
Structure, Shift in Redox Potential and Li-Ion Diffusion Behavior in Tavorite LiFe1−xVxPO4F Solid-Solution Cathodes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Phase Structure
2.2. Powder Microstructure
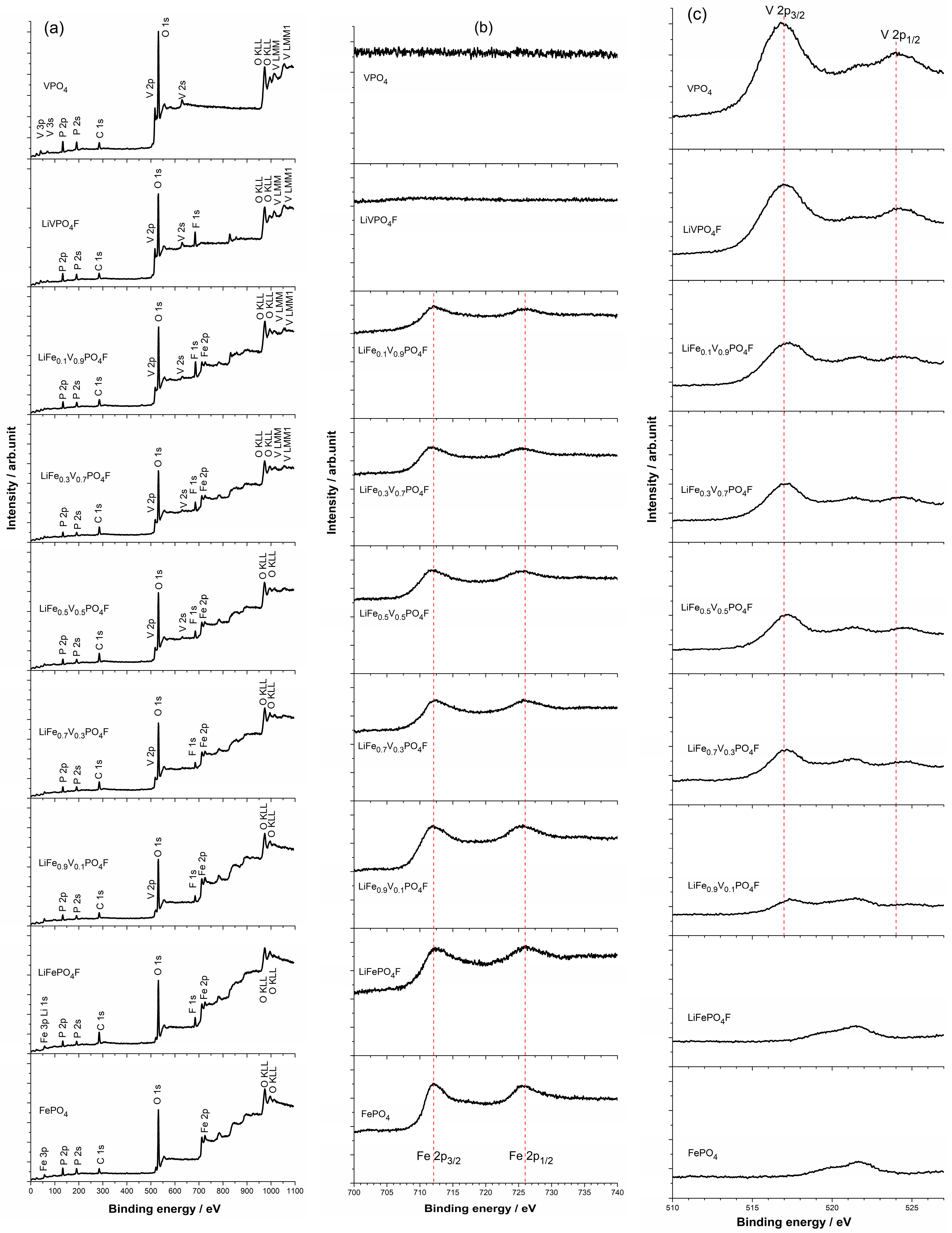
2.3. Valence States of Fe/V Components
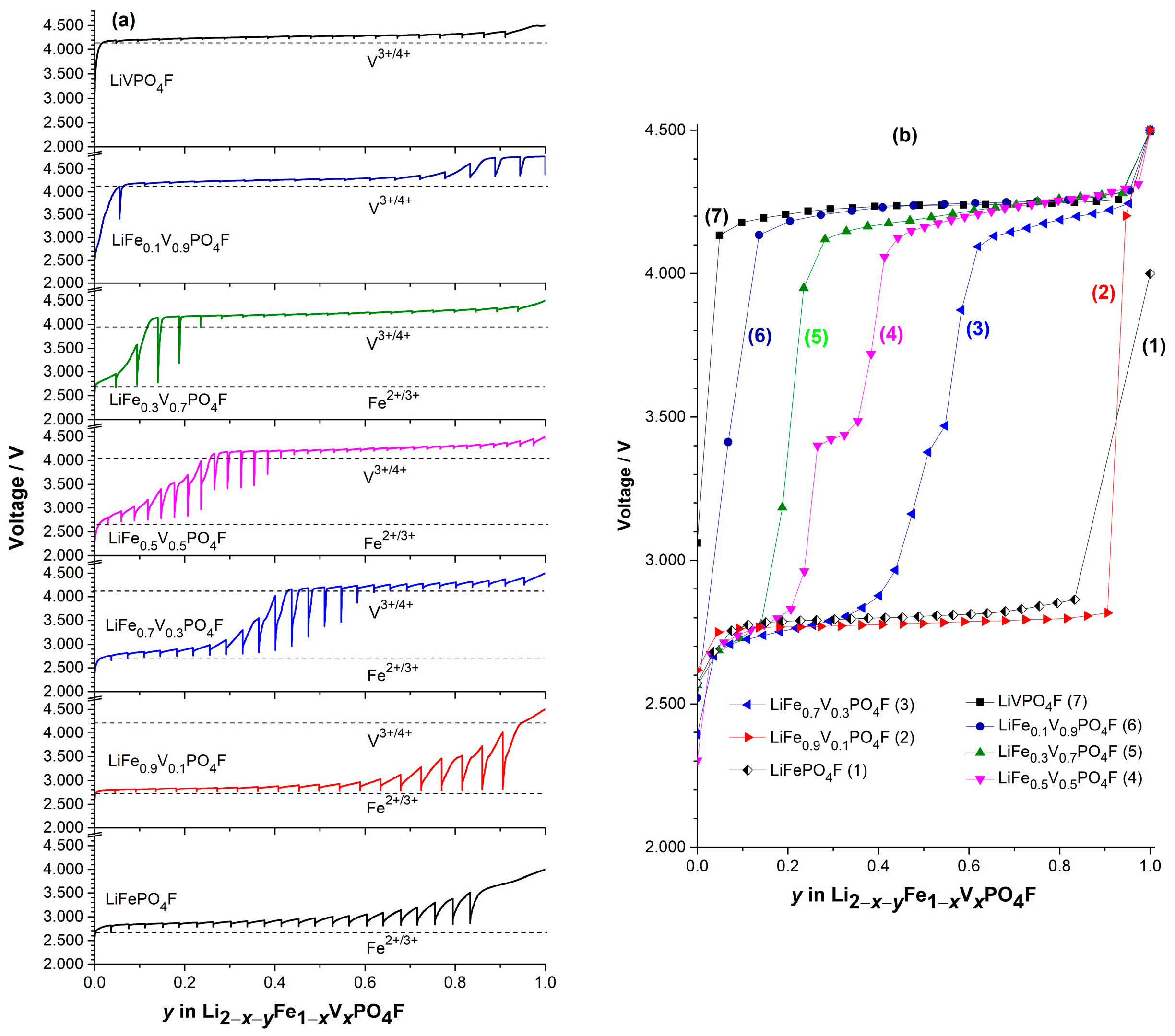
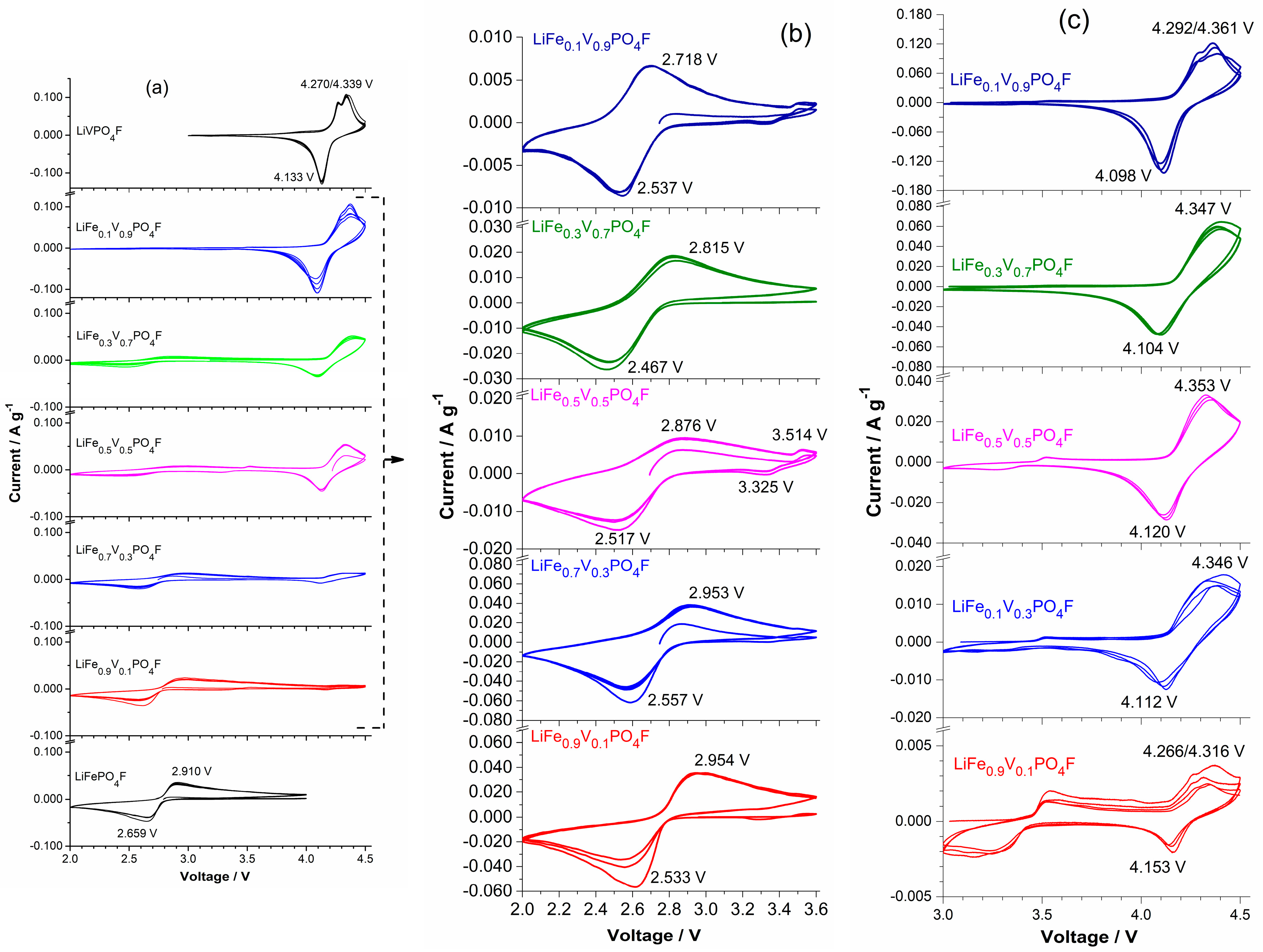
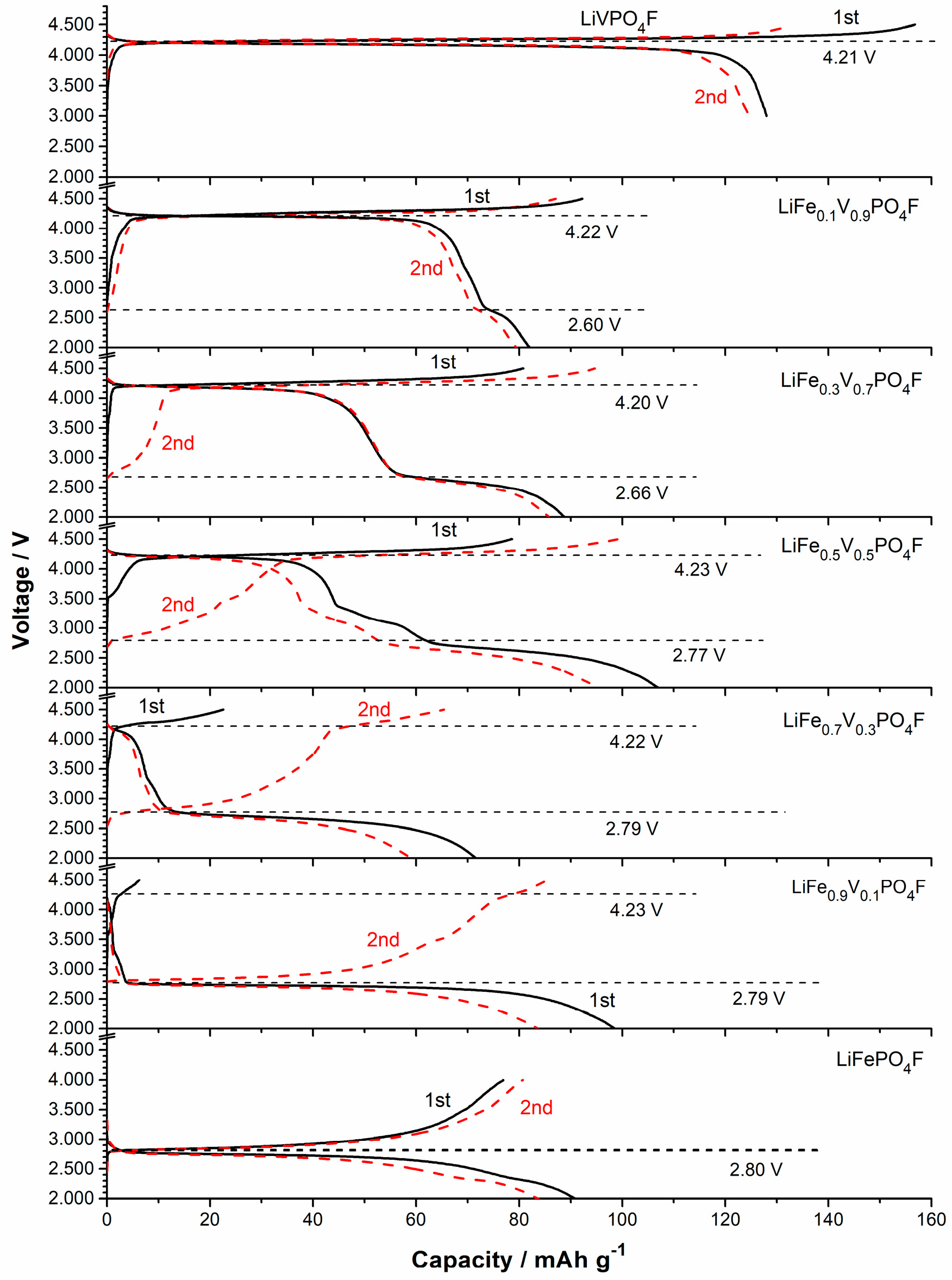
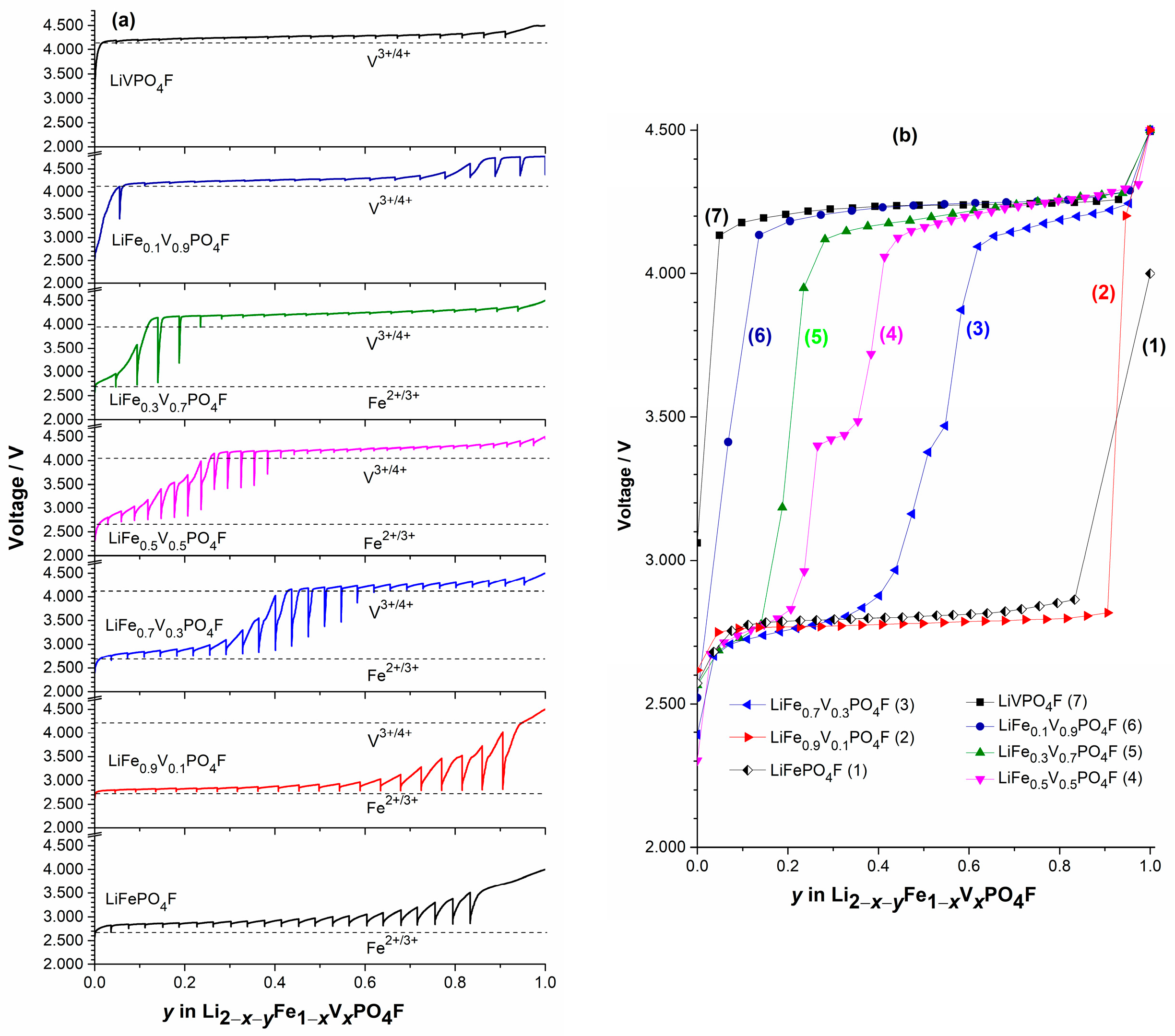
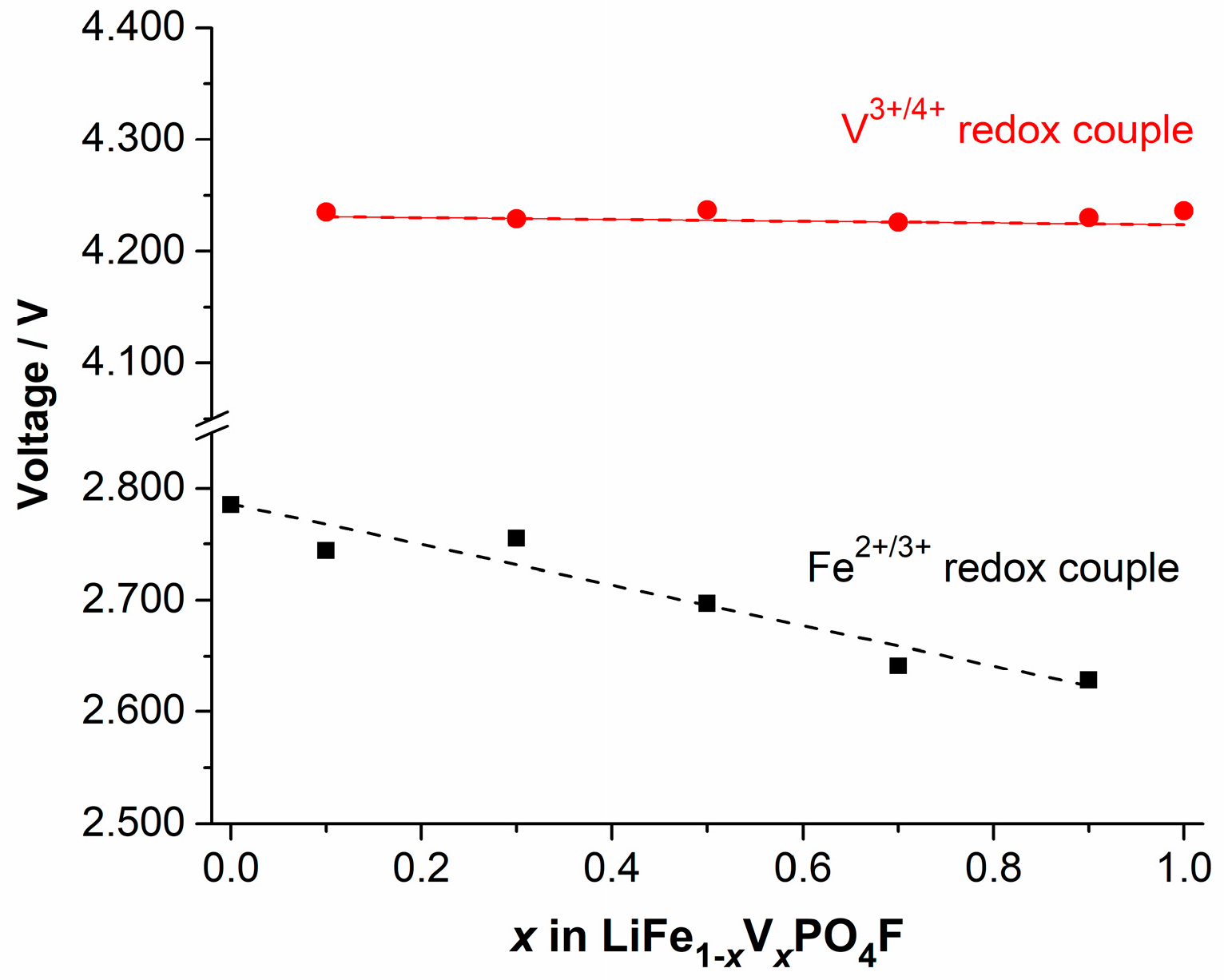
2.4. Shift in Redox Potential
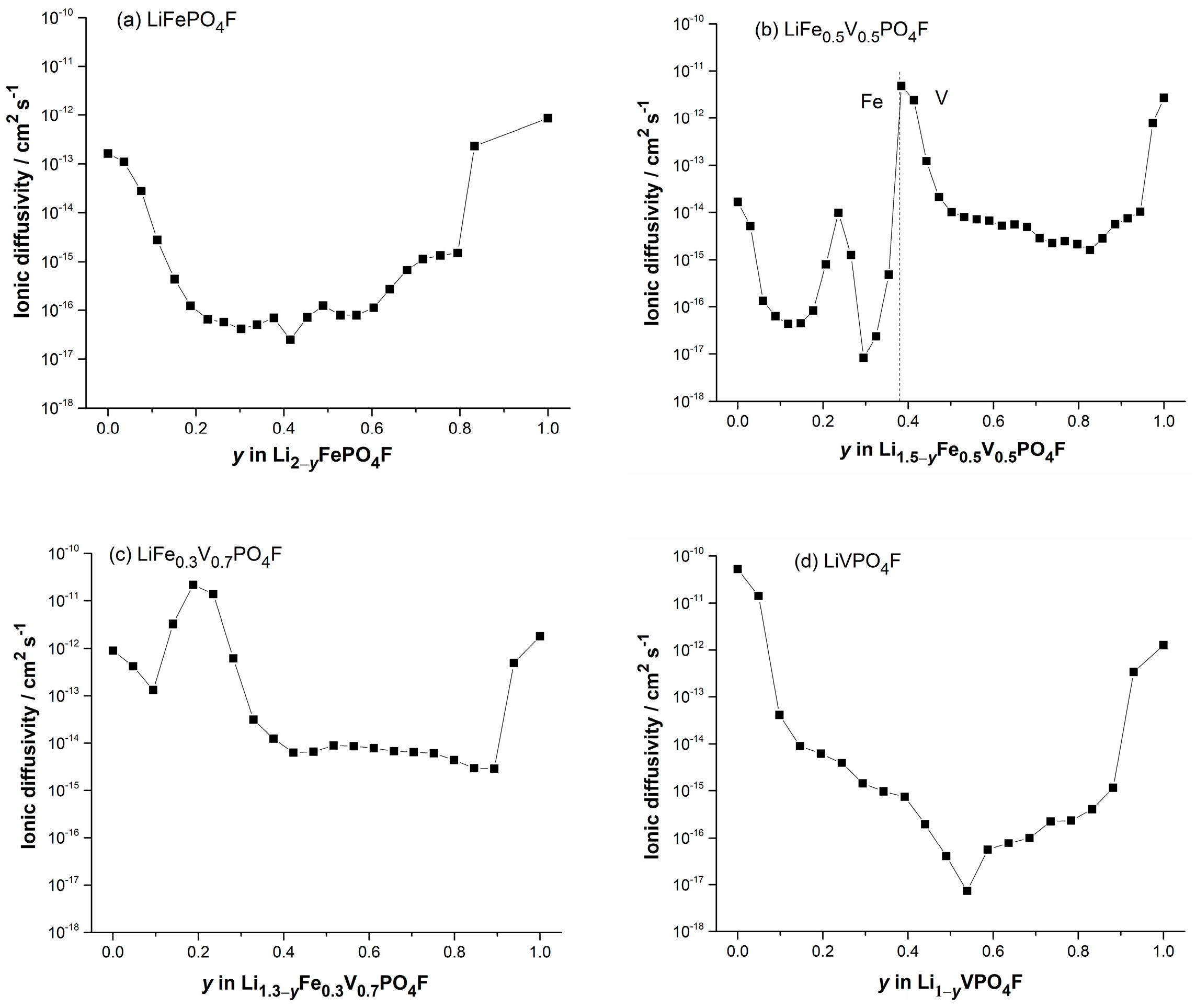
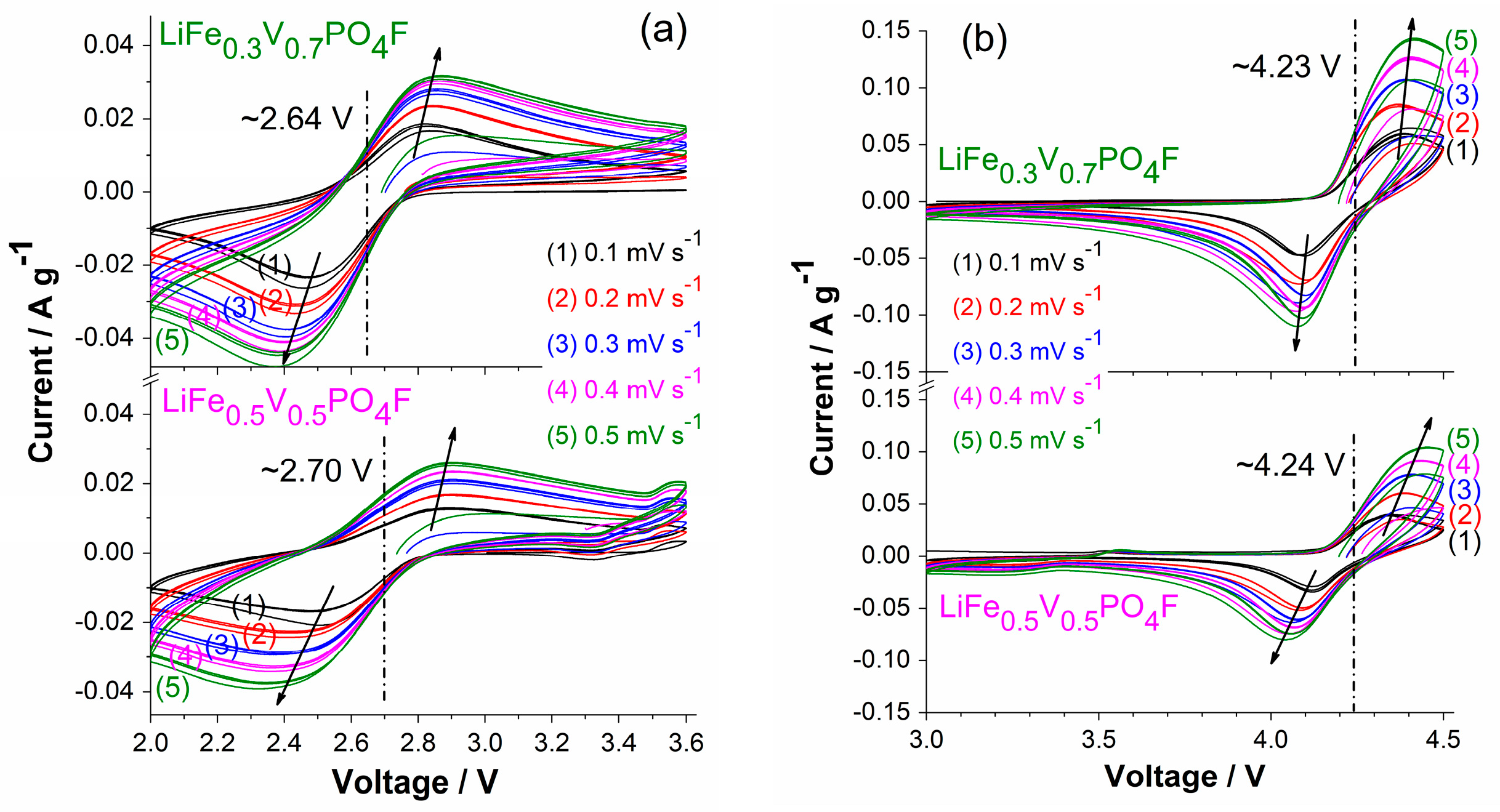
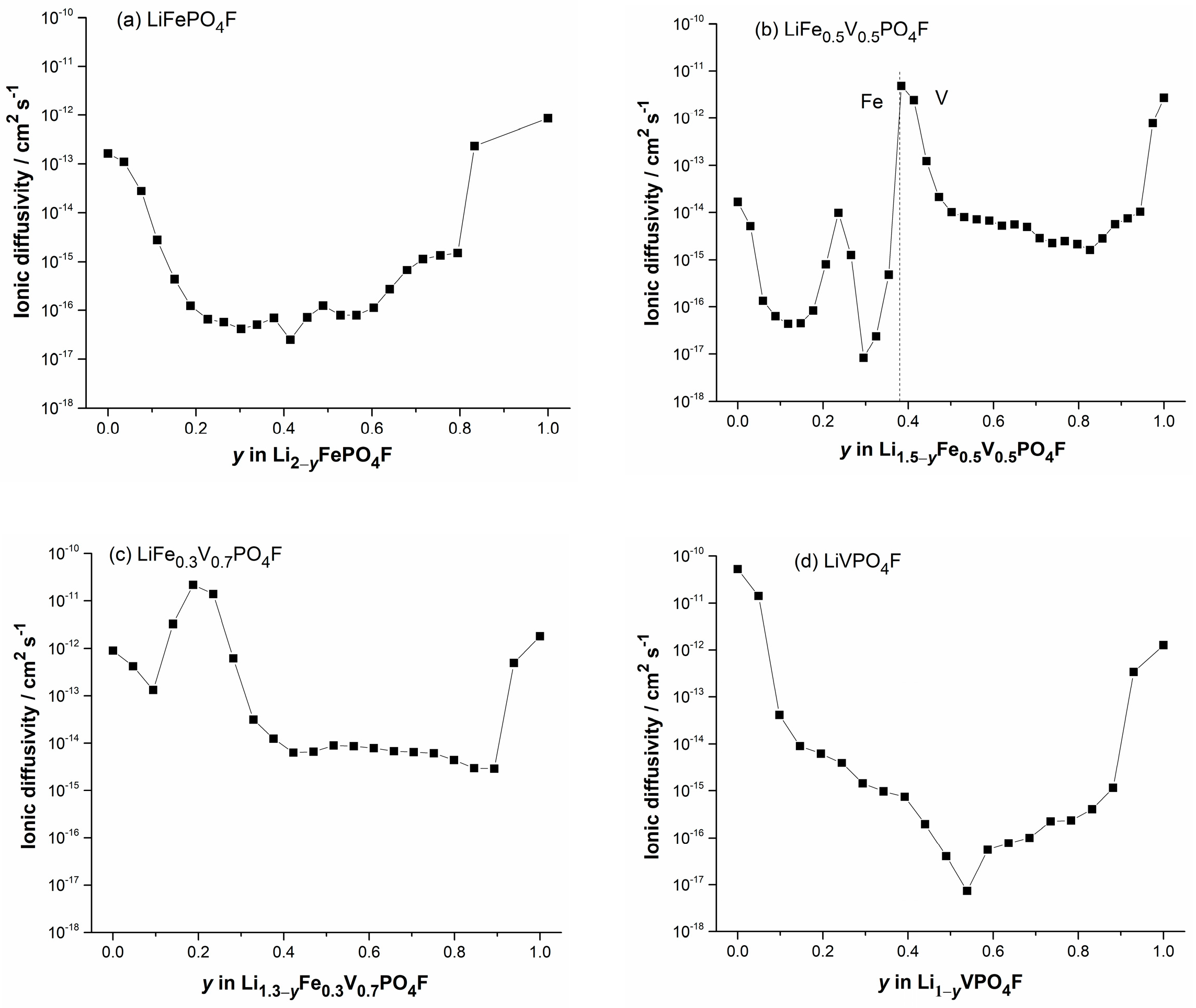
2.5. Li-Ion Diffusion Behavior
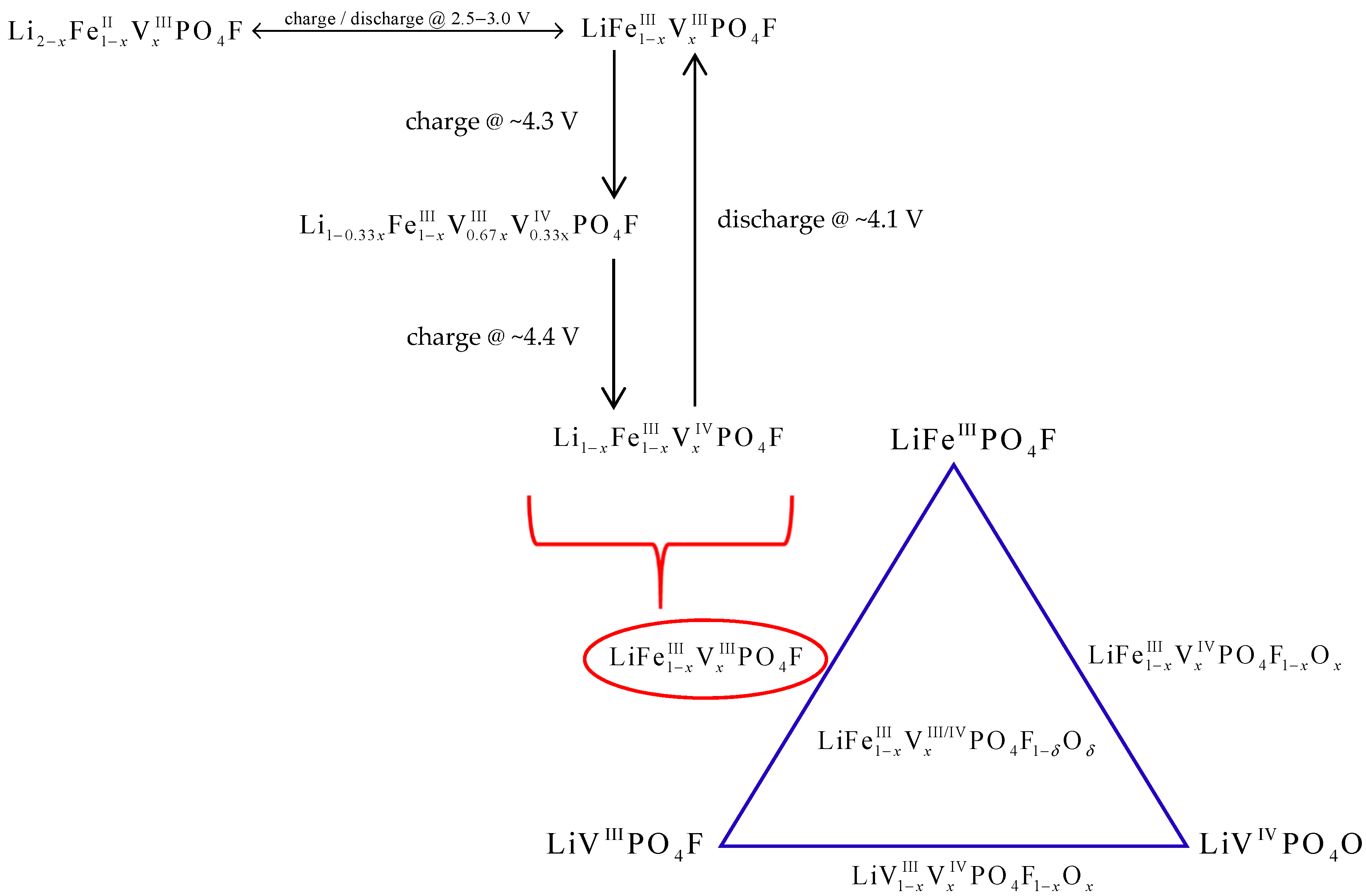
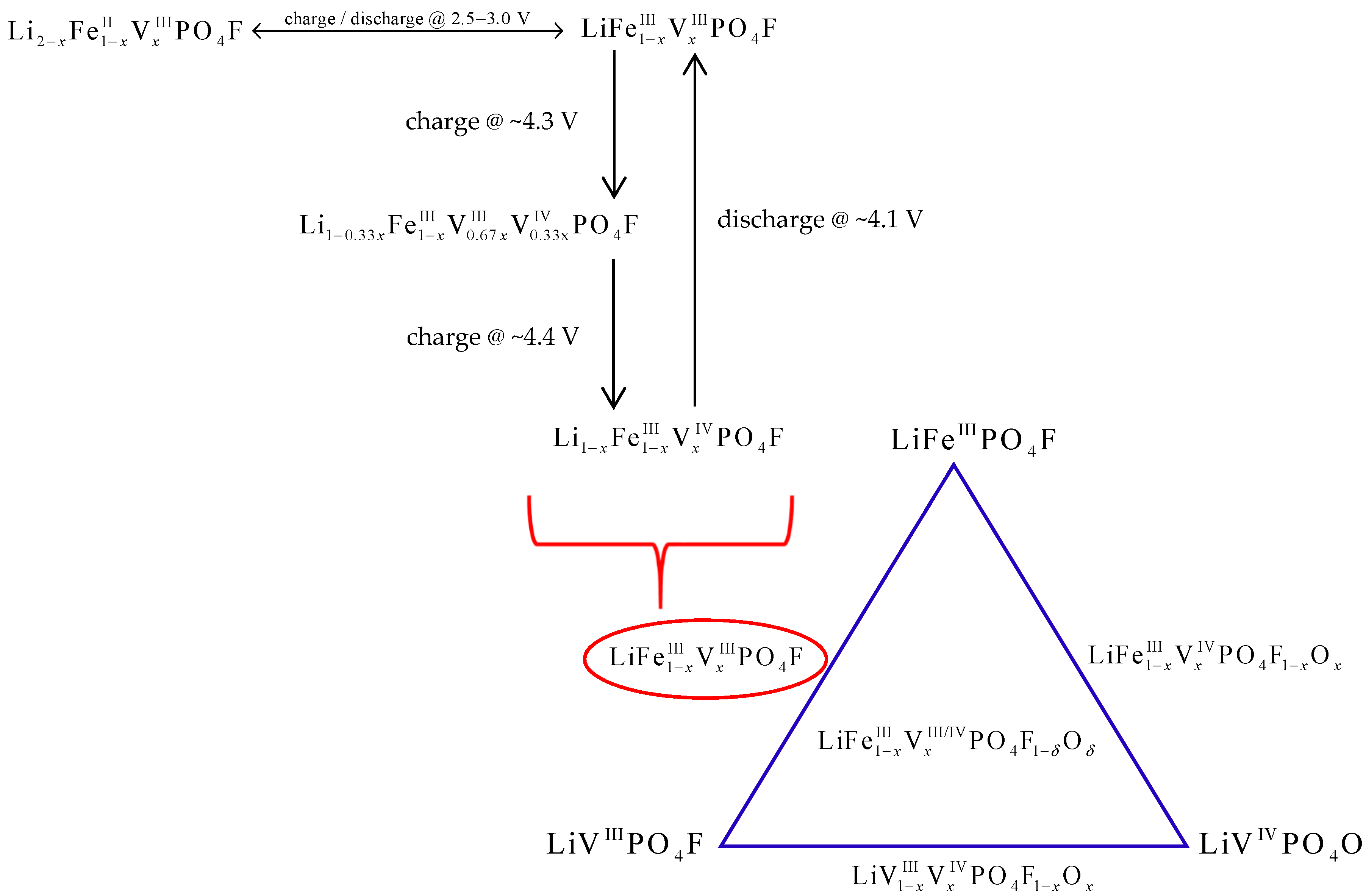
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
References
- Prabu, M.; Reddy, M.V.; Selvasekarapandian, S.; Subba-Rao, G.V.; Chowdari, B.V.R. Synthesis, impedance and electrochemical studies of lithium iron fluorophosphate, LiFePO4F cathode. Electrochim. Acta 2012, 85, 572–578. [Google Scholar] [CrossRef]
- Molenda, J.; Ojczyk, W.; Świerczek, K.; Zajac, W.; Krok, F.; Dygas, J.; Liu, R.S. Diffusional mechanism of deintercalation in LiFe1−yMnyPO4 cathode material. Solid State Ion. 2006, 177, 2617–2624. [Google Scholar] [CrossRef]
- Ramzan, M.; Lebegue, S.; Larsson, P.; Ahuja, R. Structural, magnetic, and energetic properties of Na2FePO4F, Li2FePO4F, NaFePO4F, and LiFePO4F from ab initio calculations. J. Appl. Phys. 2009, 106, 043510. [Google Scholar] [CrossRef]
- Chen, D.; Shao, G.-Q.; Li, B.; Zhao, G.-G.; Li, J.; Liu, J.-H.; Gao, Z.-S.; Zhang, H.-F. Synthesis, crystal structure and electrochemical properties of LiFePO4F cathode material for Li-ion batteries. Electrochim. Acta 2014, 147, 663–668. [Google Scholar] [CrossRef]
- Padhi, A.; Nanjundaswamy, K.; Masquelier, C.; Okada, S.; Goodenough, J. Effect of structure on the Fe3+/Fe2+ redox couple in iron phosphates. J. Electrochem. Soc. 1997, 144, 1609–1613. [Google Scholar] [CrossRef]
