Full-Dimensional Ab Initio Potential Energy Surface and Vibrational Energy Levels of Li2H


Abstract
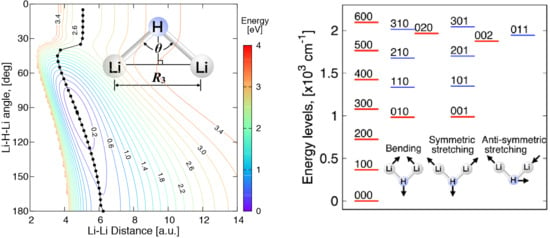
:
1. Introduction
2. Calculation Details
2.1. Ab Initio Calculations
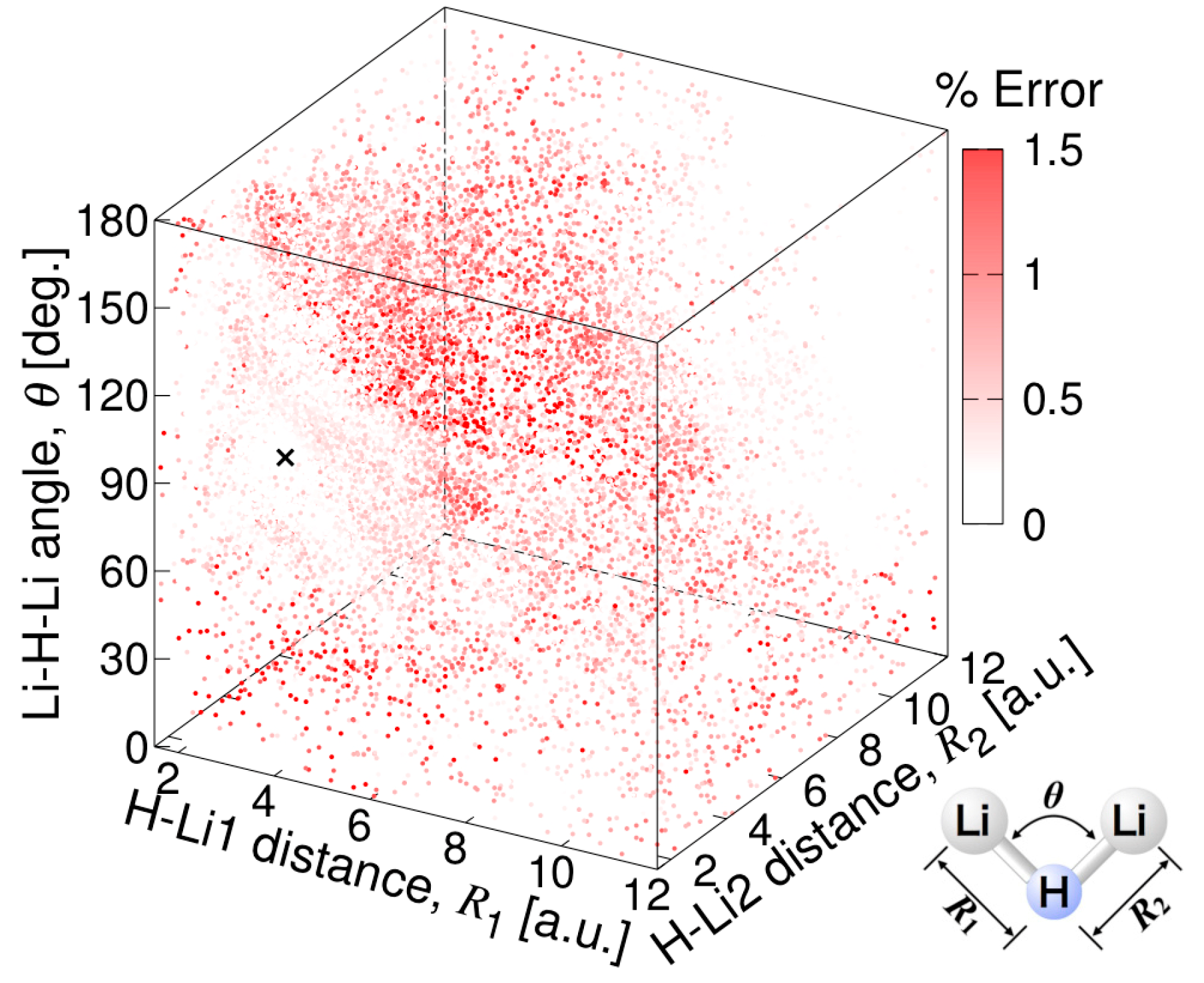
