
Peptide Conjugates with Small Molecules Designed to Enhance Efficacy and Safety

Abstract
1. Introduction
2. Why Peptide-Drug Conjugates?
3. Approved Peptide-Drug Conjugates
4. Representative Peptide-Drug Conjugates in Clinical Development
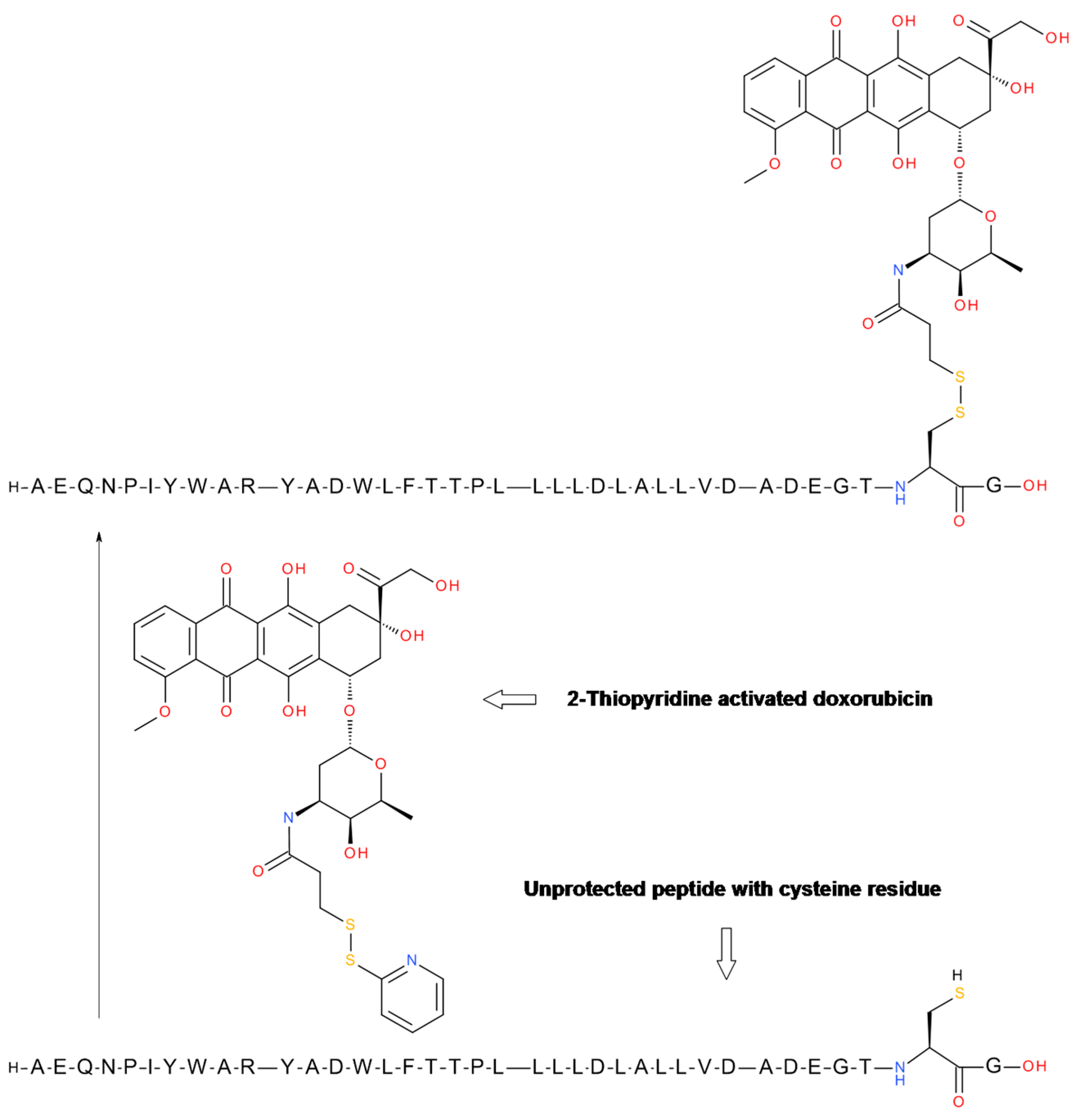
4.1. GnRH-Doxorubicin Conjugate
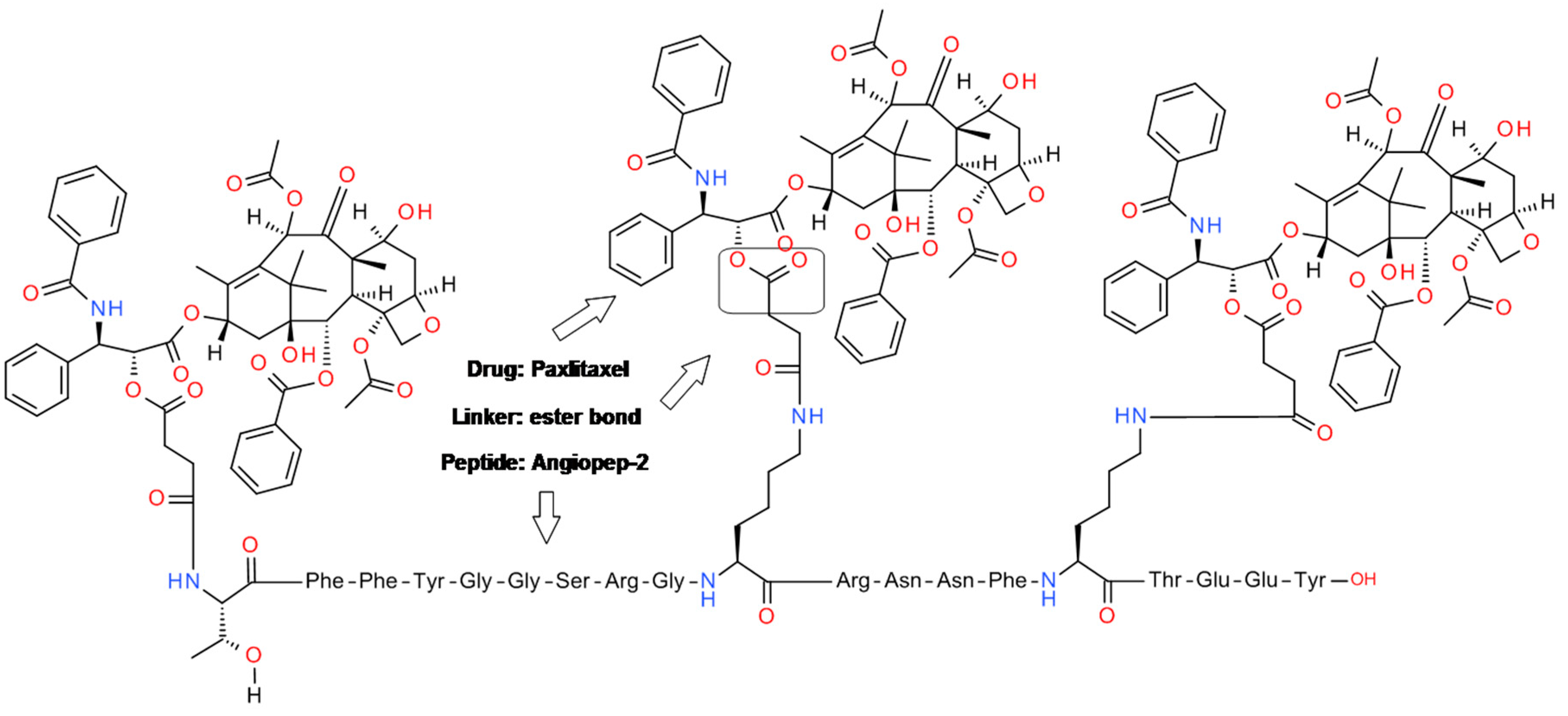
4.2. Angiopep-2-Paxlitaxel Conjugate
4.3. Tetrapeptide-Thapsigargin Conjugate
4.4. Miscellaneous Peptide-Drug Conjugates
5. Representative Peptide-Drug Conjugates in Preclinical Space
5.1. GLP-1-Estrogen Conjugate
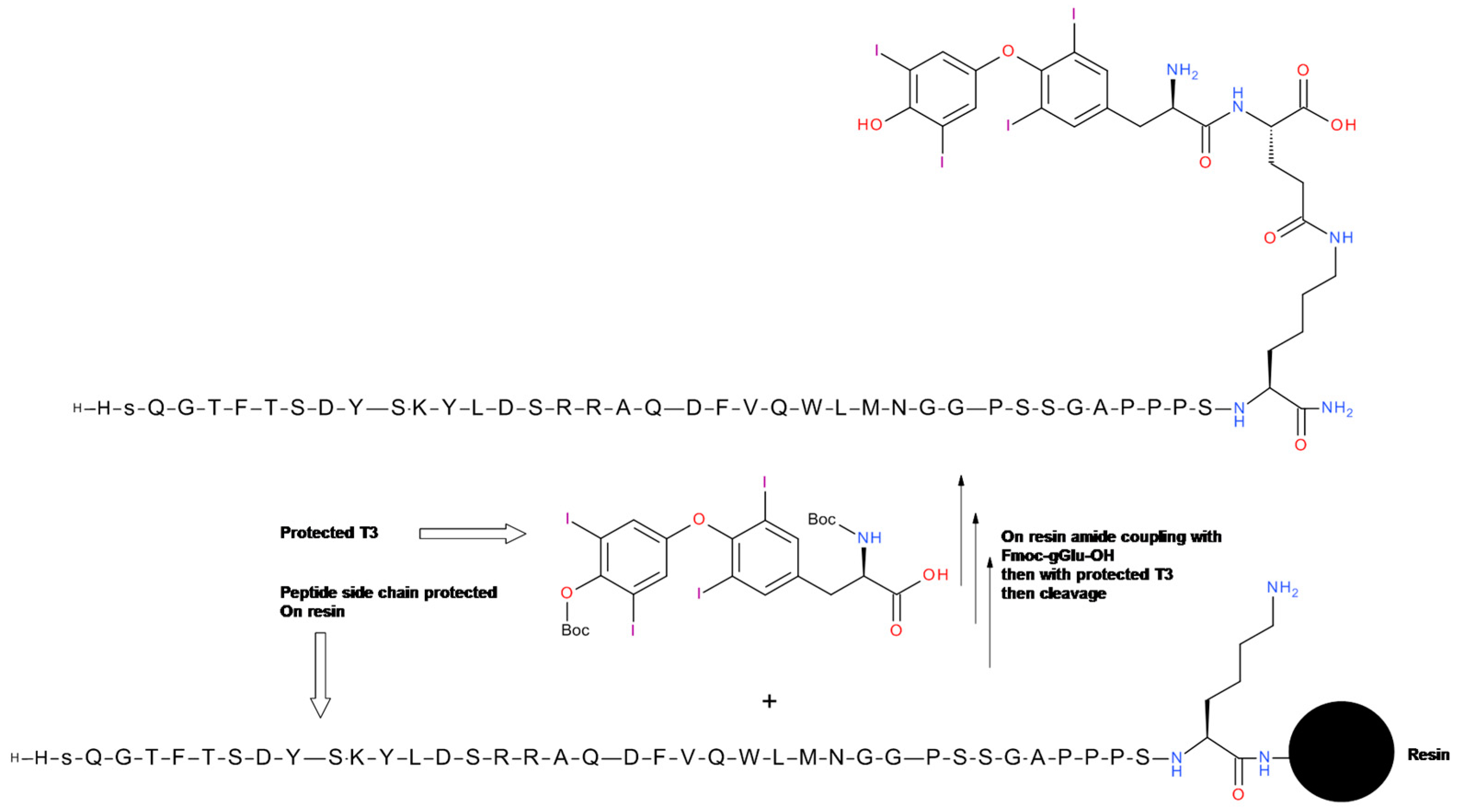
5.2. Glucagon-T3 Conjugate
5.3. Knotting Peptide Gemcitabine Conjugate
6. Linker and Conjugation Chemistry
6.1. Amide Bond Formation
6.2. Disulfide Bond Formation
6.3. Thioether Formation
6.4. Click Reaction
7. Peptide-Drug Conjugate Design Considerations
8. Outlook and Perspective
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADCs | Antibody-Drug Conjugates |
| ADT | Androgen Deprivation Therapy |
| Ala | Alanine |
| BBB | Blood–Bain–Barrier |
| Cit | Citrulline |
| DAR | Drug-Antibody Ratio |
| DDI | Drug-Drug Interactions |
| DIO | Diet-Induced Obese |
| DOTA | 1,4,7,10-Tetraazacyclododeane |
| DPP4 | Dipeptidyl Peptidase 4 |
| DTNP | 5,5′-Disulfanediylbis(2-nitrobenzoic acid) or Ellman’s reagent |
| GHIH | Growth Hormone-Inhibiting Hormone |
| GIP | Gastric Inhibitory Polypeptide |
| GLP-1 | Glucagon-Like Peptide 1 |
| GLP-2 | Glucagon-Like Peptide 2 |
| gGlu | gamma glutamic acid |
| GnRH | Gonadotropin-Releasing Hormone |
| LHRH | Luteinizing Hormone-Releasing Hormone |
| LRP1 | Low-Density Lipoprotein Receptor-Related Protein 1 |
| NSCLC | Non-Small Cell Lung Cancer |
| PABC | p-Aminobenzyl Carbamate |
| PRRT | Peptide Receptor Radionuclide Therapy |
| PSMA | Prostate Specific Membrane Antigen |
| PTH | Parathyroid Hormone |
| SERCA | the Sarco/Endoplasmic Reticulum Calcium ATPase |
| SSTR | Somatostatin Receptor |
References
- Henninot, A.; Collins, J.C.; Nuss, J.M. The Current State of Peptide Drug Discovery: Back to the Future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2017. [Google Scholar] [CrossRef]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef]
- Kaspar, A.A.; Reichert, J.M. Future directions for peptide therapeutics development. Drug Discov. Today 2013, 18, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Li, P.; Gelfanov, V.; Mayer, J.; DiMarchi, R. Synthetic Advances in Insulin-like Peptides Enable Novel Bioactivity. Acc. Chem. Res. 2017, 50, 1855–1865. [Google Scholar] [CrossRef] [PubMed]
- Mijalis, A.J.; Thomas, D.A., 3rd; Simon, M.D.; Adamo, A.; Beaumont, R.; Jensen, K.F.; Pentelute, B.L. A fully automated flow-based approach for accelerated peptide synthesis. Nat. Chem. Biol. 2017, 13, 464–466. [Google Scholar] [CrossRef] [PubMed]
- Behrendt, R.; White, P.; Offer, J. Advances in Fmoc solid-phase peptide synthesis. J. Pept. Sci. 2016, 22, 4–27. [Google Scholar] [CrossRef]
- Lau, J.; Bloch, P.; Schaffer, L.; Pettersson, I.; Spetzler, J.; Kofoed, J.; Madsen, K.; Knudsen, L.B.; McGuire, J.; Steensgaard, D.B.; et al. Discovery of the Once-Weekly Glucagon-Like Peptide-1 (GLP-1) Analogue Semaglutide. J. Med. Chem. 2015, 58, 7370–7380. [Google Scholar] [CrossRef]
- Made, V.; Els-Heindl, S.; Beck-Sickinger, A.G. Automated solid-phase peptide synthesis to obtain therapeutic peptides. Beilstein J. Org. Chem. 2014, 10, 1197–1212. [Google Scholar] [CrossRef]
- Mitchell, A.R. Bruce Merrifield and solid-phase peptide synthesis: A historical assessment. Biopolymers 2008, 90, 175–184. [Google Scholar] [CrossRef]
- Baeshen, N.A.; Baeshen, M.N.; Sheikh, A.; Bora, R.S.; Ahmed, M.M.; Ramadan, H.A.; Saini, K.S.; Redwan, E.M. Cell factories for insulin production. Microb. Cell Fact. 2014, 13, 141. [Google Scholar] [CrossRef]
- Meehl, M.A.; Stadheim, T.A. Biopharmaceutical discovery and production in yeast. Curr. Opin. Biotechnol. 2014, 30, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Thayer, A.M. Making Peptides At Large Scale. Chem. Eng. News 2011, 89, 21–25. [Google Scholar] [CrossRef]
- Mayer, J.P.; Zhang, F.; DiMarchi, R.D. Insulin structure and function. Biopolymers 2007, 88, 687–713. [Google Scholar] [CrossRef] [PubMed]
- Blazynski, C. 2016 Completed Clinical Trials: Industry Strategies Revealed and Graded; Pharma Intelligence: London, UK, 2017. [Google Scholar]
- Waring, M.J.; Arrowsmith, J.; Leach, A.R.; Leeson, P.D.; Mandrell, S.; Owen, R.M.; Pairaudeau, G.; Pennie, W.D.; Pickett, S.D.; Wang, J.; et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 2015, 14, 475–486. [Google Scholar] [CrossRef]
- Thomas, D.W.; Burns, J.; Audette, J.; Carroll, A.; Dow-Hygelund, C.; Hay, M. Clinical Development Success Rates 2006–2015; AMPLION, Biomedtracker, Biotechnology Innovation Organization (BIO): Washington, DC, USA, 2016. [Google Scholar]
- Wirtz, V.; Knox, R.; Cao, C.; Mehrtash, H.; Posner, N.W.; McClenathan, J. Insulin Market Profile; Health Action International: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Novo Nordisk Annual Report 2017; Novo Nordisk: Bagsvaerd, Denmark, 2018.
- Moya-Garcia, A.; Adeyelu, T.; Kruger, F.A.; Dawson, N.L.; Lees, J.G.; Overington, J.P.; Orengo, C.; Ranea, J.A.G. Structural and Functional View of Polypharmacology. Sci. Rep. 2017, 7, 10102. [Google Scholar] [CrossRef] [PubMed]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and opportunities in drug discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.S.; Zhang, S. Polypharmacology: Drug discovery for the future. Expert Rev. Clin. Pharm. 2013, 6, 41–47. [Google Scholar] [CrossRef]
- Tschop, M.; DiMarchi, R. Single-Molecule Combinatorial Therapeutics for Treating Obesity and Diabetes. Diabetes 2017, 66, 1766–1769. [Google Scholar] [CrossRef]
- Khajavi, N.; Biebermann, H.; Tschop, M.; DiMarchi, R. Treatment of Diabetes and Obesity by Rationally Designed Peptide Agonists Functioning at Multiple Metabolic Receptors. Endocr. Dev. 2017, 32, 165–182. [Google Scholar] [CrossRef] [PubMed]