- Barker, J.; Saidi, M.Y.; Swoyer, J.L. Electrochemical insertion properties of the novel lithium vanadium fluorophosphate, LiVPO4F. J. Electrochem. Soc. 2003, 150, A1394–A1398. [Google Scholar] [CrossRef]
- Mba, J.M.A.; Croguennec, L.; Basir, N.I.; Barker, J.; Masquelier, C. Lithium insertion or extraction from/into tavorite-type LiVPO4F: An in situ X-ray diffraction study. J. Electrochem. Soc. 2012, 159, A1171–A1175. [Google Scholar] [CrossRef]
- Fan, C.-L.; Wen, Z.; Xiao, R.-F.; Li, Q.-Y.; Gong, Y.; Zeng, T.-T.; Wei, S.; Zhang, X.; Han, S.-C. LiVPO4F/C cathode synthesized by a fast chemical reduction method for lithium-ion batteries. Mater. Lett. 2016, 170, 35–38. [Google Scholar] [CrossRef]
- Yang, B.; Yang, L. Silver-coated LiVPO4F composite with improved electrochemical performance as cathode material for lithium-ion batteries. J. Phys. Chem. Solids 2015, 87, 228–232. [Google Scholar] [CrossRef]
- Yan, H.; Wu, X.; Li, Y. Preparation and characterization of conducting polyaniline-coated LiVPO4F nanocrystals with core-shell structure and its application in lithium-ion batteries. Electrochim. Acta 2015, 182, 437–444. [Google Scholar] [CrossRef]
- Mba, J.M.A.; Masquelier, C.; Suard, E.; Croguennec, L. Synthesis and crystallographic study of homeotypic LiVPO4F and LiVPO4O. Chem. Mater. 2012, 24, 1223–1234. [Google Scholar] [CrossRef]
- Li, P.; Wang, P.; Yu, H.; Lin, X.; Shao, L.; Shui, M.; Long, N.; Shu, J. Carbothermal synthesis of LiVPO4F and its structural change in a broad potential range observed by in-situ X-ray diffraction. Ceram. Int. 2015, 41, 10766–10774. [Google Scholar] [CrossRef]
- Wang, J.-X.; Wang, Z.-X.; Li, X.-H.; Guo, H.-J.; Wu, X.-W.; Zhang, X.-P.; Xiao, W. xLi3V2(PO4)3 center dot LiVPO4F/C composite cathode materials for lithium ion batteries. Electrochim. Acta 2013, 87, 224–229. [Google Scholar] [CrossRef]
