1. Introduction
Sensitive and reliable methods for the determination of food allergens are necessary to detect the undeclared allergens resulting from contamination during food production and to enforce the regulation of allergen labeling [
1]. For these purposes, various techniques have been applied to detect either the allergen protein itself or a marker indicating its presence [
2]. Enzyme-linked immunosorbent assay (ELISA) is commonly used for allergen analysis. However, ELISA is limited by the possibility of cross-reactivity which leads to false-positive results in this antibody-based assay. DNA-based methods, which are carried out by means of detecting specific DNA fragments instead of the allergenic compounds, could not provide the exact concentration of the allergens [
1]. Because of these limitations, alternative strategies for allergen analysis are required. Mass spectrometry (MS)-based proteomic methods, providing high sensitivity and allowing the identification and quantification of allergenic protein, thus have been of great importance for allergen detection [
3].
In general, the methodology of MS-based proteomics absolute quantification is to analyze a signature peptide using a synthetic analogue with a stable isotope label as the internal standard [
4]. A signature peptide is a surrogate peptide that is unique to the target allergenic protein and fulfills certain criteria, including the length of the peptide, the accuracy of digestion, and the absence of post-translational modification [
5]. The stable isotope labels can be introduced into the signature peptide metabolically, enzymatically, chemically, or provided by synthetic peptide standard [
6]. However, many of these proposed methods suffer from several disadvantages, such as limited biological applicability for labeling in cell culture, the requirement of a certain amino acid for some chemical labeling methods, and the high cost of isobaric tags, which restrict the applicability in routine use for food samples [
7].
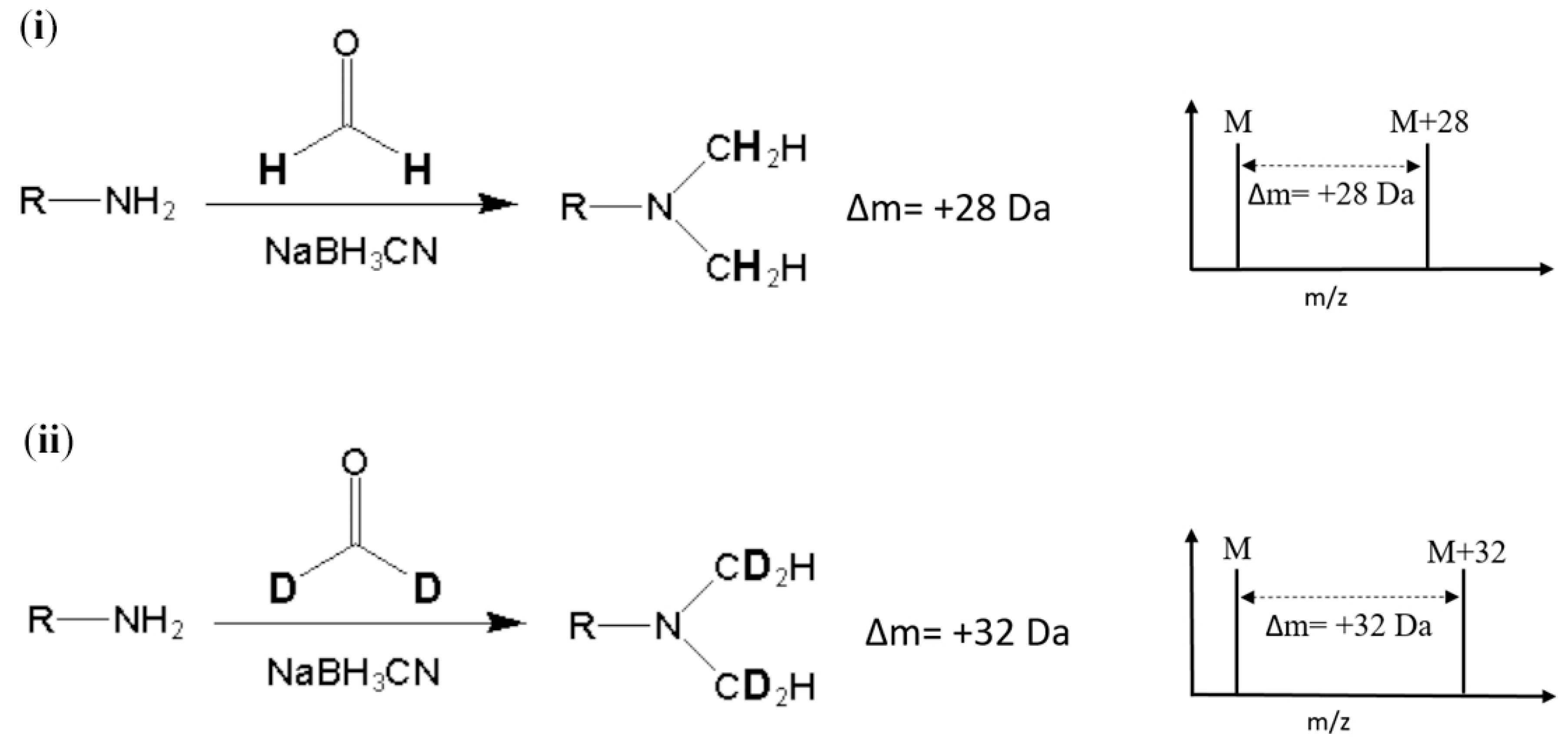
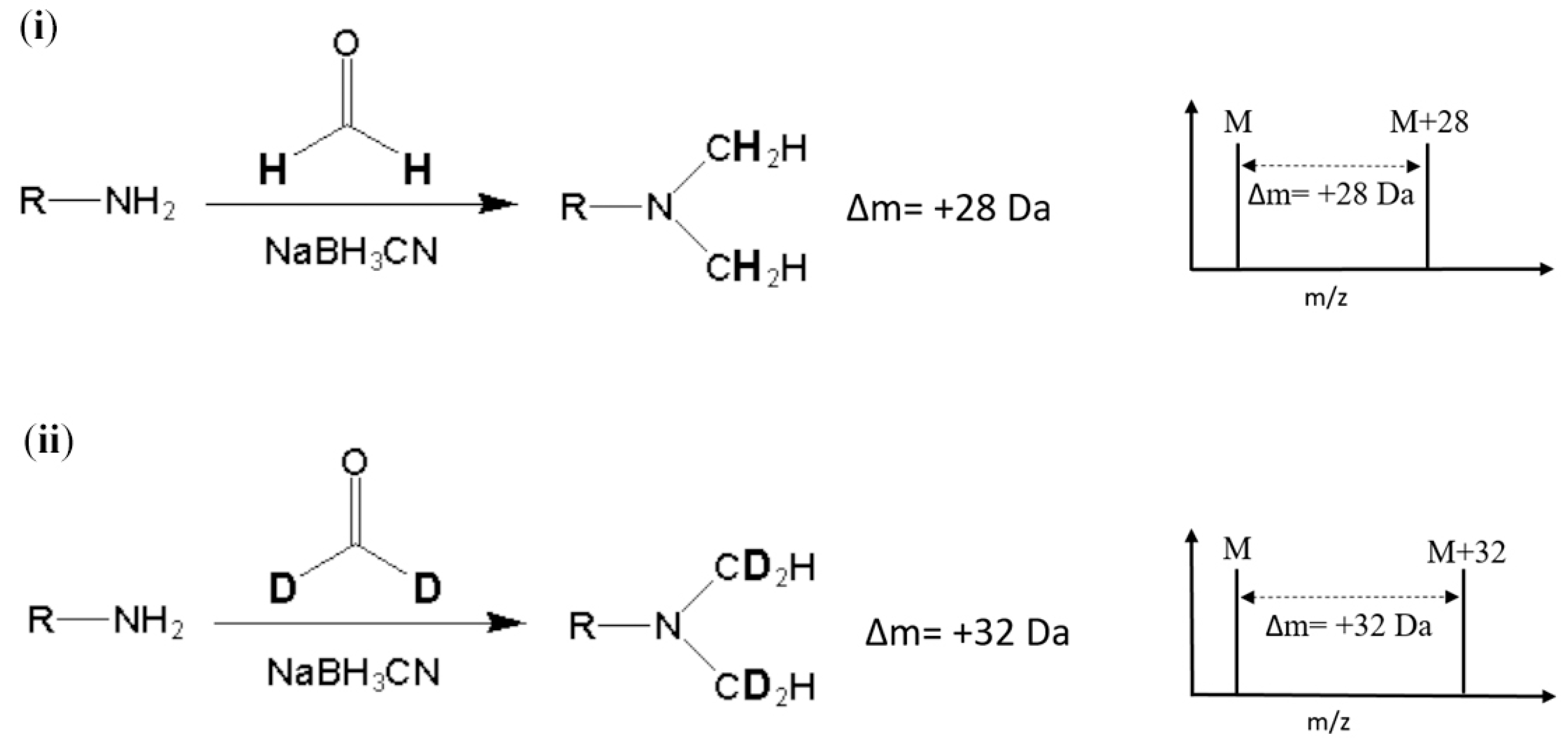
Stable-isotope dimethyl labeling, as a chemical labeling method for quantitative proteomics, is achieved by the dimethylation of a primary amine (the N-terminus and side chain of lysine residue) using different isotope forms of formaldehyde and sodium cyanoborohydride (NaBH
3CN) [
8]. The derivatization carried out with deuterated formaldehyde (CD
2O) produces a mass increase of 32 Da for each reductive site (
Figure 1). On the other hand, dimethylation performed with normal formaldehyde (CH
2O) generates a mass shift of +28 Da per primary amide. In addition, several combinations of reagents with different isotope forms were proposed to produce various mass differences [
9]. The reaction of dimethyl labeling completes in minutes without any side product, and this method is much more economical in cost than other developed methods [
9,
10]. In addition, dimethyl labeling can be practiced on protein from any species [
7]. With these advantages, stable-isotope dimethyl labeling has been applied in many quantitative proteomic studies [
11,
12,
13,