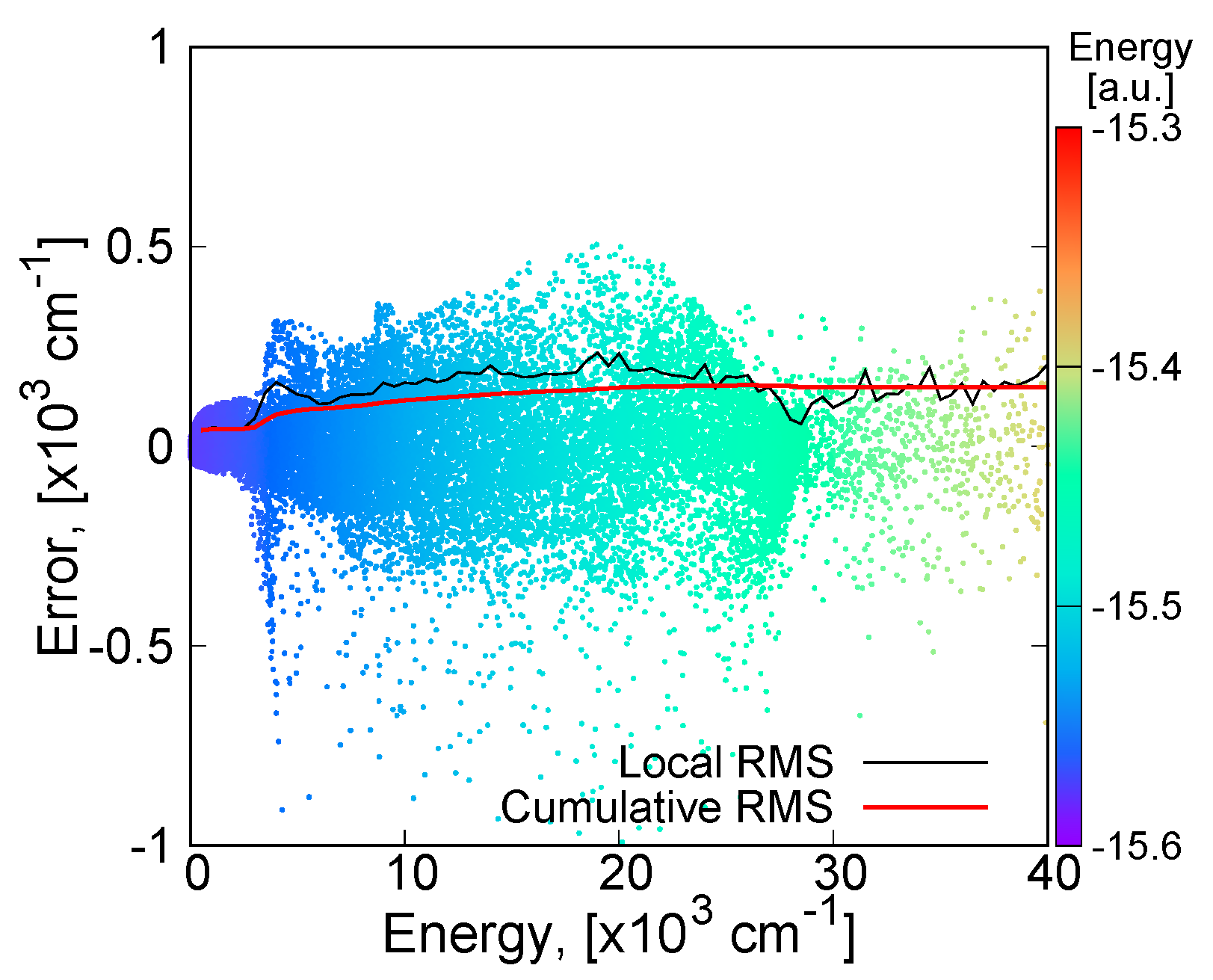
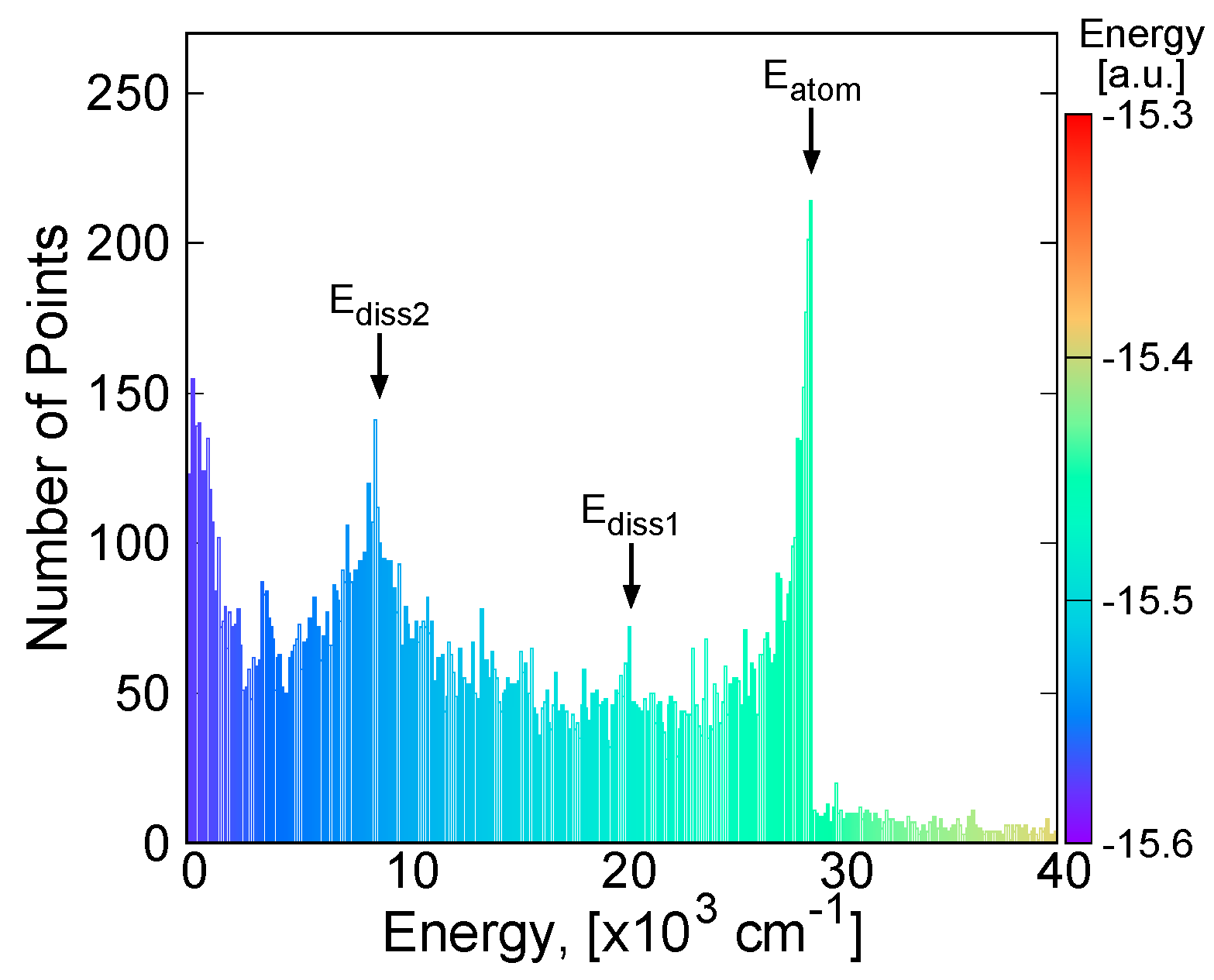
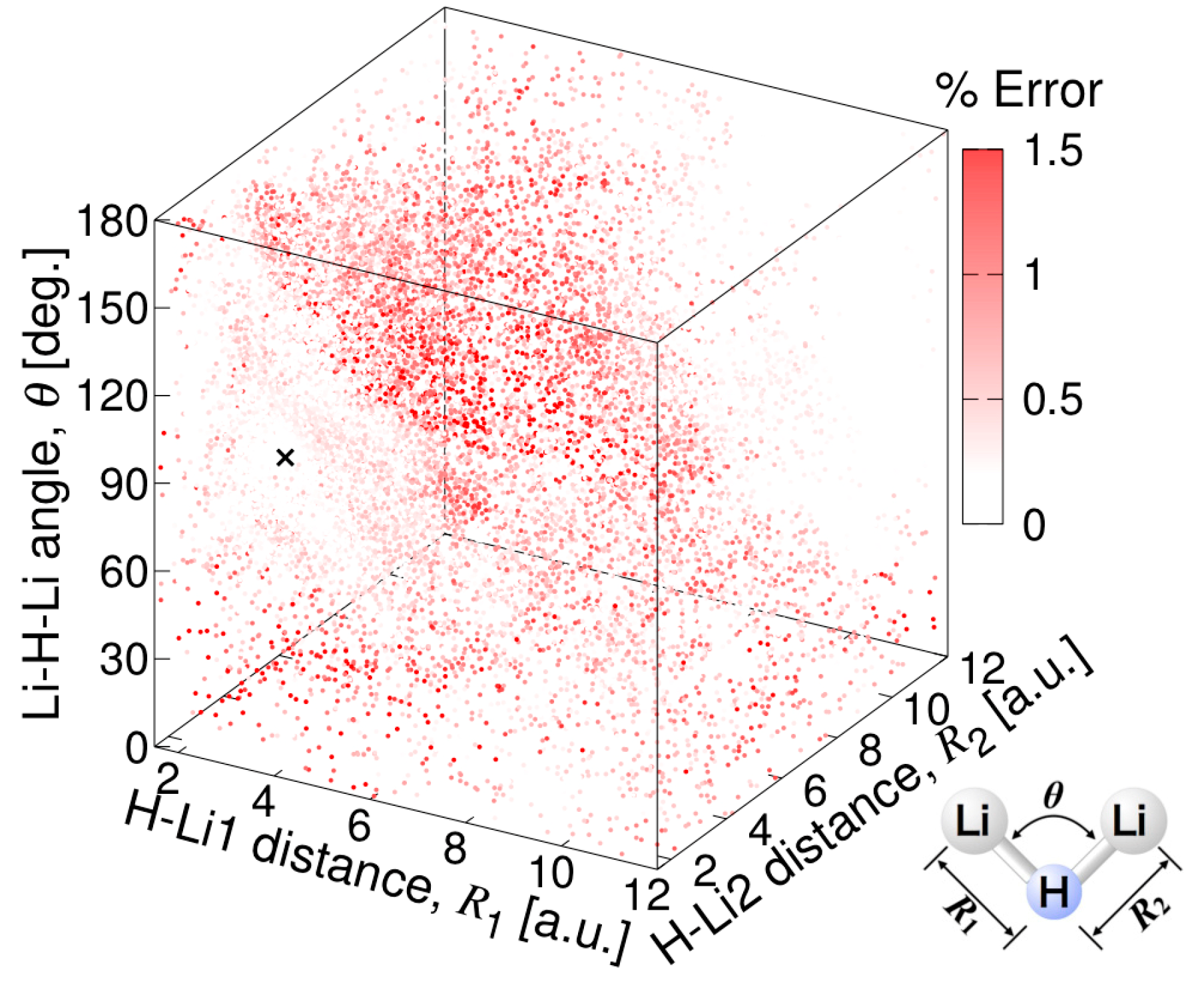