- Sadry, S.A.; Drucker, D.J. Emerging combinatorial hormone therapies for the treatment of obesity and T2DM. Nat. Rev. Endocrinol. 2013, 9, 425–433. [Google Scholar] [CrossRef]
- Finan, B.; Yang, B.; Ottaway, N.; Smiley, D.L.; Ma, T.; Clemmensen, C.; Chabenne, J.; Zhang, L.; Habegger, K.M.; Fischer, K.; et al. A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat. Med. 2015, 21, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Demartis, A.; Lahm, A.; Tomei, L.; Beghetto, E.; Di Biasio, V.; Orvieto, F.; Frattolillo, F.; Carrington, P.E.; Mumick, S.; Hawes, B.; et al. Polypharmacy through Phage Display: Selection of Glucagon and GLP-1 Receptor Co-agonists from a Phage-Displayed Peptide Library. Sci. Rep. 2018, 8, 585. [Google Scholar] [CrossRef]
- Jall, S.; Sachs, S.; Clemmensen, C.; Finan, B.; Neff, F.; DiMarchi, R.D.; Tschop, M.H.; Muller, T.D.; Hofmann, S.M. Monomeric GLP-1/GIP/glucagon triagonism corrects obesity, hepatosteatosis, and dyslipidemia in female mice. Mol. Metab. 2017, 6, 440–446. [Google Scholar] [CrossRef]
- Gault, V.A.; Bhat, V.K.; Irwin, N.; Flatt, P.R. A novel glucagon-like peptide-1 (GLP-1)/glucagon hybrid peptide with triple-acting agonist activity at glucose-dependent insulinotropic polypeptide, GLP-1, and glucagon receptors and therapeutic potential in high fat-fed mice. J. Biol. Chem. 2013, 288, 35581–35591. [Google Scholar] [CrossRef]
- Pocai, A.; Carrington, P.E.; Adams, J.R.; Wright, M.; Eiermann, G.; Zhu, L.; Du, X.; Petrov, A.; Lassman, M.E.; Jiang, G.; et al. Glucagon-like peptide 1/glucagon receptor dual agonism reverses obesity in mice. Diabetes 2009, 58, 2258–2266. [Google Scholar] [CrossRef]
- Day, J.W.; Ottaway, N.; Patterson, J.T.; Gelfanov, V.; Smiley, D.; Gidda, J.; Findeisen, H.; Bruemmer, D.; Drucker, D.J.; Chaudhary, N.; et al. A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nat. Chem. Biol. 2009, 5, 749–757. [Google Scholar] [CrossRef]
- Andreassen, K.V.; Feigh, M.; Hjuler, S.T.; Gydesen, S.; Henriksen, J.E.; Beck-Nielsen, H.; Christiansen, C.; Karsdal, M.A.; Henriksen, K. A novel oral dual amylin and calcitonin receptor agonist (KBP-042) exerts antiobesity and antidiabetic effects in rats. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E24–E33. [Google Scholar] [CrossRef] [PubMed]
- Gydesen, S.; Andreassen, K.V.; Hjuler, S.T.; Hellgren, L.I.; Karsdal, M.A.; Henriksen, K. Optimization of tolerability and efficacy of the novel dual amylin and calcitonin receptor agonist KBP-089 through dose escalation and combination with a GLP-1 analog. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E598–E607. [Google Scholar] [CrossRef] [PubMed]
- Gydesen, S.; Hjuler, S.T.; Freving, Z.; Andreassen, K.V.; Sonne, N.; Hellgren, L.I.; Karsdal, M.A.; Henriksen, K. A novel dual amylin and calcitonin receptor agonist, KBP-089, induces weight loss through a reduction in fat, but not lean mass, while improving food preference. Br. J. Pharm. 2017, 174, 591–602. [Google Scholar] [CrossRef]
- Hjuler, S.T.; Gydesen, S.; Andreassen, K.V.; Karsdal, M.A.; Henriksen, K. The Dual Amylin- and Calcitonin-Receptor Agonist KBP-042 Works as Adjunct to Metformin on Fasting Hyperglycemia and HbA1c in a Rat Model of Type 2 Diabetes. J. Pharm. Exp. 2017, 362, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. Bispecific antibody pipeline moves beyond oncology. Nat. Rev. Drug Discov. 2017, 16, 810. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.C.; Ma, B.; Trinklein, N.D.; Schellenberger, U.; Osborn, M.J.; Ouisse, L.H.; Boudreau, A.; Davison, L.M.; Harris, K.E.; Ugamraj, H.S.; et al. Multispecific Antibody Development Platform Based on Human Heavy Chain Antibodies. Front. Immunol. 2018, 9, 3037. [Google Scholar] [CrossRef]
- Sedykh, S.E.; Prinz, V.V.; Buneva, V.N.; Nevinsky, G.A. Bispecific antibodies: Design, therapy, perspectives. Drug Des. Dev. 2018, 12, 195–208. [Google Scholar] [CrossRef]
- Ma, L.; Wang, C.; He, Z.; Cheng, B.; Zheng, L.; Huang, K. Peptide-Drug Conjugate: A Novel Drug Design Approach. Curr. Med. Chem. 2017, 24, 3373–3396. [Google Scholar] [CrossRef]
- Gilad, Y.; Firer, M.; Gellerman, G. Recent Innovations in Peptide Based Targeted Drug Delivery to Cancer Cells. Biomedicines 2016, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Firer, M.A.; Gellerman, G. Targeted drug delivery for cancer therapy: The other side of antibodies. J. Hematol. Oncol. 2012, 5, 70. [Google Scholar] [CrossRef]
- Zagorodko, O.; Arroyo-Crespo, J.J.; Nebot, V.J.; Vicent, M.J. Polypeptide-Based Conjugates as Therapeutics: Opportunities and Challenges. Macromol. Biosci. 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Chilkoti, A. Protein-polymer conjugation-moving beyond PEGylation. Curr. Opin. Chem. Biol. 2015, 28, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Vhora, I.; Patil, S.; Bhatt, P.; Misra, A. Protein- and Peptide-drug conjugates: An emerging drug delivery technology. Adv. Protein Chem. Struct. Biol. 2015, 98, 1–55. [Google Scholar] [CrossRef] [PubMed]
- Staderini, M.; Megia-Fernandez, A.; Dhaliwal, K.; Bradley, M. Peptides for optical medical imaging and steps towards therapy. Bioorg. Med. Chem. 2017. [Google Scholar] [CrossRef]
- Albada, B.; Metzler-Nolte, N. Highly Potent Antibacterial Organometallic Peptide Conjugates. Acc. Chem. Res. 2017, 50, 2510–2518. [Google Scholar] [CrossRef]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Sanseau, P.; Cardon, L.R. Novelty in the target landscape of the pharmaceutical industry. Nat. Rev. Drug Discov. 2013, 12, 575–576. [Google Scholar] [CrossRef]
- Smietana, K.; Siatkowski, M.; Moller, M. Trends in clinical success rates. Nat. Rev. Drug Discov. 2016, 15, 379–380. [Google Scholar] [CrossRef]
- Bohme, D.; Beck-Sickinger, A.G. Controlling Toxicity of Peptide–Drug Conjugates by Different Chemical Linker Structures. ChemMedChem. 2015, 10, 804–814. [Google Scholar] [CrossRef]