14]. Nonetheless, to our knowledge, isotope dimethyl labeling has not been applied in any analysis method for either allergenic proteins or food samples.
Kiwifruit (
Actinidia deliciosa) is currently considered to be one of the common allergenic foods. According to the World Health Organization and International Union of Immunological Societies Allergen Nomenclature Sub-committee [
15], 13 kiwifruit allergenic proteins have been officially registered. Actinidin (Act d 1), a thiol-protease, is the major allergen of kiwifruit [
16]. Act d 2 belongs to the thaumatin-like protein family which includes several fruit allergens [
17]. Act d 3 is a glycoprotein, but its function is still unknown [
18]. Act d 4 is characterized as phytocystatin, which is a cysteine protease [
19]. Kiwellin (Act d 5), a major protein component of kiwifruit, is a cell-wall-related protein [
20]. Act d 6 and Act d 7 are a pectin methylesterase inhibitor and pectin methylesterase, respectively [
21]. Both Act d 8, a Bet v 1 homologue, and Act d 9, profilin, are pollen-related allergens [
21,
22]. Act d 10 is a lipid transfer protein, and Act d 11 belongs to ripening-related protein family [
23,
24]. Act d 12, an 11S globulin, and Act d 13, a 2S albumin, are two novel allergens that are located in the seeds [
25]. With several kiwi allergens being identified, some methods for the analysis of them have been proposed. The quantification of Act d 1 and Act d 2 was carried out by means of ELISA and liquid chromatography–ultraviolet (LC-UV) method, respectively [
26,
27]. However, these methods suffer from some disadvantages such as false-positive results caused by cross-reactivity in ELISA and overestimation of allergen contents resulting from the low selectivity in absorbance assay [
28]. Allergen detection using liquid chromatography–tandem mass spectrometry in multiple reaction monitoring mode (LC-MS-MRM) is performed by analyzing the
m/
z values and ion transitions of signature peptides for target allergens, which could improve the selectivity and prevent erroneous quantification [
28]. Moreover, higher sensitivity could be achieved within the MRM mode [
5], indicating that the LC-MS-MRM could be an appropriate method for the analysis of kiwi allergens at trace level.
In this work, we intend to propose the first-time application of stable-isotope dimethyl labeling to the trace quantification of fruit allergens using LC-MS-MRM. The identification of allergens was carried out with liquid chromatography/electrospray ionization-quadrupole-time of flight mass spectrometry (LC/ESI-Q-TOF). The identified allergens with high coverage and MASCOT scores were selected as target allergens. To develop the platform for the quantification of kiwi allergens, the external and internal standards were prepared by isotopically labeling the synthetic signature peptide standards with stable-isotope dimethyl labeling. In addition, some steps for protein extraction described in a previous study [
29] were optimized to be more time-efficient. Moreover, the established procedure was validated with certain criteria and was applied to self-made kiwi foods to examine the applicability for foodstuff analysis.
