vulgaris L.). J. Agric. Food. Chem. 2011, 59, 112–120. [Google Scholar] [CrossRef]
- Maurich, T.; Pistelli, L.; Turchi, G. Anti-clastogenic activity of two structurally related pterocarpans purified from Bituminaria bituminosa in cultured human lymphocytes. Mutat. Res. 2004, 561, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xu, F.; Wang, D.; Ye, J.; Cai, Q. Buyang Huanwu Decoction ameliorates ischemic stroke by modulating multiple targets with multiple components: In vitro evidences. Chin. J. Nat. Med. 2018, 16, 0194–0202. [Google Scholar] [CrossRef]
- Ahmad, V.U.; Iqbal, S.; Nawaz, S.A.; Choudhary, M.I.; Farooq, U.; Ali, S.T.; Ahmad, A.; Bader, S.; Kousar, F.; Arshad, S.; et al. Isolation of four new pterocarpans from Zygophyllum eurypterum (Syn. Z. atriplicoides) with enzyme-inhibition properties. Chem. Biodivers. 2006, 3, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, Y.; Zhang, H.; Liang, S. In-vivo study of metabolism of trifolirhizin in rats. Trad. Chin. Drug Res. Clin. Pharmacol. 2012, 23, 174–177. [Google Scholar]
- Hargreaves, J.A.; Mansfield, J.W.; Coxon, D.T. Identification of medicarpin as a phytoalexin in the broad bean plant (Vicia Jaba L.). Nature 1976, 262, 318–319. [Google Scholar] [CrossRef]
- Kim, J.M.; Lee, Y.M.; Lee, G.Y.; Jang, D.S.; Bae, K.H.; Kim, J.S. Constituents of the roots of Pueraria lobata inhibit formation of advanced glycation end products (AGEs). Arch. Pharm. Res. 2006, 29, 821–825. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, H.; Zhao, Y.; Wang, B.; Zhang, Q.; Zhang, L.; Tu, P. Quantification and stability studies on the flavonoids of Radix hedysari. J. Agric. Food Chem. 2006, 54, 6634–6639. [Google Scholar] [CrossRef]
- Rayanil, K.; Bunchornmaspan, P.; Tuntiwachwuttikul, P. A new phenolic compound with anticancer activity from the wood of Millettia leucantha. Arch. Pharm. Res. 2011, 34, 881–886. [Google Scholar] [CrossRef]
- Gatouillat, G.; Magid, A.A.; Bertin, E.; El Btaouri, H.; Morjani, H.; Lavaud, C.; Madoulet, C. Medicarpin and millepurpan, two flavonoids isolated from Medicago sativa, induce apoptosis and overcome multidrug resistance in leukemia P388 cells. Phytomedicine 2015, 22, 1186–1194. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Sotres, C.; López-Albarrán, P.; Cruz-de-León, J.; García-Moreno, T.; Rutiaga-Quiñones, J.G.; Vázquez-Marrufo, G.; Tamariz-Mascarúa, J.; Herrera-Bucio, R. Medicarpin, an antifungal compound identified in hexane extract of Dalbergia congestiflora Pittier heartwood. Int. Biodeterior. Biodegrad. 2012, 69, 38–40. [Google Scholar] [CrossRef]
- Promchai, T.; Janhom, P.; Maneerat, W.; Rattanajak, R.; Kamchonwongpaisan, S.; Pyne, S.G.; Limtharakul, T. Antibacterial and cytotoxic activities of phenolic constituents from the stem extracts of Spatholobus parviflorus. Nat. Prod. Res. 2018, 1–5. [Google Scholar] [CrossRef]