2.2. Potential Energy Surface Fitting
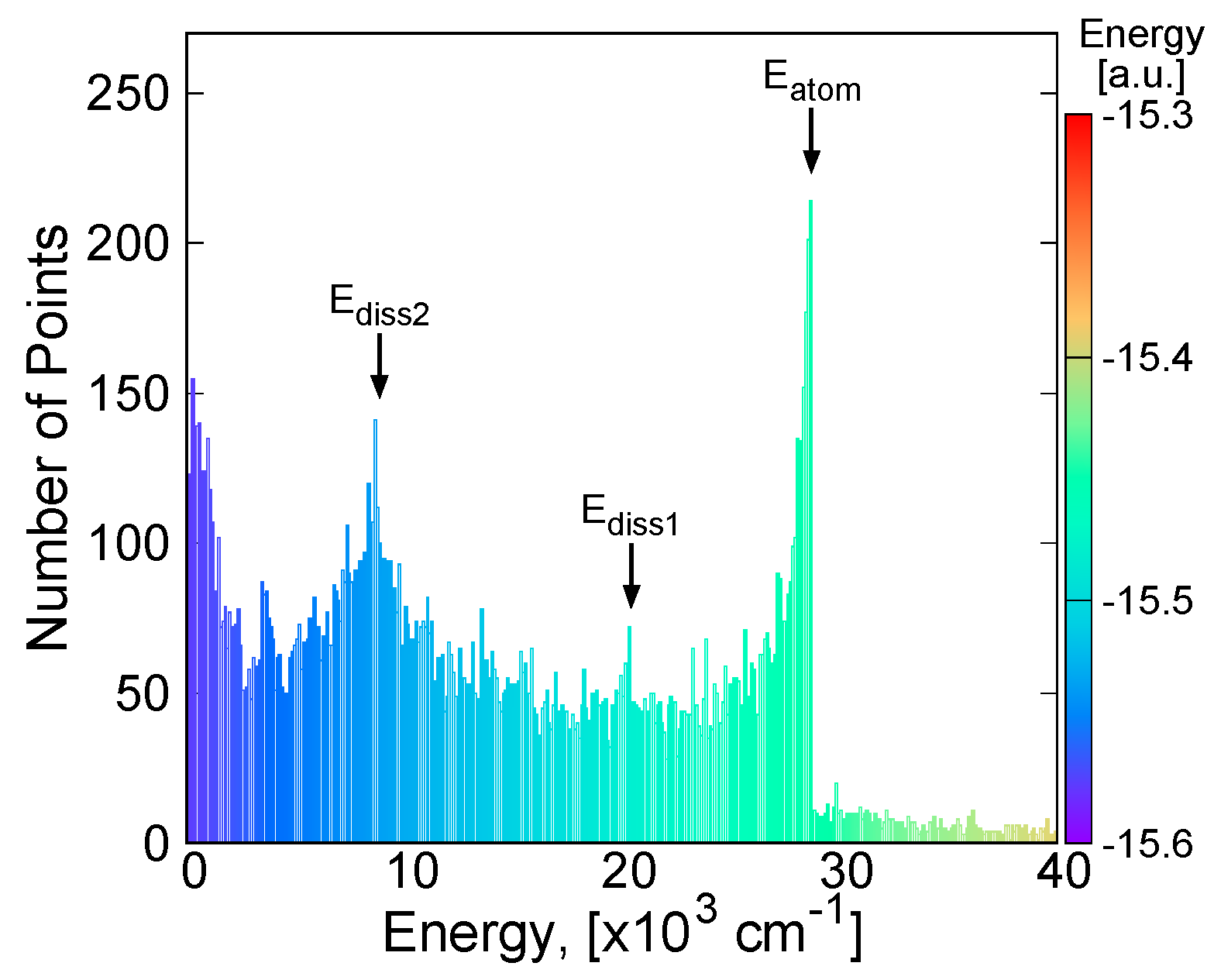
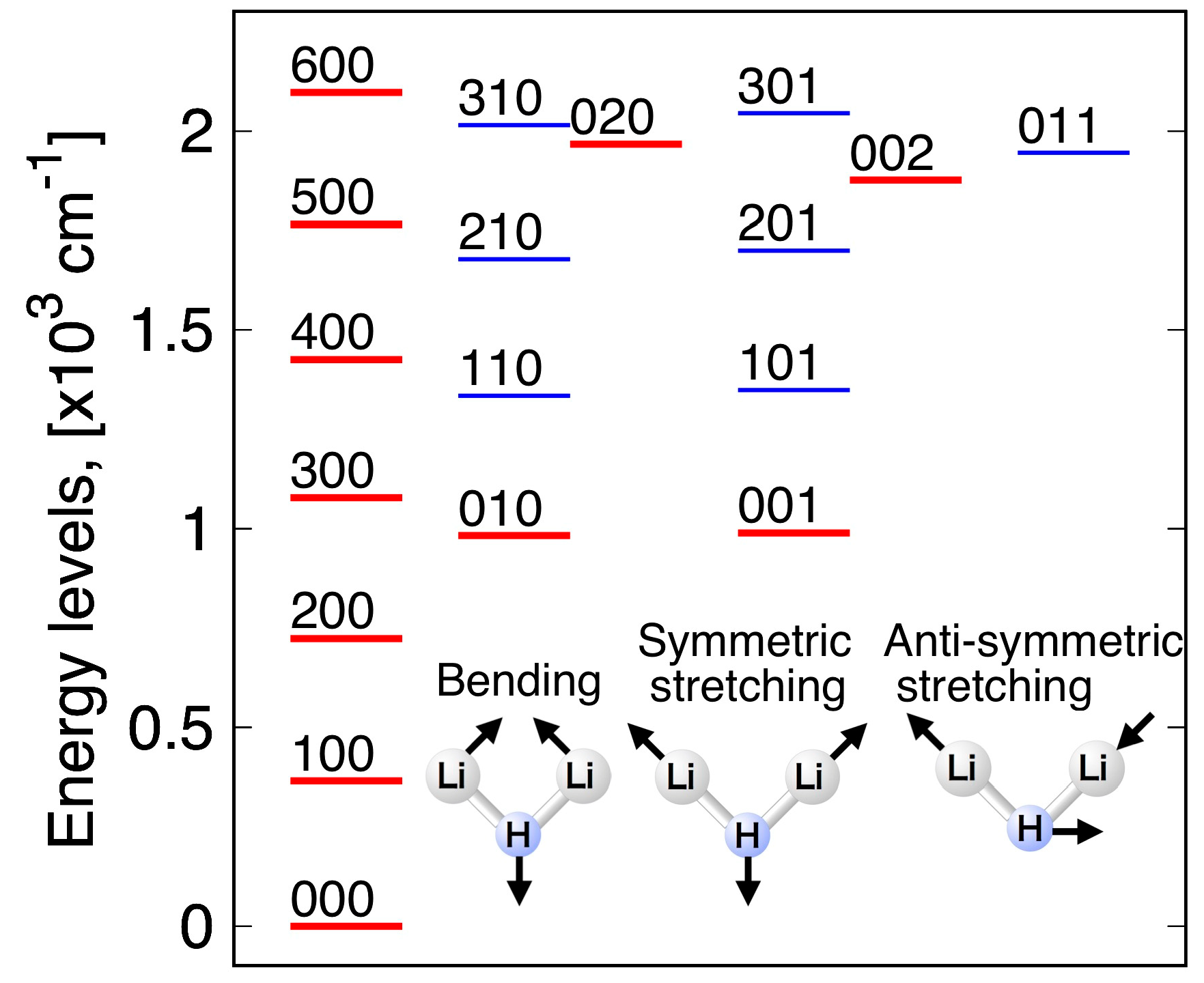