3. Discussion
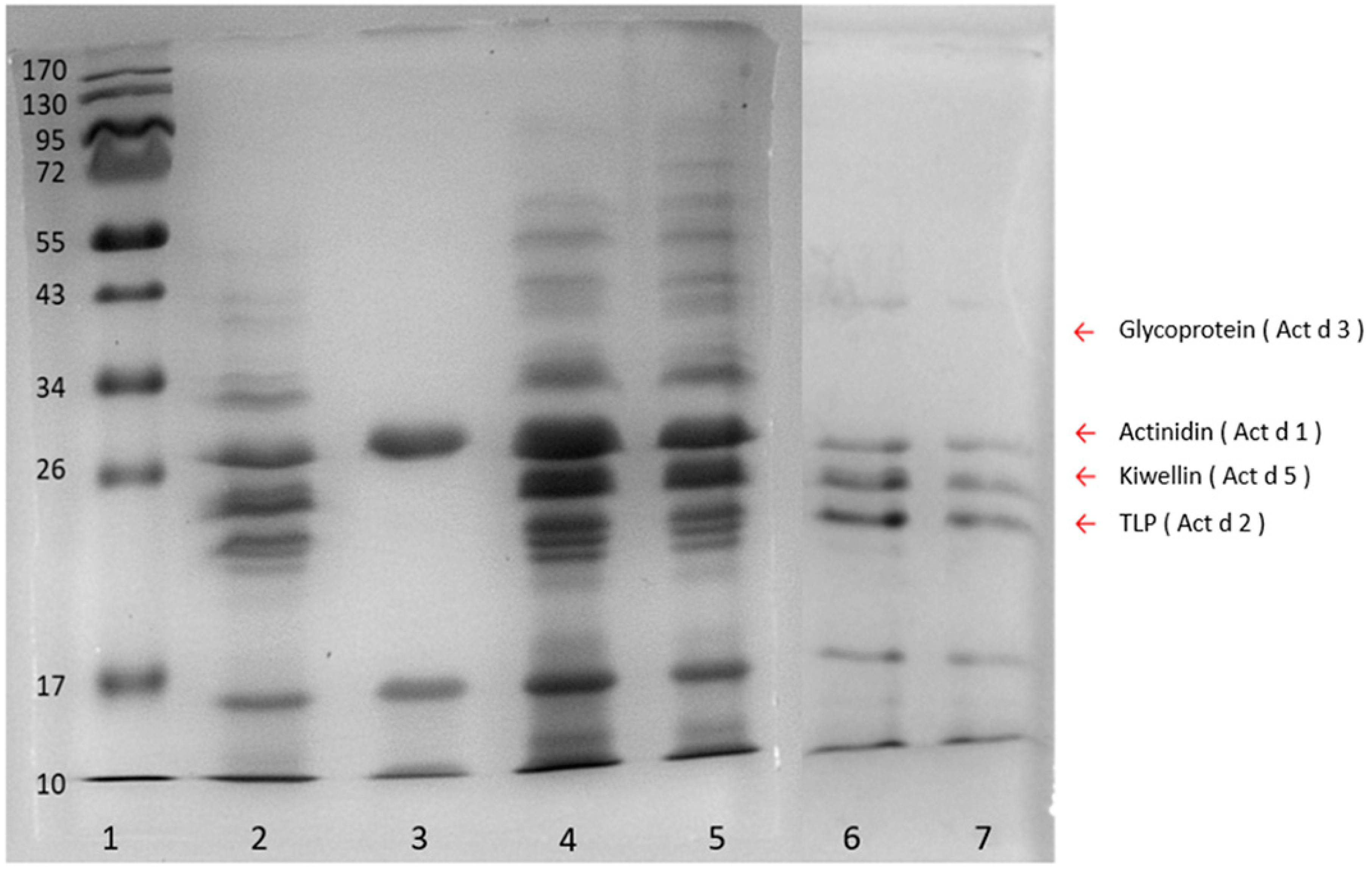
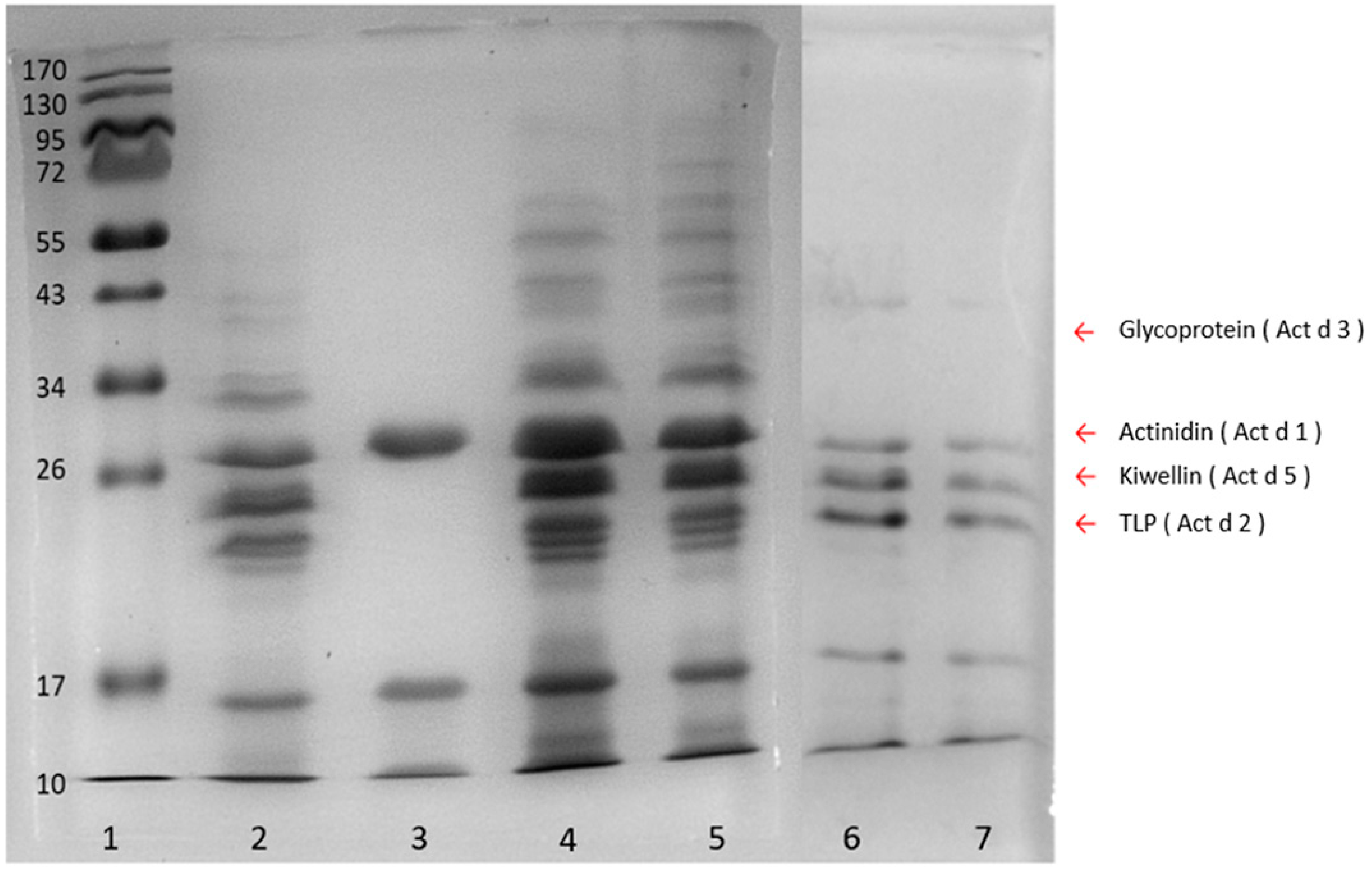
The sample preparation and protein extraction were the key steps for trace peptide determination. Compared with the ammonium sulfate precipitation and sodium chloride extraction methods, the phenol extraction coupled with methanolic ammonium acetate precipitation produced a more comprehensive population of kiwi proteins in fruit juice samples (
Figure 2). This could result from the two-step procedure in the phenol method, in which the first phenol extraction excluded interfering components such as water-soluble sugars and acids in food samples, while the following ammonium acetate precipitation increased the purity of the proteins. On the other hand, the ammonium sulfate and sodium chloride methods were direct precipitations that could obtain more hydrophilic compounds resulting in poor protein quality. In addition, the phenol method could further inhibit protein degradation and retain the protein repertoire as well [
30]. Therefore, the phenol method was suggested to extract proteins from the complex matrix and food samples with low protein content.
A signature peptide, the critical element for protein identification and quantification, should fulfill several criteria to guarantee its stability and specificity. It was reported that the peptide containing arginine and asparagine was susceptible to deamidation, which could lead to errors in MS-based quantitative methods [