- Imran, K.M.; Yoon, D.; Kim, Y. A pivotal role of AMPK signaling in medicarpin-mediated formation of brown and beige. BioFactors 2018, 44, 168–179. [Google Scholar] [CrossRef]
- Imran, K.M.; Yoon, D.; Kim, Y.S. Medicarpin induces lipolysis via activation of protein kinase A in brown adipocytes. BMB Rep. 2018, 51, 249–254. [Google Scholar] [CrossRef]
- Ma, L.; Xu, F.; Li, F.; Wang, J.; Shang, M.; Liu, G.; Cai, S. The profiling and identification of the metabolites of 8-prenylkaempferol and a study on their distribution in rats by high-performance liquid chromatography with diode array detection combined with electrospray ionization ion trap time-of-flight multista. Biomed. Chromatogr. 2016, 30, 175–190. [Google Scholar] [CrossRef]
Sample Availability: Not available. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biosample | Relative Contents (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Phase I Reaction | Phase II Reaction | Phase I Metabolites | |||||||
| -CH3 | +2H | +OH | +SO3 | +GlcUA | +Vc | +Glc | +CH3 | ||
| urine | 55.14 | 37.01 | 57.79 | 74.60 | 25.83 | 1.08 | 0.23 | 3.93 | 1.78 |
| feces | 71.51 | 44.43 | 21.55 | 73.53 | 1.62 | 15.54 | — | — 1 | 9.30 |
| plasma | 36.58 | 25.72 | 22.78 | 36.17 | 67.87 | 0.44 | — | 3.16 | — |
| colon | 57.95 | 58.48 | 28.30 | 55.81 | 16.61 | 9.23 | — | 0.78 | 17.57 |
| intestine | 39.56 | 41.46 | 36.17 | 15.62 | 78.04 | 7.60 | — | 11.23 | 0.99 |
| stomach | 39.46 | 22.38 | 26.67 | 35.00 | 54.57 | 1.23 | — | 2.50 | 9.20 |
| liver | 62.62 | 31.43 | 13.33 | 57.11 | 42.09 | — | — | 3.55 | 0.80 |
| spleen | 33.84 | 23.45 | 8.74 | 27.79 | 71.33 | — | — | 1.03 | 0.88 |
| kidney | 60.90 | 30.46 | 34.93 | 63.43 | 36.30 | 0.10 | — | 1.67 | 0.17 |
| lung | 34.25 | 22.91 | 5.29 | 28.22 | 71.78 | — | — | — | — |
| heart | 33.74 | 22.74 | 6.19 | 27.76 | 71.70 | — | — | — | 0.54 |
| brain | 57.11 | 20.86 | — | 57.11 | 42.89 | — | — | — | — |
| thymus | 25.25 | 15.66 | 9.12 | 18.96 | 80.54 | — | — | 2.01 | 0.49 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.-Y.; Li, T.; Ji, R.; Xu, F.; Liu, G.-X.; Li, Y.-L.; Shang, M.-Y.; Cai, S.-Q. Metabolites of Medicarpin and Their Distributions in Rats. Molecules 2019, 24, 1966. https://doi.org/10.3390/molecules24101966
Wang H-Y, Li T, Ji R, Xu F, Liu G-X, Li Y-L, Shang M-Y, Cai S-Q. Metabolites of Medicarpin and Their Distributions in Rats. Molecules. 2019; 24(10):1966. https://doi.org/10.3390/molecules24101966
Chicago/Turabian StyleWang, Hong-Yan, Teng Li, Rui Ji, Feng Xu, Guang-Xue Liu, Yao-Li Li, Ming-Ying Shang, and Shao-Qing Cai. 2019. "Metabolites of Medicarpin and Their Distributions in Rats" Molecules 24, no. 10: 1966. https://doi.org/10.3390/molecules24101966
APA StyleWang, H.-Y., Li, T., Ji, R., Xu, F., Liu, G.-X., Li, Y.-L., Shang, M.-Y., & Cai, S.-Q. (2019). Metabolites of Medicarpin and Their Distributions in Rats. Molecules, 24(10), 1966. https://doi.org/10.3390/molecules24101966