- Wang, J.-X.; Li, X.-H.; Wang, Z.-X.; Guo, H.-J.; Zhang, Y.-H.; Xiong, X.-H.; He, Z.-J. Synthesis and characterization of LiVPO4F/C using precursor obtained through a soft chemical route with mechanical activation assist. Electrochim. Acta 2013, 91, 75–81. [Google Scholar] [CrossRef]
- Wang, J.; Liu, Z.; Yan, G.; Li, H.; Peng, W.; Li, X.; Song, L.; Shih, K. Improving the electrochemical performance of lithium vanadium fluorophosphate cathode material: Focus on interfacial stability. J. Power Sources 2016, 329, 553–557. [Google Scholar] [CrossRef]
- Hu, G.; Gan, Z.; Cao, Y.; Du, K.; Du, Y.; Peng, Z. A three-dimensional LiVPO4F@C/MWCNTs/rGO composite with enhanced performance for high rate Li-ion batteries. Electrochim. Acta 2018, 292, 502–510. [Google Scholar] [CrossRef]
- Li, P.; Ma, R.; Lin, X.; Shao, L.; Wu, K.; Shui, M.; Long, N.; Shu, J. Impact of H2O exposure on the structure and electrochemical performance of LiVPO4F cathode material. J. Alloys Compd. 2015, 637, 20–29. [Google Scholar] [CrossRef]
- Ma, R.; Shu, J.; Shao, L.; Lin, X.; Wu, K.; Shui, M.; Li, P.; Long, N.; Ren, Y. Determination of lithium ion diffusion behaviors in tavorite LiVPO4F by galvanostatic intermittent titration technique. Ceram. Int. 2014, 40, 15113–15119. [Google Scholar] [CrossRef]
- Liu, Z.; Fan, Y.; Peng, W.; Wang, Z.; Guo, H.; Li, X.; Wang, J. Mechanical activation assisted soft chemical synthesis of Na-doped lithium vanadium fluorophosphates with improved lithium storage properties. Ceram. Int. 2015, 41, 4267–4271. [Google Scholar] [CrossRef]
- Liu, Z.; Peng, W.; Fan, Y.; Li, X.; Wang, Z.; Guo, H.; Wang, J. One-step facile synthesis of graphene-decorated LiVPO4F/C nanocomposite as cathode for high-performance lithium ion battery. Ceram. Int. 2015, 41, 9188–9192. [Google Scholar] [CrossRef]
- Ellis, B.L.; Ramesh, T.N.; Rowanweetaluktuk, W.N.; Ryan, D.H.; Nazar, L.F. Solvothermal synthesis of electroactive lithium iron tavorites and structure of Li2FePO4F. J. Mater. Chem. 2012, 22, 4759–4766. [Google Scholar] [CrossRef]