- Diaz, D.; Ford, K.A.; Hartley, D.P.; Harstad, E.B.; Cain, G.R.; Achilles-Poon, K.; Nguyen, T.; Peng, J.; Zheng, Z.; Merchant, M.; et al. Pharmacokinetic drivers of toxicity for basic molecules: Strategy to lower pKa results in decreased tissue exposure and toxicity for a small molecule Met inhibitor. Toxicol. Appl. Pharm. 2013, 266, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Palleria, C.; Di Paolo, A.; Giofre, C.; Caglioti, C.; Leuzzi, G.; Siniscalchi, A.; De Sarro, G.; Gallelli, L. Pharmacokinetic drug-drug interaction and their implication in clinical management. J. Res. Med. Sci. 2013, 18, 601–610. [Google Scholar]
- Ehrlich, P. The relationship existing between chemical constitution, distribution and pharmacological action. In Collected Studies on Immunity; Wiley & Sons: Hoboken, NJ, USA, 1906; pp. 441–450. [Google Scholar]
- Ehrlich, P. Chemotherapy. Proceedings of 17th International Congress of Medicine, in Collected Papers of Paul Ehrlich; Himmelwiet, F., Ed.; Pergamon Press: Oxford, UK, 1913; pp. 505–518. [Google Scholar]
- Mathe, G.; Loc, T.B.; Bernard, J.C.C. Effet sur la leucemie L1210 de la souris d’une combinaison par diazotation d’A-methopterine et de gamma-globulines de hamsters porteur de cette leucemie par heterogreffe. C. R. Acad. Sci. 1958, 246, 1626–1628. [Google Scholar]
- Ghose, T.; Cerini, M.; Carter, M.; Nairn, R.C. Immunoradioactive agent against cancer. Br. Med. J. 1967, 1, 90–93. [Google Scholar] [CrossRef]
- Ghose, T.; Nigam, S.P. Antibody as carrier of chlorambucil. Cancer 1972, 29, 1398–1400. [Google Scholar] [CrossRef]
- Rowland, G.F.; O’Neill, G.J.; Davies, D.A. Suppression of tumour growth in mice by a drug-antibody conjugate using a novel approach to linkage. Nature 1975, 255, 487–488. [Google Scholar] [CrossRef]
- Ford, C.H.; Newman, C.E.; Johnson, J.R.; Woodhouse, C.S.; Reeder, T.A.; Rowland, G.F.; Simmonds, R.G. Localisation and toxicity study of a vindesine-anti-CEA conjugate in patients with advanced cancer. Br. J. Cancer 1983, 47, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaia, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Perez, H.L.; Cardarelli, P.M.; Deshpande, S.; Gangwar, S.; Schroeder, G.M.; Vite, G.D.; Borzilleri, R.M. Antibody-drug conjugates: Current status and future directions. Drug Discov. Today 2014, 19, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Yasothan, U.; Kirkpatrick, P. Brentuximab vedotin. Nat. Rev. Drug Discov. 2012, 11, 19–20. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.; Chari, R.V. Ado-trastuzumab Emtansine (T-DM1): An antibody-drug conjugate (ADC) for HER2-positive breast cancer. J. Med. Chem. 2014, 57, 6949–6964. [Google Scholar] [CrossRef]
- Available online: https://www.reuters.com/article/us-pfizer-mylotarg/pfizer-pulls-leukemia-drug-from-u-s-market-idUSTRE65K5QG20100621 (accessed on 22 April 2019).
- Available online: https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm572133.htm (accessed on 22 April 2019).
- Available online: https://clinicaltrials.gov/ct2/results?intr=Antibody-Drug+Conjugate&Search=Apply&recrs=b&recrs=a&recrs=f&recrs=d&age_v=&gndr=&type=&rslt=0 (accessed on 22 April 2019).
- Diamantis, N.; Banerji, U. Antibody-drug conjugates--an emerging class of cancer treatment. Br. J. Cancer 2016, 114, 362–367. [Google Scholar] [CrossRef]
- Bouchard, H.; Viskov, C.; Garcia-Echeverria, C. Antibody-drug conjugates-a new wave of cancer drugs. Bioorg. Med. Chem. Lett. 2014, 24, 5357–5363. [Google Scholar] [CrossRef]
- Sun, X.; Ponte, J.F.; Yoder, N.C.; Laleau, R.; Coccia, J.; Lanieri, L.; Qiu, Q.; Wu, R.; Hong, E.; Bogalhas, M.; et al. Effects of Drug-Antibody Ratio on Pharmacokinetics, Biodistribution, Efficacy, and Tolerability of Antibody-Maytansinoid Conjugates. Bioconj. Chem. 2017, 28, 1371–1381. [Google Scholar] [CrossRef]
- Weckbecker, G.; Lewis, I.; Albert, R.; Schmid, H.A.; Hoyer, D.; Bruns, C. Opportunities in somatostatin research: Biological, chemical and therapeutic aspects. Nat. Rev. Drug Discov. 2003, 2, 999–1017. [Google Scholar] [CrossRef]
- Keskin, O.; Yalcin, S. A review of the use of somatostatin analogs in oncology. Onco Targets 2013, 6, 471–483. [Google Scholar] [CrossRef][Green Version]
- Spada, F.; Valente, M. Review of recents advances in medical treatment for neuroendocrine neoplasms: Somatostatin analogs and chemotherapy. J. Cancer Metastasis Treat. 2016, 2, 313–320. [Google Scholar] [CrossRef][Green Version]
- Moller, L.N.; Stidsen, C.E.; Hartmann, B.; Holst, J.J. Somatostatin receptors. Biochim. Biophys. Acta 2003, 1616, 1–84. [Google Scholar] [CrossRef]
- De Jong, M.; Valkema, R.; Jamar, F.; Kvols, L.K.; Kwekkeboom, D.J.; Breeman, W.A.; Bakker, W.H.; Smith, C.; Pauwels, S.; Krenning, E.P. Somatostatin receptor-targeted radionuclide therapy of tumors: Preclinical and clinical findings. Semin. Nucl. Med. 2002, 32, 133–140. [Google Scholar] [CrossRef]
- Pinato, D.J.; Black, J.R.; Ramaswami, R.; Tan, T.M.; Adjogatse, D.; Sharma, R. Peptide receptor radionuclide therapy for metastatic paragangliomas. Med. Oncol. 2016, 33, 47. [Google Scholar] [CrossRef]
- PRRT. Available online: http://www.prrtinfo.org/prrt (accessed on 22 April 2019).
- Krenning, E.P.; Kooij, P.P.; Bakker, W.H.; Breeman, W.A.; Postema, P.T.; Kwekkeboom, D.J.; Oei, H.Y.; de Jong, M.; Visser, T.J.; Reijs, A.E.; et al. Radiotherapy with a radiolabeled somatostatin analogue, [111In-DTPA-DPhe1]- octreotide: A case history. Ann. N. Y. Acad. Sci. 1994, 733, 496–506. [Google Scholar] [CrossRef]
- Hörsch, D.; Ezziddin, S.; Haug, A.; Gratz, K.F.; Dunkelmann, S.; Miederer, M.; Schreckenberger, M.; Krause, B.J.; Bengel, F.M.; Bartenstein, P.; et al. Effectiveness and side-effects of peptide receptor radionuclide therapy for neuroendocrine neoplasms in Germany: A multi-institutional registry study with prospective follow-up. Eur. J. Cancer 2016, 58, 41–51. [Google Scholar] [CrossRef]
- FDA Approves Lutetium Lu 177 Dotatate for Treatment of GEP-NETS. Available online: https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm594105.htm (accessed on 22 April 2019).
- Lutathera. Available online: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/004123/human_med_002163.jsp&mid=WC0b01ac058001d1244 (accessed on 22 April 2019).