2.3. Vibrational Energy Calculation
3. Results and Discussion
3.1. Ab Initio Calculations for Atomic and Diatomic Species
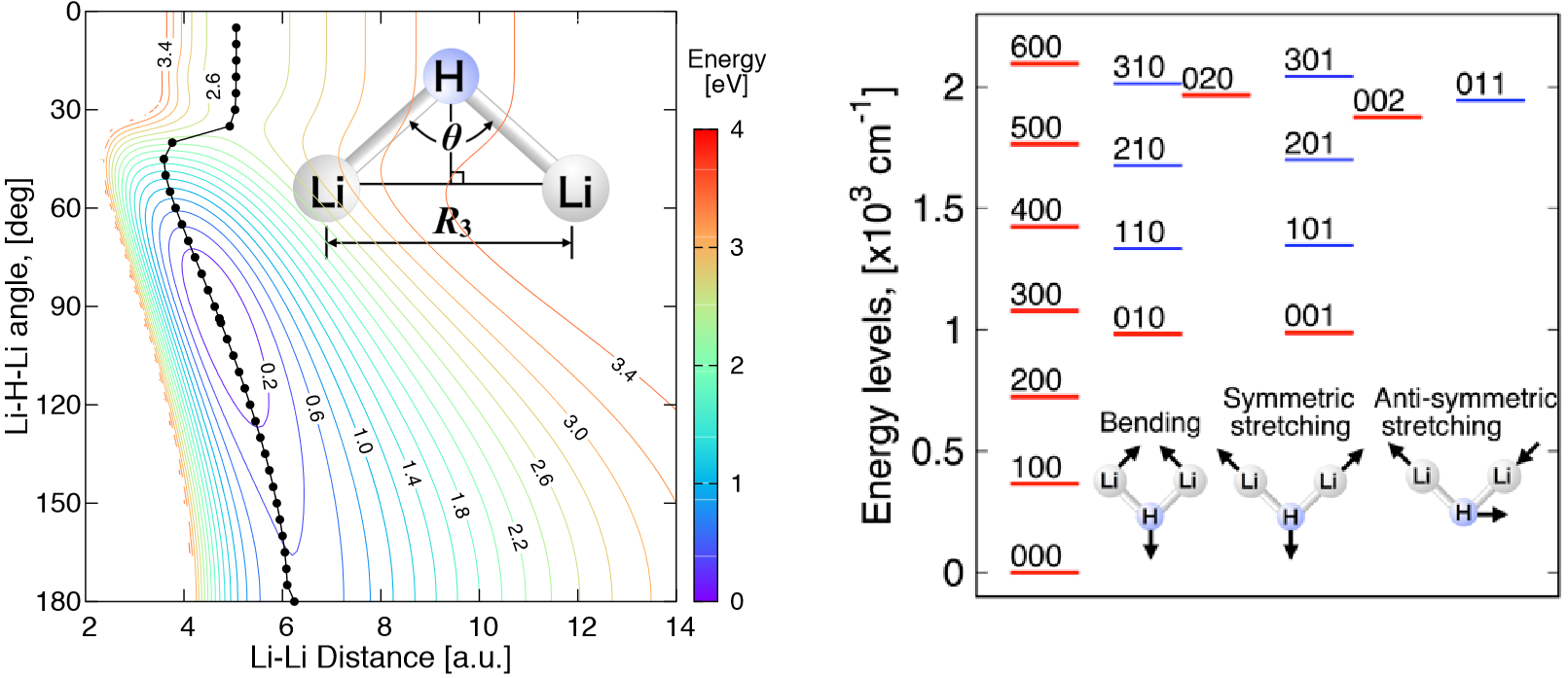
3.2. Ab Initio Calculations for the Li2H Triatomic Complex
3.3. Global Analytical PES of the Ground 