- Recham, N.; Chotard, J.N.; Jumas, J.C.; Laffont, L.; Armand, M.; Tarascon, J.M. Ionothermal synthesis of Li-based fluorophosphates electrodes. Chem. Mater. 2010, 22, 1142–1148. [Google Scholar] [CrossRef]
- Ramesh, T.N.; Lee, K.T.; Ellis, B.L.; Nazar, L.F. Tavorite lithium iron fluorophosphate cathode materials: Phase transition and electrochemistry of LiFePO4F-Li2FePO4F. Electrochem. Solid-State Lett. 2010, 13, A43–A47. [Google Scholar] [CrossRef]
- Lee, E.; Persson, K.A. Solid-solution Li intercalation as a function of cation order/disorder in the high-voltage LixNi0.5Mn1.5O4 Spinel. Chem. Mater. 2013, 25, 2885–2889. [Google Scholar] [CrossRef]
- Huang, Z.-D.; Orikasa, Y.; Masese, T.; Yamamoto, K.; Mori, T.; Minato, T.; Uchimoto, Y. A novel cationic-ordering fluoro-polyanionic cathode LiV0.5Fe0.5PO4F and its single phase Li+ insertion/extraction behaviour. RSC Adv. 2013, 3, 22935–22939. [Google Scholar] [CrossRef]
- Fan, S.-H.; Shao, G.-Q.; Zhu, C.; Ma, F.-F.; Mao, J.-W.; Zhang, A.-L.; Xie, G.-Z.; Yan, J.-L.; Zhang, Y. Crystal structure and electrochemical properties of LiFe1−xVxPO4F1−δOδ cathode materials for lithium-ion batteries. Electrochim. Acta 2018, 280, 248–257. [Google Scholar] [CrossRef]
- Bamine, T.; Boivin, E.; Boucher, F.; Messinger, R.J.; Salager, E.; Deschamps, M.; Masquelier, C.; Croguennec, L.; Ménétrier, M.; Carlier, D. Understanding local defects in Li-ion battery electrodes through combined DFT/NMR studies: Application to LiVPO4F. J. Phys. Chem. C 2017, 121, 3219–3227. [Google Scholar] [CrossRef]
- Kim, M.; Lee, S.; Kang, B. High energy density polyanion electrode material: LiVPO4O1−xFx (x ≈ 0.25) with tavorite structure. Chem. Mater. 2017, 29, 4690–4699. [Google Scholar] [CrossRef]
- Boivin, E.; Chotard, J.N.; Ménétrier, M.; Bourgeois, L.; Bamine, T.; Carlier, D.; Fauth, F.; Masquelier, C.; Croguennec, L. Oxidation under air of tavorite LiVPO4F: Influence of vanadyl-type defects on its electrochemical properties. J. Phys. Chem. C 2016, 120, 26187–26198. [Google Scholar] [CrossRef]