- Emmett, L.; Willowson, K.; Violet, J.; Shin, J.; Blanksby, A.; Lee, J. Lutetium (177) PSMA radionuclide therapy for men with prostate cancer: A review of the current literature and discussion of practical aspects of therapy. J. Med. Radiat. Sci. 2017, 64, 52–60. [Google Scholar] [CrossRef]
- Bodei, L.; Mueller-Brand, J.; Baum, R.P.; Pavel, M.E.; Horsch, D.; O’Dorisio, M.S.; O’Dorisio, T.M.; Howe, J.R.; Cremonesi, M.; Kwekkeboom, D.J.; et al. The joint IAEA, EANM, and SNMMI practical guidance on peptide receptor radionuclide therapy (PRRNT) in neuroendocrine tumours. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 800–816. [Google Scholar] [CrossRef]
- Van Essen, M.; Krenning, E.P.; De Jong, M.; Valkema, R.; Kwekkeboom, D.J. Peptide Receptor Radionuclide Therapy with radiolabelled somatostatin analogues in patients with receptor positive tumours. Acta Oncol. 2007, 46, 723–734. [Google Scholar] [CrossRef]
- Jamous, M.; Haberkorn, U.; Mier, W. Synthesis of peptide radiopharmaceuticals for the therapy and diagnosis of tumor diseases. Molecules 2013, 18, 3379–3409. [Google Scholar] [CrossRef]
- Reubi, J.C.; Schär, J.C.; Waser, B.; Wenger, S.; Heppeler, A.; Schmitt, J.S.; Mäcke, H.R. Affinity profiles for human somatostatin receptor subtypes SST1-SST5 of somatostatin radiotracers selected for scintigraphic and radiotherapeutic use. Eur. J. Nucl. Med. 2000, 27, 273–282. [Google Scholar] [CrossRef]
- Kwekkeboom, D.J.; de Herder, W.W.; Kam, B.L.; van Eijck, C.H.; van Essen, M.; Kooij, P.P.; Feelders, R.A.; van Aken, M.O.; Krenning, E.P. Treatment with the radiolabeled somatostatin analog [177 Lu-DOTA 0,Tyr3]octreotate: Toxicity, efficacy, and survival. J. Clin. Oncol. 2008, 26, 2124–2130. [Google Scholar] [CrossRef]
- Strosberg, J.; El-Haddad, G.; Wolin, E.; Hendifar, A.; Yao, J.; Chasen, B.; Mittra, E.; Kunz, P.L.; Kulke, M.H.; Jacene, H.; et al. Phase 3 Trial of (177)Lu-Dotatate for Midgut Neuroendocrine Tumors. N. Engl. J. Med. 2017, 376, 125–135. [Google Scholar] [CrossRef]
- Schneider, F.; Tomek, W.; Grundker, C. Gonadotropin-releasing hormone (GnRH) and its natural analogues: A review. Theriogenology 2006, 66, 691–709. [Google Scholar] [CrossRef]
- Millar, R.P.; Lu, Z.L.; Pawson, A.J.; Flanagan, C.A.; Morgan, K.; Maudsley, S.R. Gonadotropin-releasing hormone receptors. Endocr. Rev. 2004, 25, 235–275. [Google Scholar] [CrossRef]
- Madhunapantula, S.V.; Mosca, P.; Robertson, G.P. Steroid hormones drive cancer development. Cancer Biol. 2010, 10, 765–766. [Google Scholar] [CrossRef]
- Capper, C.P.; Rae, J.M.; Auchus, R.J. The Metabolism, Analysis, and Targeting of Steroid Hormones in Breast and Prostate Cancer. Horm. Cancer 2016, 7, 149–164. [Google Scholar] [CrossRef]
- Perrett, R.M.; McArdle, C.A. Molecular mechanisms of gonadotropin-releasing hormone signaling integrating cyclic nucleotides into the network. Front. Endocrinol. (Lausanne) 2013, 4, 180. [Google Scholar] [CrossRef]
- Bolton, E.M.; Lynch, T.H. Are all gonadotropin-releasing hormone agonists equivalent for the treatment of prostate cancer? A systematic review. BJU Int. 2018. [Google Scholar] [CrossRef]
- Cheng, C.K.; Leung, P.C. Molecular biology of gonadotropin-releasing hormone (GnRH)-I, GnRH-II, and their receptors in humans. Endocr. Rev. 2005, 26, 283–306. [Google Scholar] [CrossRef]
- Westphalen, S.; Kotulla, G.; Kaiser, F.; Krauss, W.; Werning, G.; Elsasser, H.P.; Nagy, A.; Schulz, K.D.; Grundker, C.; Schally, A.V.; et al. Receptor mediated antiproliferative effects of the cytotoxic LHRH agonist AN-152 in human ovarian and endometrial cancer cell lines. Int. J. Oncol. 2000, 17, 1063–1069. [Google Scholar] [CrossRef]
- Letsch, M.; Schally, A.V.; Szepeshazi, K.; Halmos, G.; Nagy, A. Preclinical evaluation of targeted cytotoxic luteinizing hormone-releasing hormone analogue AN-152 in androgen-sensitive and insensitive prostate cancers. Clin. Cancer Res. 2003, 9, 4505–4513. [Google Scholar]
- Liu, S.V.; Tsao-Wei, D.D.; Xiong, S.; Groshen, S.; Dorff, T.B.; Quinn, D.I.; Tai, Y.C.; Engel, J.; Hawes, D.; Schally, A.V.; et al. Phase I, dose-escalation study of the targeted cytotoxic LHRH analog AEZS-108 in patients with castration- and taxane-resistant prostate cancer. Clin. Cancer Res. 2014, 20, 6277–6283. [Google Scholar] [CrossRef]
- Emons, G.; Kaufmann, M.; Gorchev, G.; Tsekova, V.; Grundker, C.; Gunthert, A.R.; Hanker, L.C.; Velikova, M.; Sindermann, H.; Engel, J.; et al. Dose escalation and pharmacokinetic study of AEZS-108 (AN-152), an LHRH agonist linked to doxorubicin, in women with LHRH receptor-positive tumors. Gynecol. Oncol. 2010, 119, 457–461. [Google Scholar] [CrossRef]
- Engel, J.; Emons, G.; Pinski, J.; Schally, A.V. AEZS-108: A targeted cytotoxic analog of LHRH for the treatment of cancers positive for LHRH receptors. Expert Opin. Investig. Drugs 2012, 21, 891–899. [Google Scholar] [CrossRef]
- Yu, S.S.; Athreya, K.; Liu, S.V.; Schally, A.V.; Tsao-Wei, D.; Groshen, S.; Quinn, D.I.; Dorff, T.B.; Xiong, S.; Engel, J.; et al. A Phase II Trial of AEZS-108 in Castration- and Taxane-Resistant Prostate Cancer. Clin. Genitourin. Cancer 2017, 15, 742–749. [Google Scholar] [CrossRef]
- Emons, G.; Gorchev, G.; Harter, P.; Wimberger, P.; Stahle, A.; Hanker, L.; Hilpert, F.; Beckmann, M.W.; Dall, P.; Grundker, C.; et al. Efficacy and safety of AEZS-108 (LHRH agonist linked to doxorubicin) in women with advanced or recurrent endometrial cancer expressing LHRH receptors: A multicenter phase 2 trial (AGO-GYN5). Int. J. Gynecol. Cancer 2014, 24, 260–265. [Google Scholar] [CrossRef]
- Emons, G.; Gorchev, G.; Sehouli, J.; Wimberger, P.; Stahle, A.; Hanker, L.; Hilpert, F.; Sindermann, H.; Grundker, C.; Harter, P. Efficacy and safety of AEZS-108 (INN: Zoptarelin doxorubicin acetate) an LHRH agonist linked to doxorubicin in women with platinum refractory or resistant ovarian cancer expressing LHRH receptors: A multicenter phase II trial of the ago-study group (AGO GYN 5). Gynecol. Oncol. 2014, 133, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Zoptarelin Doxorubicin Falls Short in Phase III Endometrial Cancer Trial. Available online: https://www.onclive.com/web-exclusives/zoptarelin-doxorubicin-falls-short-in-phase-iii-endometrial-cancer-trial (accessed on 22 April 2019).
- Hennenfent, K.L.; Govindan, R. Novel formulations of taxanes: A review. Old wine in a new bottle? Ann. Oncol. 2006, 17, 735–749. [Google Scholar] [CrossRef]
- Spencer, C.M.; Faulds, D. Paclitaxel. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic potential in the treatment of cancer. Drugs 1994, 48, 794–847. [Google Scholar] [CrossRef] [PubMed]
- Demeule, M.; Regina, A.; Che, C.; Poirier, J.; Nguyen, T.; Gabathuler, R.; Castaigne, J.P.; Beliveau, R. Identification and design of peptides as a new drug delivery system for the brain. J. Pharm. Exp. 2008, 324, 1064–1072. [Google Scholar] [CrossRef]
- Shao, K.; Huang, R.; Li, J.; Han, L.; Ye, L.; Lou, J.; Jiang, C. Angiopep-2 modified PE-PEG based polymeric micelles for amphotericin B delivery targeted to the brain. J. Control. Release 2010, 147, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Regina, A.; Demeule, M.; Che, C.; Lavallee, I.; Poirier, J.; Gabathuler, R.; Beliveau, R.; Castaigne, J.P. Antitumour activity of ANG1005, a conjugate between paclitaxel and the new brain delivery vector Angiopep-2. Br. J. Pharm. 2008, 155, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Kurzrock, R.; Gabrail, N.; Chandhasin, C.; Moulder, S.; Smith, C.; Brenner, A.; Sankhala, K.; Mita, A.; Elian, K.; Bouchard, D.; et al. Safety, pharmacokinetics, and activity of GRN1005, a novel conjugate of angiopep-2, a peptide facilitating brain penetration, and paclitaxel, in patients with advanced solid tumors. Mol. Cancer Ther. 2012, 11, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Drappatz, J.; Brenner, A.; Wong, E.T.; Eichler, A.; Schiff, D.; Groves, M.D.; Mikkelsen, T.; Rosenfeld, S.; Sarantopoulos, J.; Meyers, C.A.; et al. Phase I study of GRN1005 in recurrent malignant glioma. Clin. Cancer Res. 2013, 19, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, Y.; Currie, J.C.; Poirier, J.; Demeule, M.; Abulrob, A.; Fatehi, D.; Stanimirovic, D.; Sartelet, H.; Castaigne, J.P.; Beliveau, R. Influence of glioma tumour microenvironment on the transport of ANG1005 via low-density lipoprotein receptor-related protein 1. Br. J. Cancer 2011, 105, 1697–1707. [Google Scholar] [CrossRef]
- Li, F.; Tang, S.C. Targeting metastatic breast cancer with ANG1005, a novel peptide-paclitaxel conjugate that crosses the blood-brain-barrier (BBB). Genes Dis. 2017, 4, 1–3. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov/ct2/results?term=ANG1005&age_v=&gndr=&type=&rslt=&phase=1&Search=Apply (accessed on 22 April 2019).
- Angiochem’s ANG1005 Received Orphan Drug Designation from FDA for the Treatment of Glioblastoma Multiform. Available online: http://angiochem.com/angiochem%E2%80%99s-ang1005-received-orphan-drug-designation-fda-treatment-glioblastoma-multiform (accessed on 22 April 2019).
- Available online: https://clinicaltrials.gov/ct2/results?cond=&term=NCT03613181&cntry=&state=&city=&dist= (accessed on 22 April 2019).
- Quynh Doan, N.T.; Christensen, S.B. Thapsigargin, Origin, Chemistry, Structure-Activity Relationships and Prodrug Development. Curr. Pharm. Des. 2015, 21, 5501–5517. [Google Scholar] [CrossRef]
- Denmeade, S.R.; Isaacs, J.T. Engineering enzymatically activated “molecular grenades” for cancer. Oncotarget 2012, 3, 666–667. [Google Scholar] [CrossRef] [PubMed]
- Denmeade, S.R.; Mhaka, A.M.; Rosen, D.M.; Brennen, W.N.; Dalrymple, S.; Dach, I.; Olesen, C.; Gurel, B.; Demarzo, A.M.; Wilding, G.; et al. Engineering a prostate-specific membrane antigen-activated tumor endothelial cell prodrug for cancer therapy. Sci. Transl. Med. 2012, 4, 140ra186. [Google Scholar] [CrossRef]
- Andersen, T.B.; Lopez, C.Q.; Manczak, T.; Martinez, K.; Simonsen, H.T. Thapsigargin--from Thapsia L. to mipsagargin. Molecules 2015, 20, 6113–6127. [Google Scholar] [CrossRef]
- Silver, D.A.; Pellicer, I.; Fair, W.R.; Heston, W.D.; Cordon-Cardo, C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin. Cancer Res. 1997, 3, 81–85. [Google Scholar] [PubMed]
- Kinoshita, Y.; Kuratsukuri, K.; Landas, S.; Imaida, K.; Rovito, P.M., Jr.; Wang, C.Y.; Haas, G.P. Expression of prostate-specific membrane antigen in normal and malignant human tissues. World J. Surg. 2006, 30, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Wilding, G.; Denmeade, S.; Sarantopoulas, J.; Cosgrove, D.; Cetnar, J.; Azad, N.; Bruce, J.; Kurman, M.; Allgood, V.E.; et al. Mipsagargin, a novel thapsigargin-based PSMA-activated prodrug: Results of a first-in-man phase I clinical trial in patients with refractory, advanced or metastatic solid tumours. Br. J. Cancer 2016, 114, 986–994. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov/ct2/results?cond=&term=G202&cntry=&state=&city=&dist= (accessed on 22 April 2019).