2A1 State of Li2H
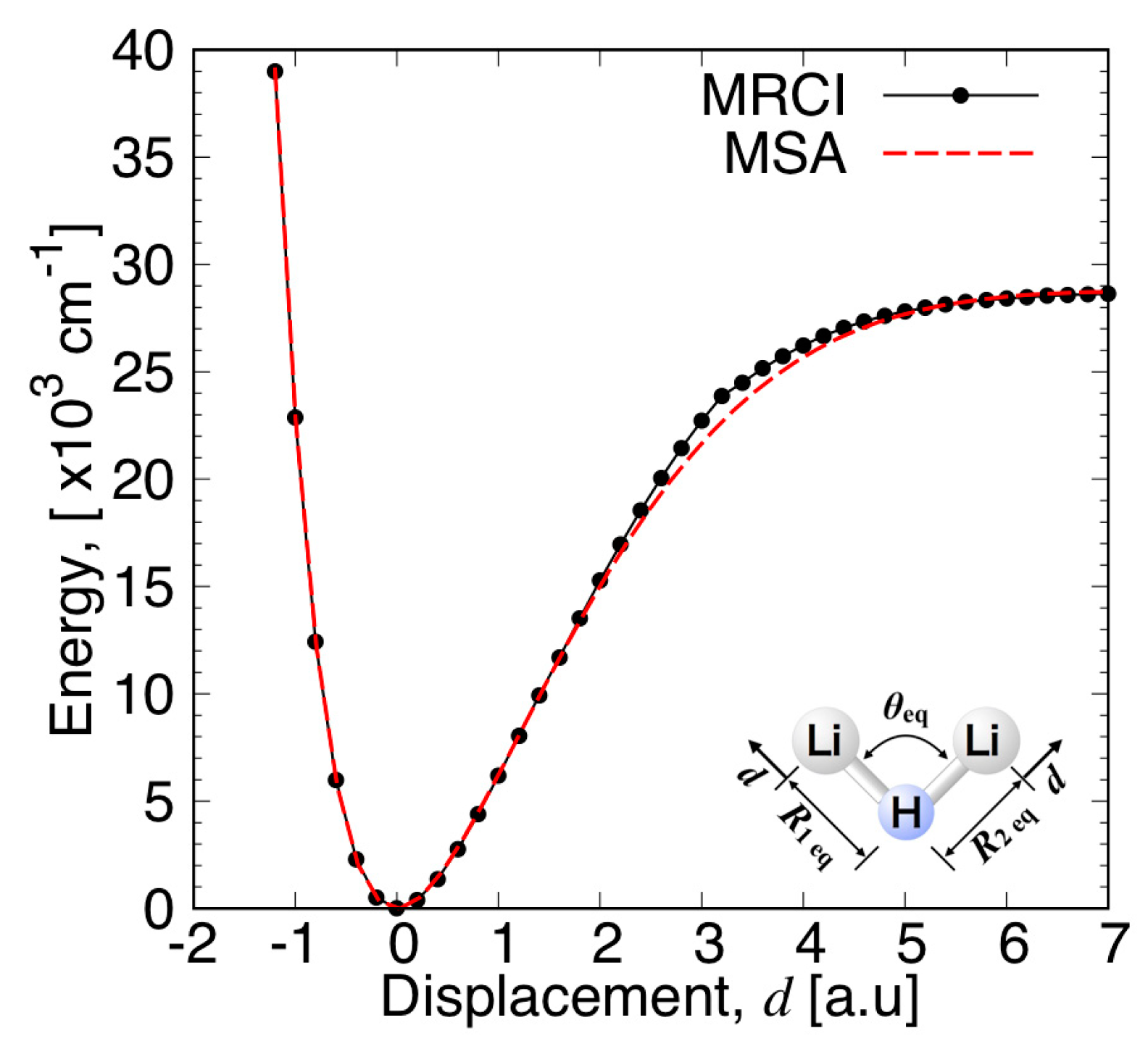
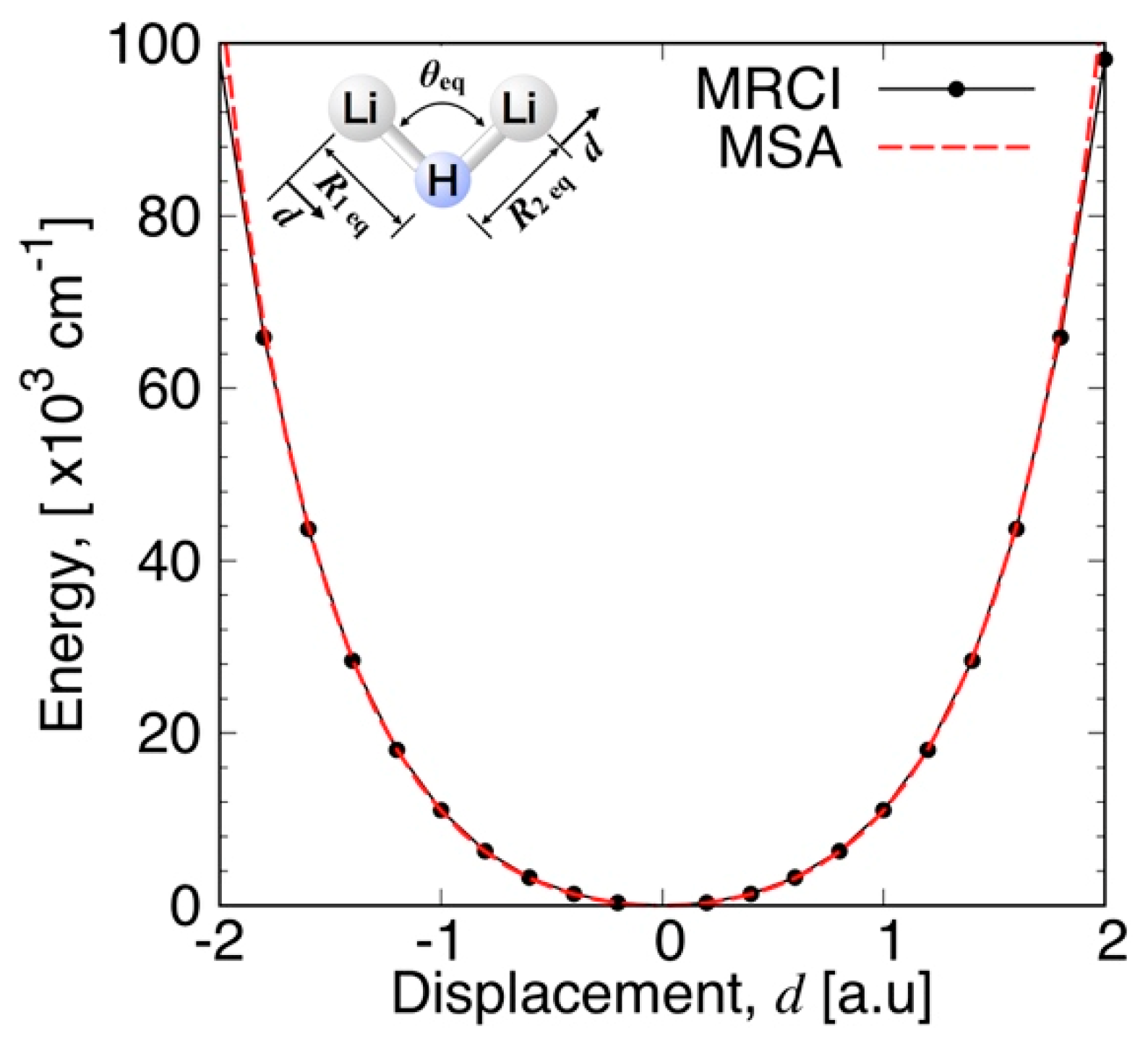
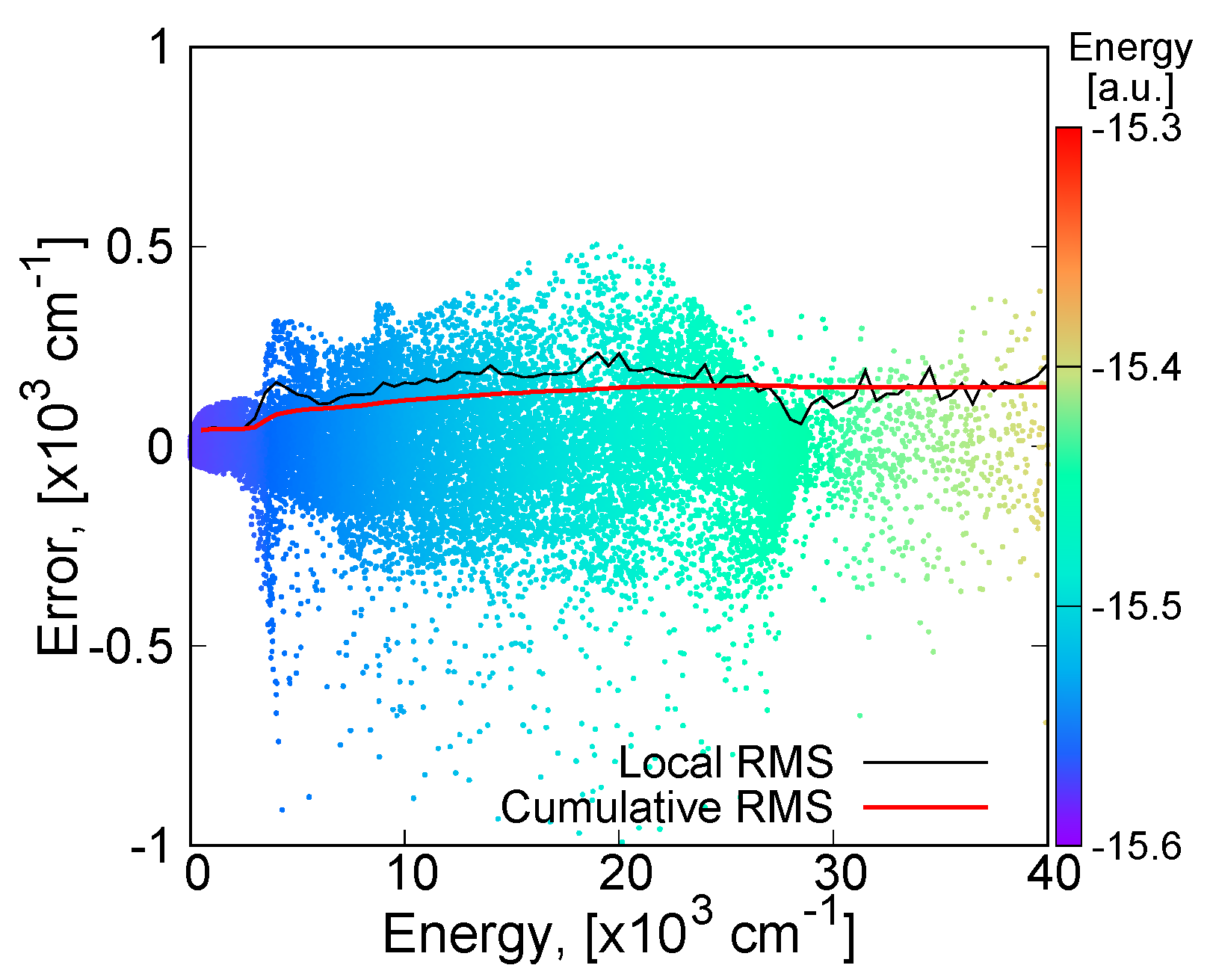