31]. However, in this study, the quantification of SPA5-H, which contains three asparagines was not influenced by deamidation, where high recovery and low coefficient of variaon were observed in the validation assessment for SPA5-H (
Table 2). This is probably because SPA5-D, as the chemically analogous internal standard, would show almost the same degree of deamidation as the SPA5-H and thus the variation resulting from deamidation was adjusted. In other studies using an isotope internal standard strategy, good robustness could be achieved as well when analyzing signature peptides containing arginine and asparagine [
32,
33], which supported our contention that the isotope internal standard could correct the errors caused by deamidation and indicated that a peptide being prone to deamidation could be selected as a signature peptide for protein quantification with the employment of an isotope internal standard.
For accurate and precise quantification, a chemically analogous isotope internal standard was essential to compensate for recovery loss and systematic bias within the analysis procedure. In this study, the artificial synthetic signature peptides were derivatized with CD
2O as the isotope internal standard. The peptides methylated with CH
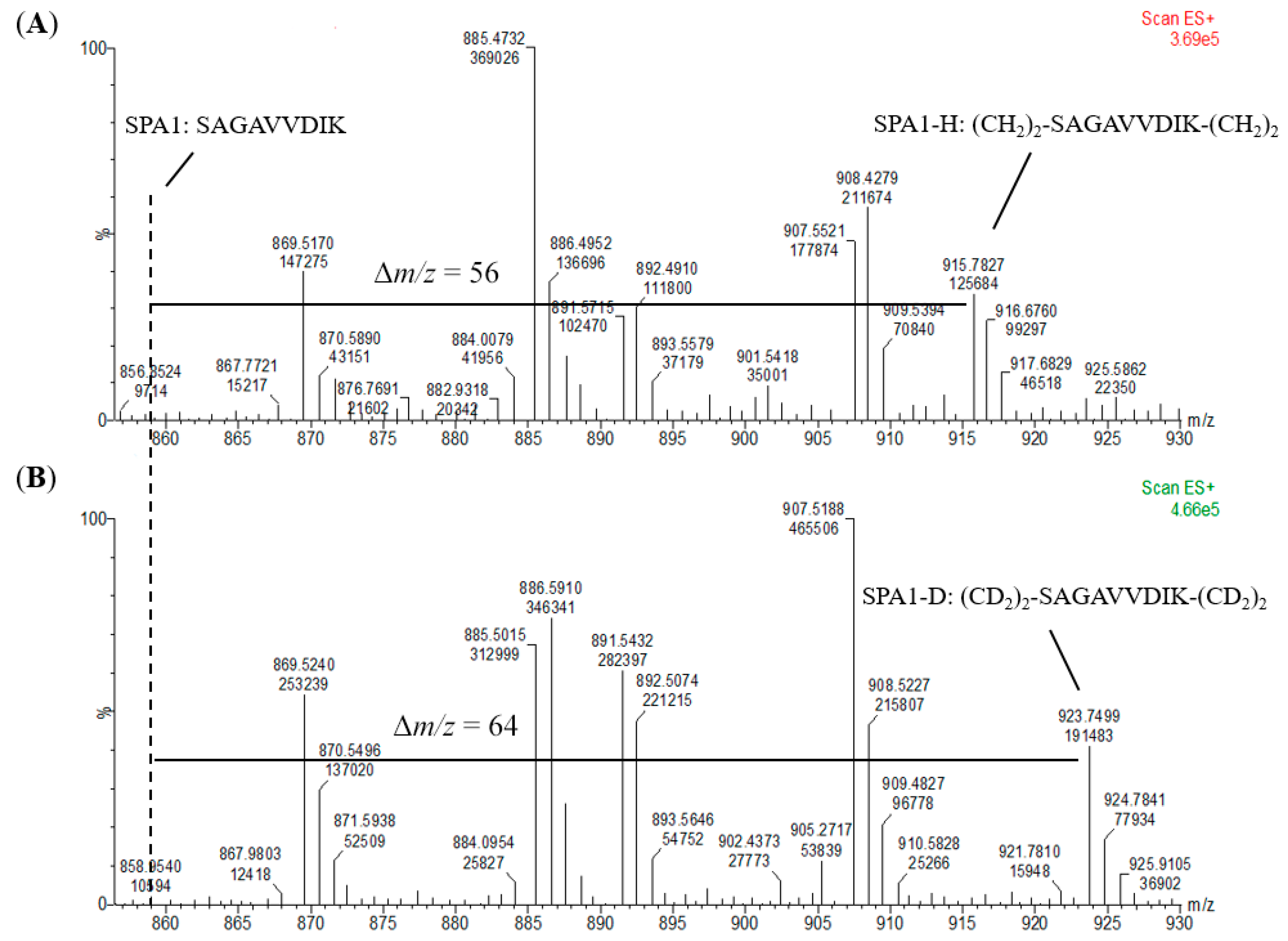
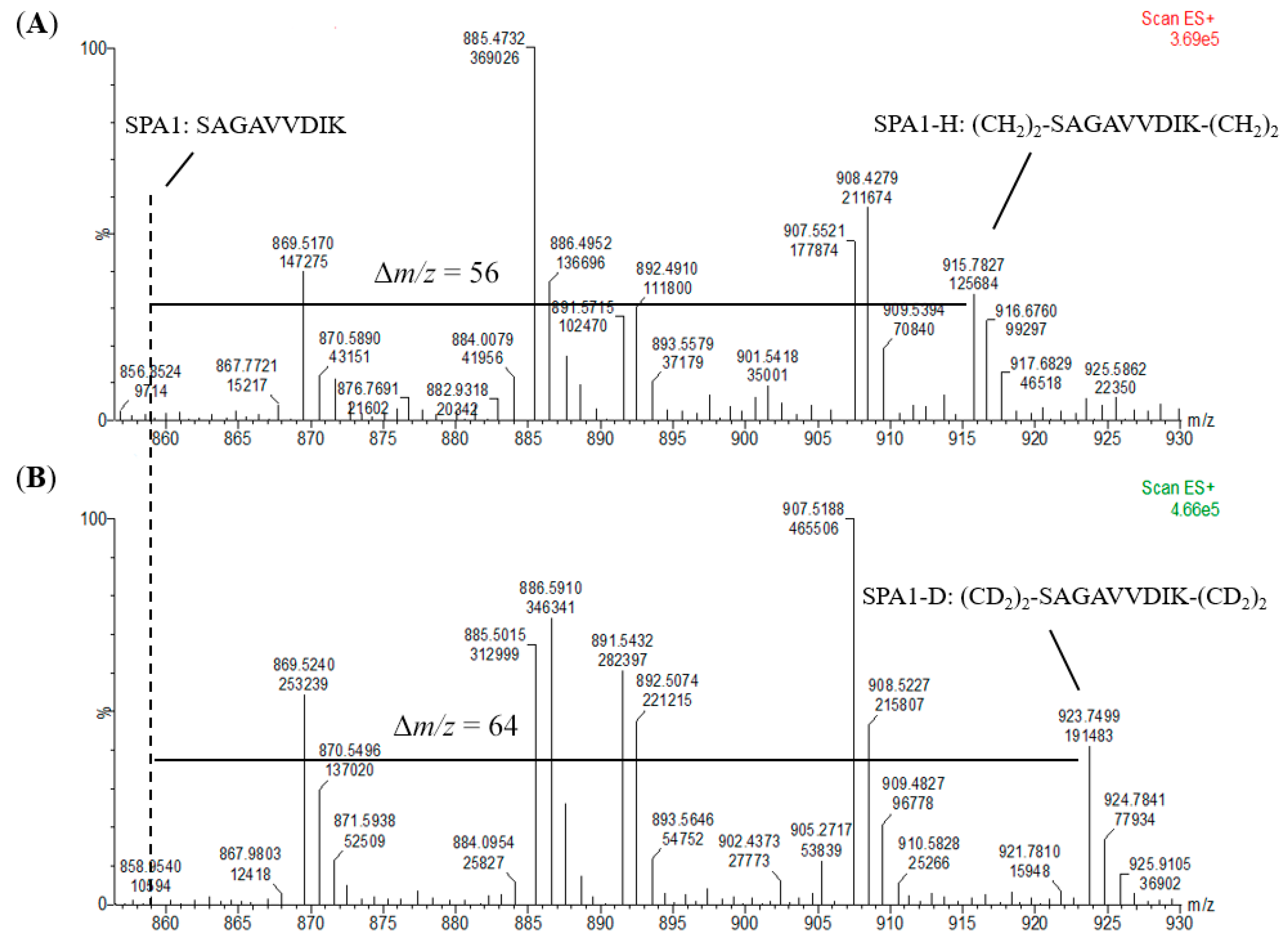
2O served as the standards for calibration curves. It was observed that the dimethylation of the signature peptide standards had been fully completed without any unlabeled peptide detected (
Figure 4 and
Figure S1), and highly linear calibration curves were established. In addition, all the signature peptides in the kiwi food samples were labeled with CH
2O as well to minimize possible variation and errors resulting from the incomplete reaction and to correct and maintain the recovery in sample preparation and the derivatization process. Fortunately, there were no unreacted allergen peptides detected within food samples after dimethylation, which indicated that the reaction of food samples was highly effective and efficient. Moreover, the isobaric tag for relative and absolute quantification (iTRAQ) which labels isotopic
N-methylpiperazine onto peptides is commercially available, and it has been widely utilized for various types of sample matrices such as blood, plant tissues, and food samples [
34,
35,
36]. Unlike the quantification by dimethylation which labeled the precursor ions of H- and D-labeled peptides and their sequence fragment ions, the iTRAQ method was carried out by means of detecting the
N-methylpiperazine tag as the reporter ion of the peptides. Hence, the efficiency of the labeling reaction would highly influence the number of reporter ions and the recovery of target proteins. The results in which no unlabeled peptides were detected in the MS spectra (
Figure 4 and
Figure S1) revealed that formaldehyde methylation was an effective and efficient derivatization and thus suggested that dimethyl labeling could be an alternative method to iTRAQ.