- Onoda, M.; Ishibashi, T. Phase transition and spin dynamics of the LiVPO4F insertion electrode with the S = 1 linear chain and the development of F–O mixed system. J. Phys. Soc. Jpn. 2015, 84, 044802. [Google Scholar] [CrossRef]
- Ma, R.; Shao, L.; Wu, K.; Shui, M.; Wang, D.; Long, N.; Ren, Y.; Shu, J. Effects of oxidation on structure and performance of LiVPO4F as cathode material for lithium-ion batteries. J. Power Sources 2014, 248, 874–885. [Google Scholar] [CrossRef]
- Channu, V.S.R.; Thanedar, S. LiVxFeyPO4F nanostructure cathodes for lithium ion batteries. In Proceedings of the 230th ECS Meeting, Honolulu, HI, USA, 2–7 October 2016; Abstract MA2016–02, 402. The Electrochemical Society: Pennington, NJ, USA, 2016. [Google Scholar]
- Li, B. Preparation and Electrochemical Properties of LiVPO4F Cathode Material for Li-ion Batteries. Master’s Thesis, Wuhan University of Technology, Wuhan, China, 2012. [Google Scholar]
- Ahrens, L.H. The use of ionization potentials Part 1. Ionic radii of the elements. Geochim. Cosmochim. Acta 1952, 2, 155–169. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chaleogenides. Acta Cryst. 1976, A32, 751–767. [Google Scholar] [CrossRef]
- Ellis, B.L.; Ramesh, T.N.; Davis, L.J.M.; Goward, G.R.; Nazar, L.F. Structure and electrochemistry of two-electron redox couples in lithium metal fluorophosphates based on the tavorite structure. Chem. Mater. 2011, 23, 5138–5148. [Google Scholar] [CrossRef]
- Yang, Y.; Fang, H.; Zheng, J.; Li, L.; Li, G.; Yan, G. Towards the understanding of poor electrochemical activity of triclinic LiVOPO4: Experimental characterization and theoretical investigations. Solid State Sci. 2008, 10, 1292–1298. [Google Scholar] [CrossRef]
- Lavrov, A.V.; Nikolaev, V.P.; Sadikov, G.G.; Poraikoshits, M.A. Synthesis and crystal structure of mixed lithium vanadyl orthophosphate. Doklady Akademii Nauk SSSR 1982, 266, 343–346. [Google Scholar]