- Dissanayake, S.; Denny, W.A.; Gamage, S.; Sarojini, V. Recent developments in anticancer drug delivery using cell penetrating and tumor targeting peptides. J. Control. Release 2017, 250, 62–76. [Google Scholar] [CrossRef]
- Kebebe, D.; Liu, Y.; Wu, Y.; Vilakhamxay, M.; Liu, Z.; Li, J. Tumor-targeting delivery of herb-based drugs with cell-penetrating/tumor-targeting peptide-modified nanocarriers. Int. J. Nanomed. 2018, 13, 1425–1442. [Google Scholar] [CrossRef]
- Timur, S.S.; Bhattarai, P.; Gursoy, R.N.; Vural, I.; Khaw, B.A. Design and In Vitro Evaluation of Bispecific Complexes and Drug Conjugates of Anticancer Peptide, LyP-1 in Human Breast Cancer. Pharm. Res. 2017, 34, 352–364. [Google Scholar] [CrossRef]
- Engel, J.B.; Schally, A.V.; Dietl, J.; Rieger, L.; Honig, A. Targeted therapy of breast and gynecological cancers with cytotoxic analogues of peptide hormones. Mol. Pharm. 2007, 4, 652–658. [Google Scholar] [CrossRef]
- Schally, A.V.; Nagy, A. Cancer chemotherapy based on targeting of cytotoxic peptide conjugates to their receptors on tumors. Eur. J. Endocrinol. 1999, 141, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Schally, A.V.; Halmos, G.; Armatis, P.; Cai, R.Z.; Csernus, V.; Kovacs, M.; Koppan, M.; Szepeshazi, K.; Kahan, Z. Synthesis and biological evaluation of cytotoxic analogs of somatostatin containing doxorubicin or its intensely potent derivative, 2-pyrrolinodoxorubicin. Proc. Natl. Acad. Sci. USA 1998, 95, 1794–1799. [Google Scholar] [CrossRef] [PubMed]
- Engel, J.B.; Schally, A.V.; Halmos, G.; Baker, B.; Nagy, A.; Keller, G. Targeted cytotoxic bombesin analog AN-215 effectively inhibits experimental human breast cancers with a low induction of multi-drug resistance proteins. Endocr. Relat. Cancer 2005, 12, 999–1009. [Google Scholar] [CrossRef]
- Nagy, A.; Armatis, P.; Cai, R.Z.; Szepeshazi, K.; Halmos, G.; Schally, A.V. Design, synthesis, and in vitro evaluation of cytotoxic analogs of bombesin-like peptides containing doxorubicin or its intensely potent derivative, 2-pyrrolinodoxorubicin. Proc. Natl. Acad. Sci. USA 1997, 94, 652–656. [Google Scholar] [CrossRef]
- Ibanez-Costa, A.; Lopez-Sanchez, L.M.; Gahete, M.D.; Rivero-Cortes, E.; Vazquez-Borrego, M.C.; Galvez, M.A.; de la Riva, A.; Venegas-Moreno, E.; Jimenez-Reina, L.; Moreno-Carazo, A.; et al. BIM-23A760 influences key functional endpoints in pituitary adenomas and normal pituitaries: Molecular mechanisms underlying the differential response in adenomas. Sci. Rep. 2017, 7, 42002. [Google Scholar] [CrossRef] [PubMed]
- Florio, T.; Barbieri, F.; Spaziante, R.; Zona, G.; Hofland, L.J.; van Koetsveld, P.M.; Feelders, R.A.; Stalla, G.K.; Theodoropoulou, M.; Culler, M.D.; et al. Efficacy of a dopamine-somatostatin chimeric molecule, BIM-23A760, in the control of cell growth from primary cultures of human non-functioning pituitary adenomas: A multi-center study. Endocr. Relat. Cancer 2008, 15, 583–596. [Google Scholar] [CrossRef]
- Jaquet, P.; Gunz, G.; Saveanu, A.; Dufour, H.; Taylor, J.; Dong, J.; Kim, S.; Moreau, J.P.; Enjalbert, A.; Culler, M.D. Efficacy of chimeric molecules directed towards multiple somatostatin and dopamine receptors on inhibition of GH and prolactin secretion from GH-secreting pituitary adenomas classified as partially responsive to somatostatin analog therapy. Eur. J. Endocrinol. 2005, 153, 135–141. [Google Scholar] [CrossRef]
- Bakker, W.H.; Albert, R.; Bruns, C.; Breeman, W.A.; Hofland, L.J.; Marbach, P.; Pless, J.; Pralet, D.; Stolz, B.; Koper, J.W.; et al. [111In-DTPA-D-Phe1]-octreotide, a potential radiopharmaceutical for imaging of somatostatin receptor-positive tumors: Synthesis, radiolabeling and in vitro validation. Life Sci. 1991, 49, 1583–1591. [Google Scholar] [CrossRef]
- Forssell-Aronsson, E.; Bernhardt, P.; Nilsson, O.; Tisell, L.E.; Wangberg, B.; Ahlman, H. Biodistribution data from 100 patients i.v. injected with 111In-DTPA-D-Phe1-octreotide. Acta Oncol. 2004, 43, 436–442. [Google Scholar] [CrossRef]
- Northfelt, D.W.; Allred, J.B.; Liu, H.; Hobday, T.J.; Rodacker, M.W.; Lyss, A.P.; Fitch, T.R.; Perez, E.A.; North Central Cancer Treatment, G. Phase 2 trial of paclitaxel polyglumex with capecitabine for metastatic breast cancer. Am. J. Clin. Oncol. 2014, 37, 167–171. [Google Scholar] [CrossRef]
- Singer, J.W. Paclitaxel poliglumex (XYOTAX, CT-2103): A macromolecular taxane. J. Control. Release 2005, 109, 120–126. [Google Scholar] [CrossRef]
- O’Brien, M.E.; Socinski, M.A.; Popovich, A.Y.; Bondarenko, I.N.; Tomova, A.; Bilynsky, B.T.; Hotko, Y.S.; Ganul, V.L.; Kostinsky, I.Y.; Eisenfeld, A.J.; et al. Randomized phase III trial comparing single-agent paclitaxel Poliglumex (CT-2103, PPX) with single-agent gemcitabine or vinorelbine for the treatment of PS 2 patients with chemotherapy-naive advanced non-small cell lung cancer. J. Thorac. Oncol. 2008, 3, 728–734. [Google Scholar] [CrossRef]
- Langer, C.J.; O’Byrne, K.J.; Socinski, M.A.; Mikhailov, S.M.; Lesniewski-Kmak, K.; Smakal, M.; Ciuleanu, T.E.; Orlov, S.V.; Dediu, M.; Heigener, D.; et al. Phase III trial comparing paclitaxel poliglumex (CT-2103, PPX) in combination with carboplatin versus standard paclitaxel and carboplatin in the treatment of PS 2 patients with chemotherapy-naive advanced non-small cell lung cancer. J. Thorac. Oncol. 2008, 3, 623–630. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Ross, H.; O’Brien, M.; Riviere, A.; Gatzemeier, U.; Von Pawel, J.; Kaukel, E.; Freitag, L.; Digel, W.; Bischoff, H.; et al. Phase III trial comparing paclitaxel poliglumex vs. docetaxel in the second-line treatment of non-small-cell lung cancer. Br. J. Cancer 2008, 98, 1608–1613. [Google Scholar] [CrossRef]
- Curtis, K.K.; Sarantopoulos, J.; Northfelt, D.W.; Weiss, G.J.; Barnhart, K.M.; Whisnant, J.K.; Leuschner, C.; Alila, H.; Borad, M.J.; Ramanathan, R.K. Novel LHRH-receptor-targeted cytolytic peptide, EP-100: First-in-human phase I study in patients with advanced LHRH-receptor-expressing solid tumors. Cancer Chemother. Pharm. 2014, 73, 931–941. [Google Scholar] [CrossRef]
- Leuschner, C.; Coulter, A.; Keener, J.; Alila, H. Targeted Oncolytic Peptide for Treatment of Ovarian Cancers. Int. J. Cancer Res. Mol. Mech. 2017, 3. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov/ct2/results?cond=&term=NCT01485848&cntry=&state=&city=&dist= (accessed on 22 April 2019).
- Rothbard, J.B.; Garlington, S.; Lin, Q.; Kirschberg, T.; Kreider, E.; McGrane, P.L.; Wender, P.A.; Khavari, P.A. Conjugation of arginine oligomers to cyclosporin A facilitates topical delivery and inhibition of inflammation. Nat. Med. 2000, 6, 1253–1257. [Google Scholar] [CrossRef]
- Ciclosporin–Cellgate. Available online: https://adisinsight.springer.com/drugs/800018283 (accessed on 18 January 2019).
- KAI Pharmaceuticals Initiates Phase 1 Trial of KAI-1455 for Ischemic Injury. Available online: https://www.businesswire.com/news/home/20070504005145/en/KAI-Pharmaceuticals-Initiates-Phase-1-Trial-KAI-1455 (accessed on 22 April 2019).
- Moodie, J.E.; Bisley, E.J.; Huang, S.; Pickthorn, K.; Bell, G. A single-center, randomized, double-blind, active, and placebo-controlled study of KAI-1678, a novel PKC-epsilon inhibitor, in the treatment of acute postoperative orthopedic pain. Pain Med. 2013, 14, 916–924. [Google Scholar] [CrossRef]
- Cousins, M.J.; Pickthorn, K.; Huang, S.; Critchley, L.; Bell, G. The safety and efficacy of KAI-1678- an inhibitor of epsilon protein kinase C (epsilonPKC)-versus lidocaine and placebo for the treatment of postherpetic neuralgia: A crossover study design. Pain Med. 2013, 14, 533–540. [Google Scholar] [CrossRef][Green Version]
- Available online: https://clinicaltrials.gov/ct2/results?cond=&term=kai-1678&cntry=&state=&city=&dist= (accessed on 18 January 2019).
- Miyaji, Y.; Walter, S.; Chen, L.; Kurihara, A.; Ishizuka, T.; Saito, M.; Kawai, K.; Okazaki, O. Distribution of KAI-9803, a novel delta-protein kinase C inhibitor, after intravenous administration to rats. Drug Metab. Dispos. 2011, 39, 1946–1953. [Google Scholar] [CrossRef]
- Direct Inhibition of delta-Protein Kinase C Enzyme to Limit Total Infarct Size in Acute Myocardial Infarction (DELTA MI) Investigators; Bates, E.; Bode, C.; Costa, M.; Gibson, C.M.; Granger, C.; Green, C.; Grimes, K.; Harrington, R.; Huber, K.; et al. Intracoronary KAI-9803 as an adjunct to primary percutaneous coronary intervention for acute ST-segment elevation myocardial infarction. Circulation 2008, 117, 886–896. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov/ct2/results?cond=&term=KAI-9803&cntry=&state=&city=&dist= (accessed on 22 April 2019).
- Available online: https://clinicaltrials.gov/ct2/results?cond=&term=XG-102&cntry=&state=&city=&dist= (accessed on 22 April 2019).
- Available online: http://www.xigenpharma.com/clinical-trials (accessed on 22 April 2019).