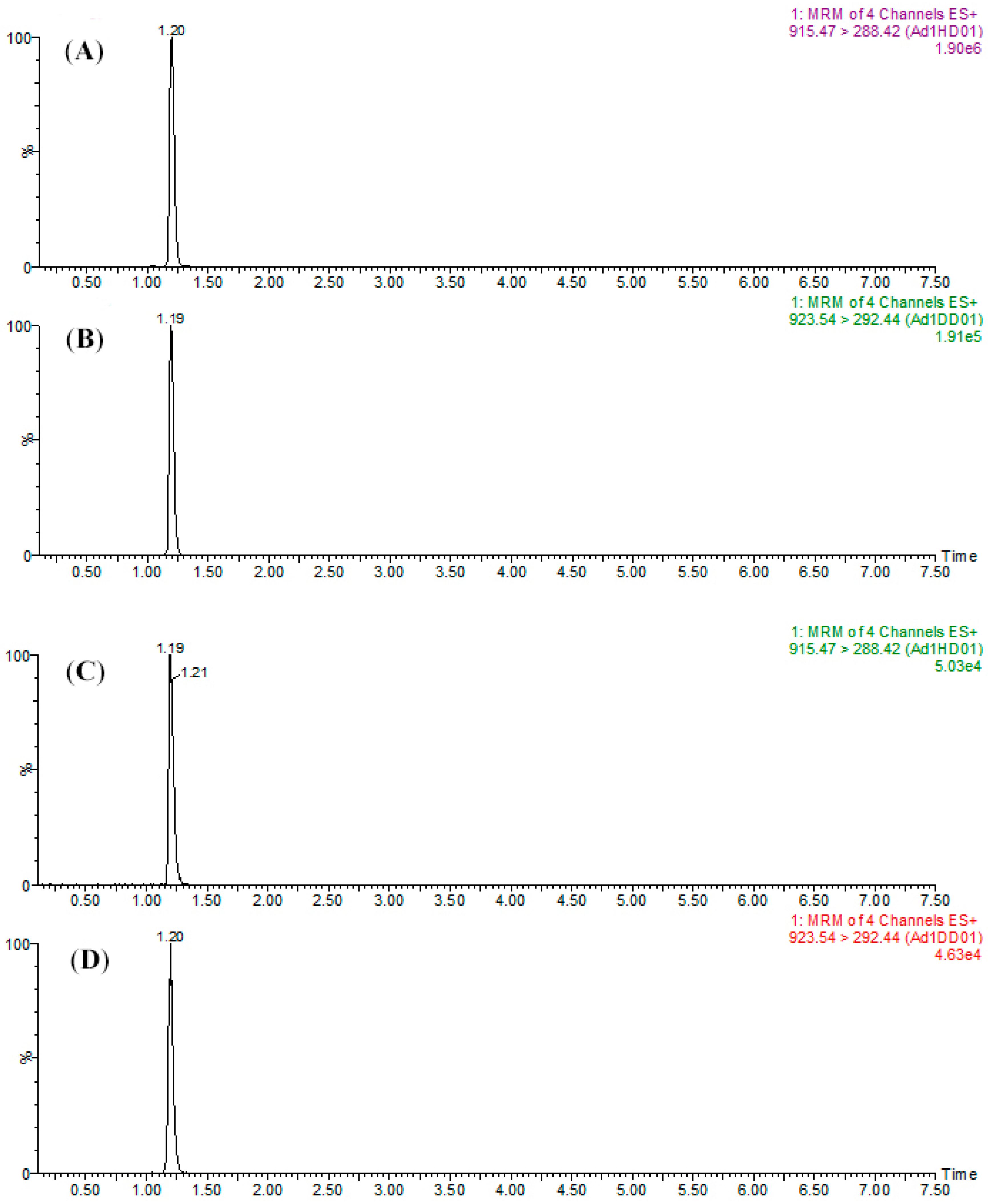
To assure the reliability of this study, certain parameters such as precision and accuracy needed be evaluated. In the validation experiments, the H-labeled peptide standards and the D-labeled internal standards were spiked before the trypsin digestion to examine the recovery and reproducibility for sample preparation. After the experiments, good robustness could be observed when analyzing SPA1-H and SPA5-H (
Table 2). These results showed that, with respective labeling of CH
2O and CD
2O on the analytes and the internal standards, the systematic errors such as recovery loss within the desalting process and inconsistent ionization efficiency for analytes could be adjusted, and accurate and precise quantification could be achieved. Furthermore, compared with other commonly used labeling approaches such as metabolic labeling and the iTRAQ method, dimethyl labeling was more convenient and accessible due to its low cost and ease of operation [
9]. Taken together, the dimethyl labeling method could be a recommended strategy for routine analysis.
The analytical procedure was validated for SPA1-H and SPA5-H, however, a lower S/N ratio and a high coefficient of variation were observed when it came to SPA11-H. This result could be attributed to the poor performance of SPA11-H on charge competition during the ionization within ESI and led to poor ionization. As reported previously, the nonpolar residues contributed to the fraction of peptides from ESI droplets, and hydrophobic residues could reduce the ability of peptides in charge competition [
37]. Thus, SPA11-H containing only two nonpolar amino acids could have been less successful in competing for charges with the matrix and led to unstable ionization efficiency. To prevent this disadvantage, an amino acid with hydrophilicity was suggested to be taken into consideration in the selection of the signature peptide for quantitative protein analysis using dimethyl labeling coupled with LC/ESI-MS/MS.
The workflow in this study was finally applied to kiwi foods, which were low in protein content. This extended the applicability of stable-isotope dimethyl labeling to the MS-based quantification of kiwi allergens at trace level and revealed the potential of this labeling method in protein analysis of food samples. Unlike blood or urine samples, in which the matrix is normally composed of water, proteins, and electrolytes, the matrix of food samples varies based on the food materials. In fruits and vegetables, there would be more cellulose and low proteins, but carbohydrates, lipids, and proteins would be the major components when it comes to soybean seeds. For meats and seafood, the samples could have high protein and lipid content. Therefore, the sample extraction and preparation steps would be critical for different food samples to remove compounds that could lead to interference in the following sample preparation and instrumental analysis. With an appropriate extraction procedure such as the phenol method in this study that removed the contaminants, a complete derivatization reaction could be achieved which resulted in accurate analysis with the stable-isotope dimethyl labeling method.
In conclusion, we first introduced the stable-isotope dimethyl labeling method to develop a reliable and economical LC-MS/MS quantitative procedure for kiwi allergens. The high efficiency of the dimethyl labeling was proved, and the developed procedure was validated and used on kiwi food samples for trace allergen analysis. This dimethyl labeling strategy could be further utilized and applied to the quantification of allergens in different food matrices and could have potential for the analysis of bioactive peptides and of peptide markers for food authenticity.