- Castro, L.; Dedryvère, R.; El Khalifi, M.; Lippens, P.E.; Bréger, J.; Tessier, C.; Gonbeau, D. The spin-polarized electronic structure of LiFePO4 and FePO4 evidenced by in-lab XPS. J. Phys. Chem. C 2010, 114, 17995–18000. [Google Scholar] [CrossRef]
- Yamashita, T.; Hayes, P. Analysis of XPS spectra of Fe2+ and Fe3+ ions in oxide materials. Appl. Surf. Sci. 2008, 254, 2441–2449. [Google Scholar] [CrossRef]
- Grosvenor, A.P.; Kobe, B.A.; Biesinger, M.C.; McIntyre, N.S. Investigation of multiplet splitting of Fe 2p XPS spectra and bonding in iron compounds. Surf. Interface Anal. 2004, 36, 1564–1574. [Google Scholar] [CrossRef]
- Dedryvère, R.; Maccario, M.; Croguennec, L.; Le Cras, F.; Delmas, C.; Gonbeau, D. X-ray photoelectron spectroscopy investigations of carbon-coated LixFePO4 materials. Chem. Mater. 2008, 20, 7164–7170. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S.C. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Sc, Ti, V., Cu and Zn. Appl. Surf. Sci. 2010, 257, 887–898. [Google Scholar] [CrossRef]
- Zhang, B.; Han, Y.-D.; Zheng, J.-C.; Shen, C.; Ming, L.; Zhang, J.-F. A novel lithium vanadium fluorophosphate nanosheet with uniform carbon coating as a cathode material for lithium-ion batteries. J. Power Sources 2014, 264, 123–127. [Google Scholar] [CrossRef]
- Zheng, J.-C.; Zhang, B.; Yang, Z.-H. Novel synthesis of LiVPO4F cathode material by chemical lithiation and postannealing. J. Power Sources 2012, 202, 380–383. [Google Scholar] [CrossRef]
- Reddy, M.V.; Rao, G.V.S.; Chowdari, B.V.R. Long-term cycling studies on 4V-cathode, lithium vanadium fluorophosphate. J. Power Sources 2010, 195, 5768–5774. [Google Scholar] [CrossRef]
- Barker, J.; Gover, R.K.B.; Burns, P.; Bryan, A.; Saidi, M.Y.; Swoyer, J.L. Structural and electrochemical properties of lithium vanadium fluorophosphate, LiVPO4F. J. Power Sources 2005, 146, 516–520. [Google Scholar] [CrossRef]
- Barker, J.; Saidi, M.Y.; Swoyer, J.L. A comparative investigation of the Li insertion properties of the novel fluorophosphate phases, NaVPO4F and LiVPO4F. J. Electrochem. Soc. 2004, 151, A1670–A1677. [Google Scholar] [CrossRef]