- Chiquet, C.; Aptel, F.; Creuzot-Garcher, C.; Berrod, J.P.; Kodjikian, L.; Massin, P.; Deloche, C.; Perino, J.; Kirwan, B.A.; de Brouwer, S.; et al. Postoperative Ocular Inflammation: A Single Subconjunctival Injection of XG-102 Compared to Dexamethasone Drops in a Randomized Trial. Am. J. Ophthalmol. 2017, 174, 76–84. [Google Scholar] [CrossRef][Green Version]
- Beydoun, T.; Deloche, C.; Perino, J.; Kirwan, B.A.; Combette, J.M.; Behar-Cohen, F. Subconjunctival injection of XG-102, a JNK inhibitor peptide, in patients with intraocular inflammation: A safety and tolerability study. J. Ocul. Pharm. 2015, 31, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Coriat, R.; Faivre, S.J.; Mir, O.; Dreyer, C.; Ropert, S.; Bouattour, M.; Desjardins, R.; Goldwasser, F.; Raymond, E. Pharmacokinetics and safety of DTS-108, a human oligopeptide bound to SN-38 with an esterase-sensitive cross-linker in patients with advanced malignancies: A Phase I study. Int. J. Nanomed. 2016, 11, 6207–6216. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Losic, F.; Nicolazzi, C.; Quinonero, J.; Ribes, F.; Michel, M.; Dubois, V.; de Coupade, C.; Boukaissi, M.; Chene, A.S.; Tranchant, I.; et al. DTS-108, a novel peptidic prodrug of SN38: In vivo efficacy and toxicokinetic studies. Clin. Cancer Res. 2008, 14, 2145–2153. [Google Scholar] [CrossRef] [PubMed]
- Schoffski, P.; Delord, J.P.; Brain, E.; Robert, J.; Dumez, H.; Gasmi, J.; Trouet, A. First-in-man phase I study assessing the safety and pharmacokinetics of a 1-h intravenous infusion of the doxorubicin prodrug DTS-201 every 3 weeks in patients with advanced or metastatic solid tumours. Eur. J. Cancer 2017, 86, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Ravel, D.; Dubois, V.; Quinonero, J.; Meyer-Losic, F.; Delord, J.; Rochaix, P.; Nicolazzi, C.; Ribes, F.; Mazerolles, C.; Assouly, E.; et al. Preclinical toxicity, toxicokinetics, and antitumoral efficacy studies of DTS-201, a tumor-selective peptidic prodrug of doxorubicin. Clin. Cancer Res. 2008, 14, 1258–1265. [Google Scholar] [CrossRef]
- Available online: https://www.clinicaltrialsregister.eu/ctr-search/search?query=DTS-201 (accessed on 22 April 2019).
- Available online: https://clinicaltrials.gov/ct2/show/NCT03486730 (accessed on 22 April 2019).
- Available online: https://clinicaltrials.gov/ct2/show/NCT03511664 (accessed on 22 April 2019).
- Gupte, A.A.; Pownall, H.J.; Hamilton, D.J. Estrogen: An emerging regulator of insulin action and mitochondrial function. J. Diabetes Res. 2015, 2015, 916585. [Google Scholar] [CrossRef]
- Mauvais-Jarvis, F. Estrogen and androgen receptors: Regulators of fuel homeostasis and emerging targets for diabetes and obesity. Trends Endocrinol. Metab. 2011, 22, 24–33. [Google Scholar] [CrossRef]
- Pereira, R.I.; Casey, B.A.; Swibas, T.A.; Erickson, C.B.; Wolfe, P.; Van Pelt, R.E. Timing of Estradiol Treatment After Menopause May Determine Benefit or Harm to Insulin Action. J. Clin. Endocrinol. Metab. 2015, 100, 4456–4462. [Google Scholar] [CrossRef] [PubMed]
- Bonds, D.E.; Lasser, N.; Qi, L.; Brzyski, R.; Caan, B.; Heiss, G.; Limacher, M.C.; Liu, J.H.; Mason, E.; Oberman, A.; et al. The effect of conjugated equine oestrogen on diabetes incidence: The Women’s Health Initiative randomised trial. Diabetologia 2006, 49, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Rossouw, J.E.; Anderson, G.L.; Prentice, R.L.; LaCroix, A.Z.; Kooperberg, C.; Stefanick, M.L.; Jackson, R.D.; Beresford, S.A.; Howard, B.V.; Johnson, K.C.; et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: Principal results From the Women’s Health Initiative randomized controlled trial. JAMA 2002, 288, 321–333. [Google Scholar] [PubMed]
- Kim, J.H.; Meyers, M.S.; Khuder, S.S.; Abdallah, S.L.; Muturi, H.T.; Russo, L.; Tate, C.R.; Hevener, A.L.; Najjar, S.M.; Leloup, C.; et al. Tissue-selective estrogen complexes with bazedoxifene prevent metabolic dysfunction in female mice. Mol. Metab. 2014, 3, 177–190. [Google Scholar] [CrossRef]
- Barrera, J.; Chambliss, K.L.; Ahmed, M.; Tanigaki, K.; Thompson, B.; McDonald, J.G.; Mineo, C.; Shaul, P.W. Bazedoxifene and conjugated estrogen prevent diet-induced obesity, hepatic steatosis, and type 2 diabetes in mice without impacting the reproductive tract. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E345–E354. [Google Scholar] [CrossRef] [PubMed]
- Hansdottir, H. Raloxifene for older women: A review of the literature. Clin. Interv. Aging 2008, 3, 45–50. [Google Scholar] [CrossRef]
- Barrett-Connor, E.; Mosca, L.; Collins, P.; Geiger, M.J.; Grady, D.; Kornitzer, M.; McNabb, M.A.; Wenger, N.K.; Investigators, R.U.f.T.H.R.T. Effects of raloxifene on cardiovascular events and breast cancer in postmenopausal women. N. Engl. J. Med. 2006, 355, 125–137. [Google Scholar] [CrossRef]
- Finan, B.; Yang, B.; Ottaway, N.; Stemmer, K.; Muller, T.D.; Yi, C.X.; Habegger, K.; Schriever, S.C.; Garcia-Caceres, C.; Kabra, D.G.; et al. Targeted estrogen delivery reverses the metabolic syndrome. Nat. Med. 2012, 18, 1847–1856. [Google Scholar] [CrossRef]
- Bullock, B.P.; Heller, R.S.; Habener, J.F. Tissue distribution of messenger ribonucleic acid encoding the rat glucagon-like peptide-1 receptor. Endocrinology 1996, 137, 2968–2978. [Google Scholar] [CrossRef]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000112164-GLP1R/tissue (accessed on 22 April 2019).
- Cao, X.; Xu, P.; Oyola, M.G.; Xia, Y.; Yan, X.; Saito, K.; Zou, F.; Wang, C.; Yang, Y.; Hinton, A., Jr.; et al. Estrogens stimulate serotonin neurons to inhibit binge-like eating in mice. J. Clin. Investig. 2014, 124, 4351–4362. [Google Scholar] [CrossRef]
- Vogel, H.; Wolf, S.; Rabasa, C.; Rodriguez-Pacheco, F.; Babaei, C.S.; Stober, F.; Goldschmidt, J.; DiMarchi, R.D.; Finan, B.; Tschop, M.H.; et al. GLP-1 and estrogen conjugate acts in the supramammillary nucleus to reduce food-reward and body weight. Neuropharmacology 2016, 110, 396–406. [Google Scholar] [CrossRef]
- Tiano, J.P.; Tate, C.R.; Yang, B.S.; DiMarchi, R.; Mauvais-Jarvis, F. Effect of targeted estrogen delivery using glucagon-like peptide-1 on insulin secretion, insulin sensitivity and glucose homeostasis. Sci. Rep. 2015, 5, 10211. [Google Scholar] [CrossRef]
- Schwenk, R.W.; Baumeier, C.; Finan, B.; Kluth, O.; Brauer, C.; Joost, H.G.; DiMarchi, R.D.; Tschop, M.H.; Schurmann, A. GLP-1-oestrogen attenuates hyperphagia and protects from beta cell failure in diabetes-prone New Zealand obese (NZO) mice. Diabetologia 2015, 58, 604–614. [Google Scholar] [CrossRef]
- Quarta, C.; Clemmensen, C.; Zhu, Z.; Yang, B.; Joseph, S.S.; Lutter, D.; Yi, C.X.; Graf, E.; Garcia-Caceres, C.; Legutko, B.; et al. Molecular Integration of Incretin and Glucocorticoid Action Reverses Immunometabolic Dysfunction and Obesity. Cell Metab. 2017, 26, 620–632 e626. [Google Scholar] [CrossRef] [PubMed]
- Grozinsky-Glasberg, S.; Fraser, A.; Nahshoni, E.; Weizman, A.; Leibovici, L. Thyroxine-triiodothyronine combination therapy versus thyroxine monotherapy for clinical hypothyroidism: Meta-analysis of randomized controlled trials. J. Clin. Endocrinol. Metab. 2006, 91, 2592–2599. [Google Scholar] [CrossRef]
- Mullur, R.; Liu, Y.Y.; Brent, G.A. Thyroid hormone regulation of metabolism. Physiol. Rev. 2014, 94, 355–382. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.A.; Singh, B.K.; Yen, P.M. Thyroid hormone regulation of hepatic lipid and carbohydrate metabolism. Trends Endocrinol. Metab. 2014, 25, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Ochs, N.; Auer, R.; Bauer, D.C.; Nanchen, D.; Gussekloo, J.; Cornuz, J.; Rodondi, N. Meta-analysis: Subclinical thyroid dysfunction and the risk for coronary heart disease and mortality. Ann. Intern. Med. 2008, 148, 832–845. [Google Scholar] [CrossRef] [PubMed]
- Baxter, J.D.; Webb, P. Thyroid hormone mimetics: Potential applications in atherosclerosis, obesity and type 2 diabetes. Nat. Rev. Drug Discov. 2009, 8, 308–320. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.D.; Finan, B.; Clemmensen, C.; DiMarchi, R.D.; Tschop, M.H. The New Biology and Pharmacology of Glucagon. Physiol. Rev. 2017, 97, 721–766. [Google Scholar] [CrossRef]
- Finan, B.; Clemmensen, C.; Zhu, Z.; Stemmer, K.; Gauthier, K.; Muller, L.; De Angelis, M.; Moreth, K.; Neff, F.; Perez-Tilve, D.; et al. Chemical Hybridization of Glucagon and Thyroid Hormone Optimizes Therapeutic Impact for Metabolic Disease. Cell 2016, 167, 843–857 e814. [Google Scholar] [CrossRef]
- Cox, D.; Brennan, M.; Moran, N. Integrins as therapeutic targets: Lessons and opportunities. Nat. Rev. Drug Discov. 2010, 9, 804–820. [Google Scholar] [CrossRef]
- Rathinam, R.; Alahari, S.K. Important role of integrins in the cancer biology. Cancer Metastasis Rev. 2010, 29, 223–237. [Google Scholar] [CrossRef]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Rivera-Nieves, J.; Sandborn, W.J.; Shattil, S. Integrin-based therapeutics: Biological basis, clinical use and new drugs. Nat. Rev. Drug Discov. 2016, 15, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Cox, N.; Kintzing, J.R.; Smith, M.; Grant, G.A.; Cochran, J.R. Integrin-Targeting Knottin Peptide-Drug Conjugates Are Potent Inhibitors of Tumor Cell Proliferation. Angew. Chem. 2016, 55, 9894–9897. [Google Scholar] [CrossRef] [PubMed]