4. Materials and Methods
4.1. Materials and Reagents
Dithiothreitol (DTT), iodoacetamide (IAA), ethanol, ammonia, formic acid, formaldehyde (37% solution in H2O), formaldehyde-D2 (20% solution in H2O), phenol, sodium deoxycholate (SDC), trichloroacetic acid (TCA), ethylenediaminetetraacetic acid (EDTA), β-mercaptoethanol, and triethylammonium bicarbonate buffer (TEAB) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Acrylamide, ammonium persulfate, sodium dodecyl sulfate (SDS), N,N′-methylene-bis-acrylamide, glycine, and N,N,N′,N′-tetraethylmethylenediamine were obtained from Bio-Rad (Hercules, CA, USA). MS grade trypsin was provided by Fisher Scientific (Houston, TX, USA). Acetonitrile (MS grade), acetone, Coomassie brilliant blue, methanol, Tris-Base, and ammonium acetate were from J.T.Baker (Phillipsburg, NJ, USA). Sodium cyanoborohydride was purchased from Alfa Aesar (purity 95%, Haverhill, MA, USA). Urea was from VWR International (Radnor, PA, USA). Ammonium sulfate was obtained from Taiwan Fertilizer Co. (Hsinchu, Taiwan). The peptide standards, IVALSTGWYNGGSR (SPA1), NNIVDGSNAVWSALGLDK (SPA5), and GEHNSVTWTFHYEK (SPA11) were synthesized at the National Institute of Infectious Diseases and Vaccinology (Taipei, Taiwan). Kiwifruits were purchased from a local market in Taipei city.
4.2. Phenol Extraction
The phenol extraction method was modified according to a previously described method [
29]. Seventy μL of the extraction buffer (1.8 M sucrose, 240 mM Tris-HCl, pH 8.0, 24 mM EDTA, and 0.96% (
v/
v) β-mercaptoethanol) were added to 0.07 g peeled and homogenized kiwifruit, and 100 μL of saturated sucrose in phenol was added subsequently. After mixing for a few seconds, the mixtures were placed in an ultrasonicator bath for 20 min at 40 °C and centrifuged at 3900×
g for 15 min at 25 °C. Afterwards, the supernatants (the phenol phase) were transferred to a new tube, and the lower aqueous phase was extracted twice with the same steps as described above. Proteins were precipitated from the collected phenol phase with five volumes of 0.1 M ammonium acetate in methanol at −20 °C for 4 h and then pelleted by centrifugation at 21,100×
g for 10 min at −9 °C. The protein pellets were further washed with 0.1 M ammonium acetate in methanol, 80% ice-cold (
v/
v) acetone, and cold 70% (
v/
v) ethanol. After drying with a vacuum dryer, the protein pellets were finally stored at −20 °C.
4.3. TCA Extraction
Kiwifruit was first peeled and then homogenized by the blender. Four grams of the treated kiwi puree was added to 25 mL TCA buffer (10% (w/v) TCA and 2% (v/v) β-mercaptoethanol in acetone). Extracts were mixed for a few seconds and precipitated for 1 h at −20 °C. The precipitated proteins were pelleted by centrifugation at 5000× g for 30 min at 4 °C and washed twice with 10 mL ice-cold acetone. The pellets were dried with a vacuum dryer and resuspended in 15 mL tank buffer (25 mM Tris-base, 0.2 M glycine, and 0.1% (w/v) SDS). The extracts were centrifuged at 5000× g for 10 min at 4 °C, and the resulting supernatants were transferred to a new tube and stored at −20 °C.
4.4. Ammonium Sulfate Precipitation
Peeled kiwifruit (250 g) was blended with 250 mL of extraction buffer (100 mM Tris-HCl, 2 mM NaCl, and 10 mM EDTA). After centrifugation (4500 rpm, 15 min, 25 °C), the supernatants were mixed with ammonium sulfate to 95% saturation and precipitated overnight at 4 °C. The precipitated protein was collected by centrifugation (9300× g, 15 min, 4 °C). Afterward, the protein pellets were suspended in dialysis buffer (200 mM Tris-HCl, 20 mM EDTA, 2 mM NaCl, 0.8% (v/v) β-mercaptoethanol) and dialyzed against water for at least 24 h. The treated solution was finally stored at −80 °C.
4.5. Sodium Chloride Extraction
This method was carried out as described previously [
38]. One mL extraction solution (0.5 M NaCl, pH 8.3, 10 mM DTT) was added to 0.7 g of the peeled and homogenized kiwifruit. Extracts were placed on ice for 1 h and centrifuged at 16,000×
g for 10 min at 4 °C. The supernatants were transferred to a new tube and stored at −20 °C.
4.6. SDS-PAGE Analysis
Extracted proteins were separated via polyacrylamide gel electrophoresis (PAGE) using 12% SDS-PAGE gels at a voltage of 60–130 V. After the separation, the proteins were visualized with Coomassie blue G-250, and the gels were finally digitized with a scanner.
4.7. Tryptic Digestion
For samples subjected to LC/ESI-Q-TOF analysis, the protein pellets collected from the phenol extraction were resuspended with 0.5 mL of resuspending buffer (9 M urea in 1 M TEAB buffer). The protein solution was then diluted to a concentration range of 1.5–2.4 μg μL−1. Reduction of the disulfide bonds was performed with 0.6 μL of 100 mM DTT, and the alkylation of the cysteine residues was carried out in the dark for 30 min with 0.9 μL of 500 mM IAA. Afterward, 4 μL of 100 mM DTT was added, and the treated solution was incubated for 10 min to quench the alkylation reaction. Prior to tryptic digestion, the solution was first diluted with 182 μL of water. Finally, 2.5 μL of trypsin (0.1 mg mL−1) was added to the samples for 16-h digestion at 37 °C.
The digestion protocol for samples for LC-MRM analysis was as follows. The protein pellets from the phenol method were added to 100 μL of resuspension buffer (5% SDC in 50 mM TEAB buffer) and 1 μL of 500 mM DTT in 50 mM TEAB. The protein pellets were then resuspended by pipetting. For the protein solution, disulfide bonds were reacted with DTT for 30 min at room temperature, and the cysteine residues were alkylated with 8 μL of IAA (50 mM in TEAB buffer) for 30 min in the dark at room temperature. The alkylation reaction was quenched with 6 μL of DTT (500 mM in TEAB buffer) for 10 min at room temperature. The treated protein solution was then diluted with 885 μL of TEAB buffer. Afterwards, 35 μL of the diluted protein solution was transferred to a new tube and combined with 15 μL of internal standard solution, formed by mixing the D-labeled peptide standards, SPA1-D, SPA5-D, and SPA11-D, with a concentration of 25 μg mL−1 of each in the ratio 1:1:1 (v/v/v). The mixed solution was finally added with 2.5 μL of trypsin (0.1 mg mL−1) and incubated for 7 h at 37 °C.