- Yang, C.-Y.; Ji, X.; Fan, X.-L.; Gao, T.; Suo, L.-M.; Wang, F.; Sun, W.; Chen, J.; Chen, L.; Han, F.-D.; et al. Flexible aqueous Li-ion battery with high energy and power densities. Adv. Mater. 2017, 29, 1701972. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Yang, X.; Jiang, X.; Zhang, Y.; Wang, X. Synchronous tailoring surface structure and chemical composition of Li-rich-layered oxide for high-energy lithium-ion batteries. Adv. Funct. Mater. 2018, 28, 1803392. [Google Scholar] [CrossRef]
- Ellis, B.L.; Nazar, L.F. Anion-induced solid solution electrochemical behavior in iron tavorite phosphates. Chem. Mater. 2012, 24, 966–968. [Google Scholar] [CrossRef]
- Weppner, W.; Huggins, R.A. Electrochemical investigation of the chemical diffusion, partial ionic conductivities, and other kinetic parameters in Li3Sb and Li3Bi. J. Solid State Chem. 1977, 22, 297–308. [Google Scholar] [CrossRef]
- Xiao, P.-F.; Lai, M.-O.; Lu, L. Transport and electrochemical properties of high potential tavorite LiVPO4F. Solid State Ion. 2013, 242, 10–19. [Google Scholar] [CrossRef]
- Prosini, P.P.; Lisi, M.; Zane, D.; Pasquali, M. Determination of the chemical diffusion coefficient of lithium in LiFePO4. Solid State Ion. 2002, 148, 45–51. [Google Scholar] [CrossRef]
- Tang, K.; Yu, X.; Sun, J.; Li, H.; Huang, X. Kinetic analysis on LiFePO4 thin films by CV, GITT, and EIS. Electrochim. Acta 2011, 56, 4869–4875. [Google Scholar] [CrossRef]
- Muraliganth, T.; Manthiram, A. Understanding the shifts in the redox potentials of olivine LiM1−yMyPO4 (M = Fe, Mn, Co, and Mg) solid solution cathodes. J. Phys. Chem. C 2010, 114, 15530–15540. [Google Scholar] [CrossRef]
- Kobayashi, G.; Yamada, A.; Nishimura, S.I.; Kanno, R.; Kobayashi, Y.; Seki, S.; Ohno, Y.; Miyashiro, H. Shift of redox potential and kinetics in Lix(MnyFe1−y)PO4. J. Power Sources 2009, 189, 397–401. [Google Scholar] [CrossRef]