- Toschi, L.; Finocchiaro, G.; Bartolini, S.; Gioia, V.; Cappuzzo, F. Role of gemcitabine in cancer therapy. Future Oncol. 2005, 1, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Noble, S.; Goa, K.L. Gemcitabine. A review of its pharmacology and clinical potential in non-small cell lung cancer and pancreatic cancer. Drugs 1997, 54, 447–472. [Google Scholar] [CrossRef]
- Karampelas, T.; Skavatsou, E.; Argyros, O.; Fokas, D.; Tamvakopoulos, C. Gemcitabine Based Peptide Conjugate with Improved Metabolic Properties and Dual Mode of Efficacy. Mol. Pharm. 2017, 14, 674–685. [Google Scholar] [CrossRef]
- Jain, N.; Smith, S.W.; Ghone, S.; Tomczuk, B. Current ADC Linker Chemistry. Pharm. Res. 2015, 32, 3526–3540. [Google Scholar] [CrossRef]
- Lu, J.; Jiang, F.; Lu, A.; Zhang, G. Linkers Having a Crucial Role in Antibody-Drug Conjugates. Int. J. Mol. Sci. 2016, 17, 561. [Google Scholar] [CrossRef]
- McCombs, J.R.; Owen, S.C. Antibody drug conjugates: Design and selection of linker, payload and conjugation chemistry. Aaps J. 2015, 17, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Gebleux, R.; Casi, G. Antibody-drug conjugates: Current status and future perspectives. Pharm. Ther. 2016, 167, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.G.; Kim, K.M. Strategies and Advancement in Antibody-Drug Conjugate Optimization for Targeted Cancer Therapeutics. Biomol. Ther. (Seoul) 2015, 23, 493–509. [Google Scholar] [CrossRef] [PubMed]
- Chari, R.V.; Miller, M.L.; Widdison, W.C. Antibody-drug conjugates: An emerging concept in cancer therapy. Angew. Chem. 2014, 53, 3796–3827. [Google Scholar] [CrossRef]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2018, 9, 33–46. [Google Scholar] [CrossRef]
- Doronina, S.O.; Bovee, T.D.; Meyer, D.W.; Miyamoto, J.B.; Anderson, M.E.; Morris-Tilden, C.A.; Senter, P.D. Novel peptide linkers for highly potent antibody-auristatin conjugate. Bioconj. Chem. 2008, 19, 1960–1963. [Google Scholar] [CrossRef] [PubMed]
- Staben, L.R.; Koenig, S.G.; Lehar, S.M.; Vandlen, R.; Zhang, D.; Chuh, J.; Yu, S.F.; Ng, C.; Guo, J.; Liu, Y.; et al. Targeted drug delivery through the traceless release of tertiary and heteroaryl amines from antibody-drug conjugates. Nat. Chem. 2016, 8, 1112–1119. [Google Scholar] [CrossRef]
- Anami, Y.; Yamazaki, C.M.; Xiong, W.; Gui, X.; Zhang, N.; An, Z.; Tsuchikama, K. Glutamic acid-valine-citrulline linkers ensure stability and efficacy of antibody-drug conjugates in mice. Nat. Commun. 2018, 9, 2512. [Google Scholar] [CrossRef]
- Jeffrey, S.C.; Andreyka, J.B.; Bernhardt, S.X.; Kissler, K.M.; Kline, T.; Lenox, J.S.; Moser, R.F.; Nguyen, M.T.; Okeley, N.M.; Stone, I.J.; et al. Development and properties of beta-glucuronide linkers for monoclonal antibody-drug conjugates. Bioconj. Chem. 2006, 17, 831–840. [Google Scholar] [CrossRef]
- Burke, P.J.; Senter, P.D.; Meyer, D.W.; Miyamoto, J.B.; Anderson, M.; Toki, B.E.; Manikumar, G.; Wani, M.C.; Kroll, D.J.; Jeffrey, S.C. Design, synthesis, and biological evaluation of antibody-drug conjugates comprised of potent camptothecin analogues. Bioconj. Chem. 2009, 20, 1242–1250. [Google Scholar] [CrossRef]
- Gunnoo, S.B.; Madder, A. Chemical Protein Modification through Cysteine. ChemBioChem 2016, 17, 529–553. [Google Scholar] [CrossRef]
- Gunnoo, S.B.; Madder, A. Bioconjugation—Using selective chemistry to enhance the properties of proteins and peptides as therapeutics and carriers. Org. Biomol. Chem. 2016, 14, 8002–8013. [Google Scholar] [CrossRef]
- Su, D.; Kozak, K.R.; Sadowsky, J.; Yu, S.F.; Fourie-O’Donohue, A.; Nelson, C.; Vandlen, R.; Ohri, R.; Liu, L.; Ng, C.; et al. Modulating Antibody-Drug Conjugate Payload Metabolism by Conjugation Site and Linker Modification. Bioconj. Chem. 2018, 29, 1155–1167. [Google Scholar] [CrossRef]
- Spicer, C.D.; Davis, B.G. Selective chemical protein modification. Nat. Commun. 2014, 5, 4740. [Google Scholar] [CrossRef]
- Chudasama, V.; Maruani, A.; Caddick, S. Recent advances in the construction of antibody-drug conjugates. Nat. Chem. 2016, 8, 114–119. [Google Scholar] [CrossRef]
- Schumacher, D.; Hackenberger, C.P.; Leonhardt, H.; Helma, J. Current Status: Site-Specific Antibody Drug Conjugates. J. Clin. Immunol. 2016, 36 (Suppl. 1), 100–107. [Google Scholar] [CrossRef]
- Sochaj, A.M.; Swiderska, K.W.; Otlewski, J. Current methods for the synthesis of homogeneous antibody-drug conjugates. Biotechnol. Adv. 2015, 33, 775–784. [Google Scholar] [CrossRef]
- Calce, E.; Leone, M.; Monfregola, L.; De Luca, S. Chemical modifications of peptide sequences via S-alkylation reaction. Org. Lett. 2013, 15, 5354–5357. [Google Scholar] [CrossRef]
- Lu, Y.; Huang, F.; Wang, J.; Xia, J. Affinity-guided covalent conjugation reactions based on PDZ-peptide and SH3-peptide interactions. Bioconj. Chem. 2014, 25, 989–999. [Google Scholar] [CrossRef]
- Calce, E.; Leone, M.; Mercurio, F.A.; Monfregola, L.; De Luca, S. Solid-Phase S-Alkylation Promoted by Molecular Sieves. Org. Lett. 2015, 17, 5646–5649. [Google Scholar] [CrossRef]
- Doan, N.D.; Bourgault, S.; Letourneau, M.; Fournier, A. Effectiveness of the suzuki-miyaura cross-coupling reaction for solid-phase peptide modification. J. Comb. Chem. 2008, 10, 44–51. [Google Scholar] [CrossRef]
- Afonso, A.; Feliu, L.; Planas, M. Solid-phase synthesis of biaryl cyclic peptides by borylation and microwave-assisted intramolecular Suzuki–Miyaura reaction. Tetrahedron 2011, 67, 2238–2245. [Google Scholar] [CrossRef]
- Silvestri, A.P.; Cistrone, P.A.; Dawson, P.E. Adapting the Glaser Reaction for Bioconjugation: Robust Access to Structurally Simple, Rigid Linkers. Angew. Chem. 2017, 56, 10438–10442. [Google Scholar] [CrossRef] [PubMed]
- Pagel, M.; Meier, R.; Braun, K.; Wiessler, M.; Beck-Sickinger, A.G. On-resin Diels-Alder reaction with inverse electron demand: An efficient ligation method for complex peptides with a varying spacer to optimize cell adhesion. Org. Biomol. Chem. 2016, 14, 4809–4816. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, S.; Spring, D.R. C-H activation: Complex peptides made simple. Nat. Chem. 2016, 9, 9–10. [Google Scholar] [CrossRef][Green Version]
- Oriana, S.; Cai, Y.; Bode, J.W.; Yamakoshi, Y. Synthesis of tri-functionalized MMP2 FRET probes using a chemo-selective and late-stage modification of unprotected peptides. Org. Biomol. Chem. 2017, 15, 1792–1800. [Google Scholar] [CrossRef]
- Decostaire, I.E.; Lelievre, D.; Aucagne, V.; Delmas, A.F. Solid phase oxime ligations for the iterative synthesis of polypeptide conjugates. Org. Biomol. Chem. 2014, 12, 5536–5543. [Google Scholar] [CrossRef] [PubMed]
- VanBrunt, M.P.; Shanebeck, K.; Caldwell, Z.; Johnson, J.; Thompson, P.; Martin, T.; Dong, H.; Li, G.; Xu, H.; D’Hooge, F.; et al. Genetically Encoded Azide Containing Amino Acid in Mammalian Cells Enables Site-Specific Antibody-Drug Conjugates Using Click Cycloaddition Chemistry. Bioconj. Chem. 2015, 26, 2249–2260. [Google Scholar] [CrossRef]
- Presolski, S.I.; Hong, V.P.; Finn, M.G. Copper-Catalyzed Azide-Alkyne Click Chemistry for Bioconjugation. Curr. Protoc. Chem. Biol. 2011, 3, 153–162. [Google Scholar] [CrossRef]
- El-Faham, A.; Albericio, F. Peptide coupling reagents, more than a letter soup. Chem. Rev. 2011, 111, 6557–6602. [Google Scholar] [CrossRef]
- Li, D.; Elbert, D.L. The kinetics of the removal of the N-methyltrityl (Mtt) group during the synthesis of branched peptides. J. Pept. Res. 2002, 60, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Koniev, O.; Wagner, A. Developments and recent advancements in the field of endogenous amino acid selective bond forming reactions for bioconjugation. Chem. Soc. Rev. 2015, 44, 5495–5551. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.W.; Callahan, F.M.; Zimmerman, J.E. Synthesis of N-hydroxysuccinimide esters of acyl peptides by the mixed anhydride method. J. Am. Chem. Soc. 1967, 89, 178. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, Y.; Rappoport, S.; Wolman, Y. Use of esters of N-hydroxysuccinimide in the synthesis of N-acylamino acids. J. Lipid Res. 1967, 8, 142–145. [Google Scholar] [PubMed]
- Nagy, A.; Schally, A.V.; Armatis, P.; Szepeshazi, K.; Halmos, G.; Kovacs, M.; Zarandi, M.; Groot, K.; Miyazaki, M.; Jungwirth, A.; et al. Cytotoxic analogs of luteinizing hormone-releasing hormone containing doxorubicin or 2-pyrrolinodoxorubicin, a derivative 500–1000 times more potent. Proc. Natl. Acad. Sci. USA 1996, 93, 7269–7273. [Google Scholar] [CrossRef]
- White, C.J.; Yudin, A.K. Contemporary strategies for peptide macrocyclization. Nat. Chem. 2011, 3, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Basle, E.; Joubert, N.; Pucheault, M. Protein chemical modification on endogenous amino acids. Chem. Biol. 2010, 17, 213–227. [Google Scholar] [CrossRef]
- Geng, Q.; Sun, X.; Gong, T.; Zhang, Z.R. Peptide-drug conjugate linked via a disulfide bond for kidney targeted drug delivery. Bioconj. Chem. 2012, 23, 1200–1210. [Google Scholar] [CrossRef]
- Song, Q.; Chuan, X.; Chen, B.; He, B.; Zhang, H.; Dai, W.; Wang, X.; Zhang, Q. A smart tumor targeting peptide-drug conjugate, pHLIP-SS-DOX: Synthesis and cellular uptake on MCF-7 and MCF-7/Adr cells. Drug Deliv. 2016, 23, 1734–1746. [Google Scholar] [CrossRef]
- Witt, D. Recent developments in disulfide bond formation. Synthesis 2008, 16, 2491–2509. [Google Scholar] [CrossRef]
- Mandal, B.; Basu, B. Recent advances in S–S bond formation. Rsc Adv. 2014, 4, 13854–13881. [Google Scholar] [CrossRef]