4.8. Stable Isotope Dimethyl Labeling
This procedure was modified from the study previously proposed [
8]. For the preparation of standards, 5 μL of 4% (
v/
v) formaldehyde solution (light, CH
2O; heavy, CD
2O) was added to 25 μL of the peptide standards (1 mg mL
−1) diluted with 100 μL of TEAB buffer. Immediately after the addition of formaldehyde, 5 μL of freshly prepared 0.6 M NaBH
3CN was added. After incubation for 1 h at 20 °C, the derivatization reaction was quenched by adding 20 μL of 1% (
v/
v) ammonia. Then, 10 μL of 5% (
v/
v) formic acid was added to acidify the solution. It should be noted that the acidification needs to be performed on ice. After that, the solution of peptide standards was finally diluted to a concentration of 25 μg mL
−1 with 835 μL of water.
For the preparation of samples for LC-MS-MRM analysis, the dimethylation was carried out after the tryptic digestion. Samples undergoing tryptic digestion were added with 1.5 μL of 4% (v/v) CH2O, and 1.5 μL of freshly prepared 0.85 M sodium cyanoborohydride was then added straight after the addition of formaldehyde. The reaction was quenched with the addition of 6 μL 1% (v/v) ammonia after 1 h of incubation at 20 °C and was then acidified with 3.5 μL 5% (v/v) formic acid. The treated samples were finally centrifuged at 15,700× g for 2 min to pellet SDC, and 52 μL of the supernatants were transferred to a new tube.
4.9. Zip-Tip Desalting
Prior to LC-MS/MS analysis, samples were desalted using the Zip-Tip C18 pipette tips. The tips were pre-wetted with acetonitrile and then equilibrated with 0.1% (v/v) formic acid before use. The peptides were bound to the tips by approximately 30 times of up-down pipette draws. The tips were then washed with 0.1% (v/v) formic acid for desalting. After that, the peptides were eluted with 20 μL of 60:40 acetonitrile/0.1% formic acid (v/v) by aspirating and dispensing the elute solution for about 20 times. The eluted solution was finally diluted with 180 μL of water and stored at −20 °C for further analysis.
4.10. LC/ESI-Q-TOF Analysis
Samples treated with digestion and desalting were subjected to LC/ESI-Q-TOF analysis. The peptide mixtures were separated with a Waters Acquity solvent delivery system. Mobile phase A was 0.1% (v/v) formic acid in water whereas mobile phase B was 0.1% (v/v) formic acid in acetonitrile. The separation was carried out on an ACQUITY UPLC® Peptide BEH C18 nanoACQUITY column (1.7 μm, 75 μm × 250 mm) at a stationary flow rate at 0.25 mL min−1 with 120 min total run time, and the injection volume was 5 μL. The mobile phase gradient started from 2% to 10% mobile phase B over 0.1 min; mobile phase B was then linearly ramped 40% over 90 min; from 90.1 min to 95 min the amount of mobile phase B increased linearly from 40% to 85% and this proportion was maintained for another 5 min. Following this, mobile phase B was changed to 2% in 5 min, and the column was re-equilibrated under the initial condition for 15 min. The eluted peptides were ionized with positive-ion electrospray ionization and analyzed using a Waters nanoACQUITY-SYNAPT G2 HDMS hybrid quadrupole-time of flight mass spectrometer. The capillary voltage was set at 2.75 kV, sample cone at 40 V, desolvation temperature at 300 °C, and source temperature at 100 °C. Data of full scan and MS/MS scan were obtained in the range of m/z 350–1700 and of m/z 50–2000, respectively.
4.11. LC-MS-MRM Analysis
The samples for quantification were analyzed using a Waters ACQUITY UPLC-Quattro Primer XE mass spectrometer. The
ACQUITY UPLC
® Peptide BEH C18 nanoACQUITY column (1.7 μm, 2.1 mm × 50 mm) was run at a flow rate of 0.25 mL min
−1. Mobile phase A was 5 mM ammonium acetate in water with 0.1% (
v/
v) formic acid and mobile phase B was 0.1% (
v/
v) formic acid in acetonitrile. The column was equilibrated using 15% mobile phase B and the samples were injected with 10 μL injection volume. A linear gradient was employed from 15% to 40% mobile phase B over 3 min; mobile phase B was then ramped to 90% from 3 min to 3.25 min and was maintained for 1 min with this proportion. Afterward, mobile phase B was changed to 15% in 0.5 min and was re-equilibrated for 5 min. The mass measurement system was operated in positive mode. Solutions were sprayed through a capillary held at 2.8 kV. The source temperature was set at 120 °C, desolvation temperature at 450 °C. The quadrupoles were scanned in MRM mode with a dwell time of 0.05 s. The cone voltage and collision energy for each transition were derived from repetitive tuning to obtain the highest signal intensity and are summarized in
Table 2. Chromatograms and mass spectra were recorded and processed using MassLynx (Waters, v. 4.1, Milford, MA, USA).
4.12. Validation Experiments
The analytical method was validated for the three allergens in accordance with the International Conference on Harmonization of Technical Requirements of Registration of Pharmaceuticals for Human Use guidelines Q2 (R1).
The calibration curves were established with diluted peptide standards over a concentration range of 0.008–4000 ng mL−1 and internal standards at a concentration of 500 ng mL−1. The LOD and LOQ were determined on the basis of sign-to-noise ratio (S/N), where the LOD >3 and the LOQ >10. For the assessment of the accuracy and precision, five different concentration levels of the H-labeled peptide standards were spiked before tryptic digestion was analyzed. The spiked amounts of the quality control standards were 125–12500 ng for SPA1-H, 250–25000 ng for SPA5-H, and 12.5–1250 ng for SPA11-H.
4.13. Statistical Analysis
All the error bars of the bar charts were ±SD of at least three independent experiments by performing duplicates. The difference between experimental groups was determined with one-way analysis of variance. A p-value under 0.05 was considered a statistically significant difference.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}