- Yamada, A.; Takei, Y.; Koizumi, H.; Sonoyama, N.; Kanno, R.; Itoh, K.; Yonemura, M.; Kamiyama, T. Electrochemical, magnetic, and structural investigation of the Lix(MnyFe1−y)PO4 olivine phases. Chem. Mater. 2006, 18, 804–813. [Google Scholar] [CrossRef]
- Melot, B.C.; Tarascon, J.M. Design and preparation of materials for advanced electrochemical storage. Acc. Chem. Res. 2013, 46, 1226–1238. [Google Scholar] [CrossRef] [PubMed]
- Girish, H.N.; Shao, G.-Q. Advances in high-capacity Li2MSiO4 (M = Mn, Fe, Co, Ni, …) cathode materials for lithium-ion batteries. RSC Adv. 2015, 5, 98666–98686. [Google Scholar] [CrossRef]
- Larson, A.C.; Von Dreele, R.B.; General Structure Analysis System (GSAS). Report LAUR 86–748; Los Alamos National Laboratory: Los Alamos, NM, USA, 2004; pp. 86–748.
Sample Availability: Samples of the compounds VPO4, LiFe1−xVxPO4F (x = 0, 0.3, 1) are available from the authors. |


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, J.-L.; Shao, G.-Q.; Fan, S.-H.; Zhu, C.; Zhang, Y.; Wang, J.; Liu, Q. Structure, Shift in Redox Potential and Li-Ion Diffusion Behavior in Tavorite LiFe1−xVxPO4F Solid-Solution Cathodes. Molecules 2019, 24, 1893. https://doi.org/10.3390/molecules24101893
Yan J-L, Shao G-Q, Fan S-H, Zhu C, Zhang Y, Wang J, Liu Q. Structure, Shift in Redox Potential and Li-Ion Diffusion Behavior in Tavorite LiFe1−xVxPO4F Solid-Solution Cathodes. Molecules. 2019; 24(10):1893. https://doi.org/10.3390/molecules24101893
Chicago/Turabian StyleYan, Jia-Li, Gang-Qin Shao, Shu-Hao Fan, Can Zhu, Yong Zhang, Jun Wang, and Qi Liu. 2019. "Structure, Shift in Redox Potential and Li-Ion Diffusion Behavior in Tavorite LiFe1−xVxPO4F Solid-Solution Cathodes" Molecules 24, no. 10: 1893. https://doi.org/10.3390/molecules24101893
APA StyleYan, J.-L., Shao, G.-Q., Fan, S.-H., Zhu, C., Zhang, Y., Wang, J., & Liu, Q. (2019). Structure, Shift in Redox Potential and Li-Ion Diffusion Behavior in Tavorite LiFe1−xVxPO4F Solid-Solution Cathodes. Molecules, 24(10), 1893. https://doi.org/10.3390/molecules24101893