- Le Guern, F.; Ouk, T.S.; Ouk, C.; Vanderesse, R.; Champavier, Y.; Pinault, E.; Sol, V. Lysine Analogue of Polymyxin B as a Significant Opportunity for Photodynamic Antimicrobial Chemotherapy. ACS Med. Chem. Lett. 2018, 9, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Torres, O.B.; Matyas, G.R.; Rao, M.; Peachman, K.K.; Jalah, R.; Beck, Z.; Michael, N.L.; Rice, K.C.; Jacobson, A.E.; Alving, C.R. Heroin-HIV-1 (H2) vaccine: Induction of dual immunologic effects with a heroin hapten-conjugate and an HIV-1 envelope V2 peptide with liposomal lipid A as an adjuvant. Npj Vaccines 2017, 2, 13. [Google Scholar] [CrossRef]
- Jalah, R.; Torres, O.B.; Mayorov, A.V.; Li, F.; Antoline, J.F.; Jacobson, A.E.; Rice, K.C.; Deschamps, J.R.; Beck, Z.; Alving, C.R.; et al. Efficacy, but not antibody titer or affinity, of a heroin hapten conjugate vaccine correlates with increasing hapten densities on tetanus toxoid, but not on CRM197 carriers. Bioconj. Chem. 2015, 26, 1041–1053. [Google Scholar] [CrossRef]
- Ponte, J.F.; Sun, X.; Yoder, N.C.; Fishkin, N.; Laleau, R.; Coccia, J.; Lanieri, L.; Bogalhas, M.; Wang, L.; Wilhelm, S.; et al. Understanding How the Stability of the Thiol-Maleimide Linkage Impacts the Pharmacokinetics of Lysine-Linked Antibody-Maytansinoid Conjugates. Bioconj. Chem. 2016, 27, 1588–1598. [Google Scholar] [CrossRef]
- Dovgan, I.; Kolodych, S.; Koniev, O.; Wagner, A. 2-(Maleimidomethyl)-1,3-Dioxanes (MD): A Serum-Stable Self-hydrolysable Hydrophilic Alternative to Classical Maleimide Conjugation. Sci. Rep. 2016, 6, 30835. [Google Scholar] [CrossRef]
- Fontaine, S.D.; Reid, R.; Robinson, L.; Ashley, G.W.; Santi, D.V. Long-term stabilization of maleimide-thiol conjugates. Bioconj. Chem. 2015, 26, 145–152. [Google Scholar] [CrossRef]
- Lyon, R.P.; Setter, J.R.; Bovee, T.D.; Doronina, S.O.; Hunter, J.H.; Anderson, M.E.; Balasubramanian, C.L.; Duniho, S.M.; Leiske, C.I.; Li, F.; et al. Self-hydrolyzing maleimides improve the stability and pharmacological properties of antibody-drug conjugates. Nat. Biotechnol. 2014, 32, 1059–1062. [Google Scholar] [CrossRef]
- Schumacher, F.F.; Nunes, J.P.; Maruani, A.; Chudasama, V.; Smith, M.E.; Chester, K.A.; Baker, J.R.; Caddick, S. Next generation maleimides enable the controlled assembly of antibody-drug conjugates via native disulfide bond bridging. Org. Biomol. Chem. 2014, 12, 7261–7269. [Google Scholar] [CrossRef]
- Maruani, A.; Smith, M.E.; Miranda, E.; Chester, K.A.; Chudasama, V.; Caddick, S. A plug-and-play approach to antibody-based therapeutics via a chemoselective dual click strategy. Nat. Commun. 2015, 6, 6645. [Google Scholar] [CrossRef]
- Behrens, C.R.; Ha, E.H.; Chinn, L.L.; Bowers, S.; Probst, G.; Fitch-Bruhns, M.; Monteon, J.; Valdiosera, A.; Bermudez, A.; Liao-Chan, S.; et al. Antibody-Drug Conjugates (ADCs) Derived from Interchain Cysteine Cross-Linking Demonstrate Improved Homogeneity and Other Pharmacological Properties over Conventional Heterogeneous ADCs. Mol. Pharm. 2015, 12, 3986–3998. [Google Scholar] [CrossRef]
- Anderson, R.J.; Li, J.; Kedzierski, L.; Compton, B.J.; Hayman, C.M.; Osmond, T.L.; Tang, C.W.; Farrand, K.J.; Koay, H.F.; Almeida, C.; et al. Augmenting Influenza-Specific T Cell Memory Generation with a Natural Killer T Cell-Dependent Glycolipid-Peptide Vaccine. ACS Chem. Biol. 2017, 12, 2898–2905. [Google Scholar] [CrossRef]
- Speir, M.; Authier-Hall, A.; Brooks, C.R.; Farrand, K.J.; Compton, B.J.; Anderson, R.J.; Heiser, A.; Osmond, T.L.; Tang, C.W.; Berzofsky, J.A.; et al. Glycolipid-peptide conjugate vaccines enhance CD8(+) T cell responses against human viral proteins. Sci. Rep. 2017, 7, 14273. [Google Scholar] [CrossRef]
- Barile, E.; Wang, S.; Das, S.K.; Noberini, R.; Dahl, R.; Stebbins, J.L.; Pasquale, E.B.; Fisher, P.B.; Pellecchia, M. Design, synthesis and bioevaluation of an EphA2 receptor-based targeted delivery system. ChemMedChem 2014, 9, 1403–1412. [Google Scholar] [CrossRef]
- Wang, S.; Placzek, W.J.; Stebbins, J.L.; Mitra, S.; Noberini, R.; Koolpe, M.; Zhang, Z.; Dahl, R.; Pasquale, E.B.; Pellecchia, M. Novel targeted system to deliver chemotherapeutic drugs to EphA2-expressing cancer cells. J. Med. Chem. 2012, 55, 2427–2436. [Google Scholar] [CrossRef]
- Bonaccorso, R.L.; Chepurny, O.G.; Becker-Pauly, C.; Holz, G.G.; Doyle, R.P. Enhanced Peptide Stability Against Protease Digestion Induced by Intrinsic Factor Binding of a Vitamin B12 Conjugate of Exendin-4. Mol. Pharm. 2015, 12, 3502–3506. [Google Scholar] [CrossRef]
Sample Availability: To acquire samples of the compounds, please direct attention to primary publications and authors. |














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Generic Name | Indication | Peptide | Drug | Linker | Mechanism | Status | Reference |
|---|---|---|---|---|---|---|---|
| Lu177-dotatate | Dastroenteropancreatic neuroendocrine tumors | Somatostatin analogue Octreotide | Radio therapeutic agent Lu177 | Amide (Lu177 chelating to metalchelating molecule DOTA) | Somatostatin receptor 2 SSTR2 mediated delivery of nucleotide 177Lu | Approved by FDA and EMA | [76,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99] |
| [111In-DTPA-D-Phe1]-octreotide | Imaging/diagnostic | Somatostatin analogue Octreotide | Radio therapeutic agent 111In | Amide (111In chelating to metalchelating molecule DOTA) | Somatostatin receptor 2 SSTR2 mediated tumor scintigraphic imaging | Phase 1 completed | [136,137] |
| Zoptarelin Doxorubicin, AN-152, AEZS-108 | Endometrial cancer Ovarian cancer | GnRH/LHRH | Doxorubicin | Ester | GnRH mediated delivery of doxorubicin to cancer cells | Phase 3 completed | [96,98,99,100,101,102,103] |
| ANG1005 GRN1005 | Metastases brain cancer | Angiopep-2 | Paclitaxel | Ester | Low-density lipoprotein receptor-related protein 1 (LPR1) mediated brain uptake | Orphan drug for glioblastoma multiform, Several phase 2 ongoing | [109,110,111,112,113,114,115,116] |
| Mipsagargin G202 | Various Cancer | Tetrapeptide | Thapsigargin | Ester | Extracellularly tumor-activated prodrug of Thapsigargin | Phase 2 completed | [118,119,120,123] |
| Paclitaxel poliglumex CT2103 | Various cancer | Poliglumex | Paclitaxel | Ester | Enhanced permeability of tumor vasculature and lack of lymphatic drainage prolonged tumor exposure to the active drug while minimizing systemic exposure | Phase 3 completed | [138,139,140,141,142] |
| EP-100 | Cancer | GnRH/LHRH | CLIP71 | Amide | GnRH receptor-mediated cancer cell membrane lysis | Phase 2 completed | [143,144,145] |
| BIM-23A760 | Pituitary adenomas | Somatostatin | Dopamine | Amide/Thioether | Somatostatin/dopaminethe dual action inhibit the expression/secretion of several pituitary hormones (especially GH/PRL) | Phase 2 terminated | [133,134,135] |
| CGC 1072 | Psoriasis | Heptaarginine | Cyclosporin A | Ester | CPP mediated topical delivery and inhibition of inflammation | Phase 2, discontinued | [146,147] |
| KAI-1455 | Ischemic organ injury | TAT47-57 | εPKC activator | Disulfide | CPP mediated εPKC activator delivery | Phase 1 | [148] |
| KAI-1678 | Neuropathic and inflammatory pain | TAT47-57 | δ-Protein kinase C inhibitor peptide | Disulfide | CPP mediated εPKC inhibitor delivery | Phase 2 completed | [149,150,151] |
| KAI-9803 | Myocardial infarction & Cardiovascular disease | TAT47-57 | δ-Protein kinase C inhibitor peptide | Disulfide | CPP mediated εPKC inhibitor delivery to reduce the injury to myocardial and endothelial cells during a heart attack | Phase 2 completed | [152,153,154] |
| XG-102 | Post-cataract surgery, intraocular inflammation and Pain | Tat48-57 | 31-mer peptide JNK inhibitor | Disulfide | CPP mediated JNK inhibitor delivery to reduce pain and inflammation upon cataract surgery | Phase 3 completed | [155,156,157,158] |
| DTS-108 | Cancer | DPV1047 Vectocell peptide | SN38 | Ester | CPP DPV1047 mediated delivery of chemotherapeutic drug SN38 | Phase 1 completed | [159,160] |
| DTS-201 | Cancer | Tetra peptide | Doxorubicin | Amide | Extracellularly tumor-activated prodrug of doxorubicin | Phase 2 completed | [161,162,163] |
| BT-1718 | Cancer | Bicyclic peptide | Maytansinoid | Disulfide | Membrane type 1-matrixmetalloprotease mediated toxin delivery | Phase 1 | [164] |
| 177Lu- PSMA-617 | Cancer | Glutamate-urea-lysine | Radio therapeutic agent Lu177 | Amide (Lu177 chelating to metalchelating molecule DOTA) | Prostate-specific membrane antigen (PSMA) mediated delivery of nucleotide 177Lu | Phase 3 | [165] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, R.; Finan, B.; Mayer, J.P.; DiMarchi, R.D. Peptide Conjugates with Small Molecules Designed to Enhance Efficacy and Safety. Molecules 2019, 24, 1855. https://doi.org/10.3390/molecules24101855
He R, Finan B, Mayer JP, DiMarchi RD. Peptide Conjugates with Small Molecules Designed to Enhance Efficacy and Safety. Molecules. 2019; 24(10):1855. https://doi.org/10.3390/molecules24101855
Chicago/Turabian StyleHe, Rongjun, Brian Finan, John P. Mayer, and Richard D. DiMarchi. 2019. "Peptide Conjugates with Small Molecules Designed to Enhance Efficacy and Safety" Molecules 24, no. 10: 1855. https://doi.org/10.3390/molecules24101855
APA StyleHe, R., Finan, B., Mayer, J. P., & DiMarchi, R. D. (2019). Peptide Conjugates with Small Molecules Designed to Enhance Efficacy and Safety. Molecules, 24(10), 1855. https://doi.org/10.3390/molecules24101855