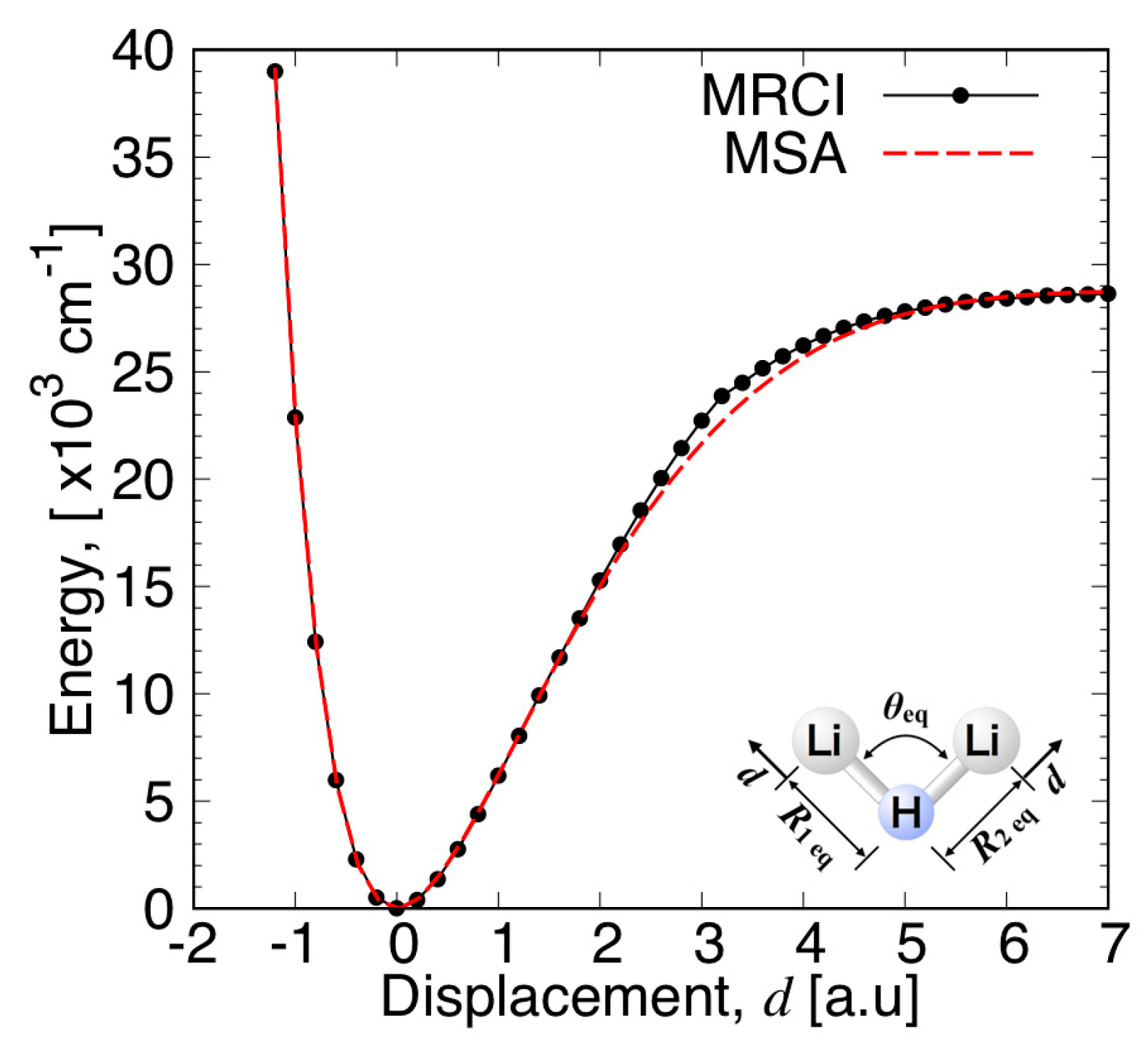
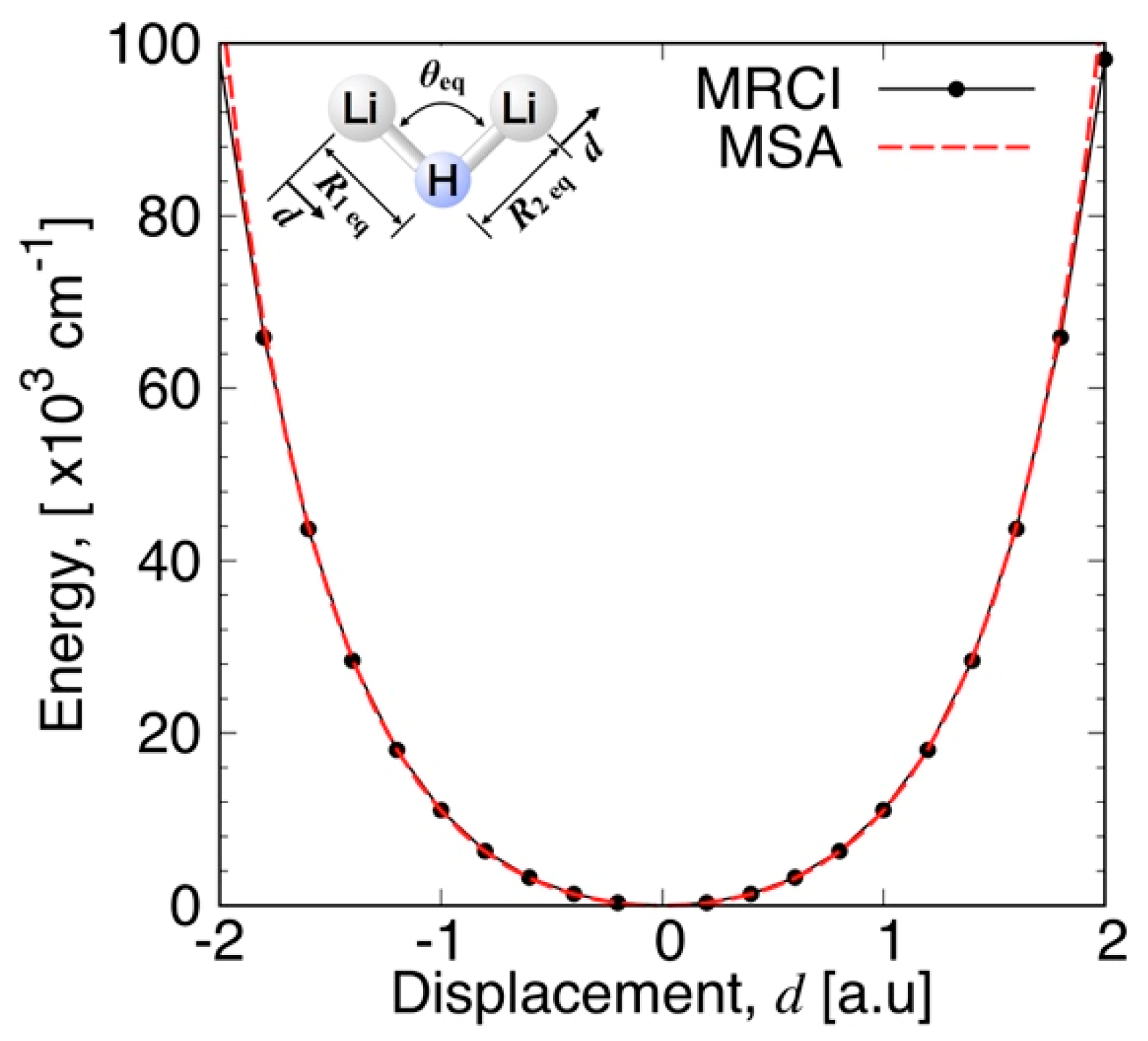
3.3.1. MSA Fitting of PES
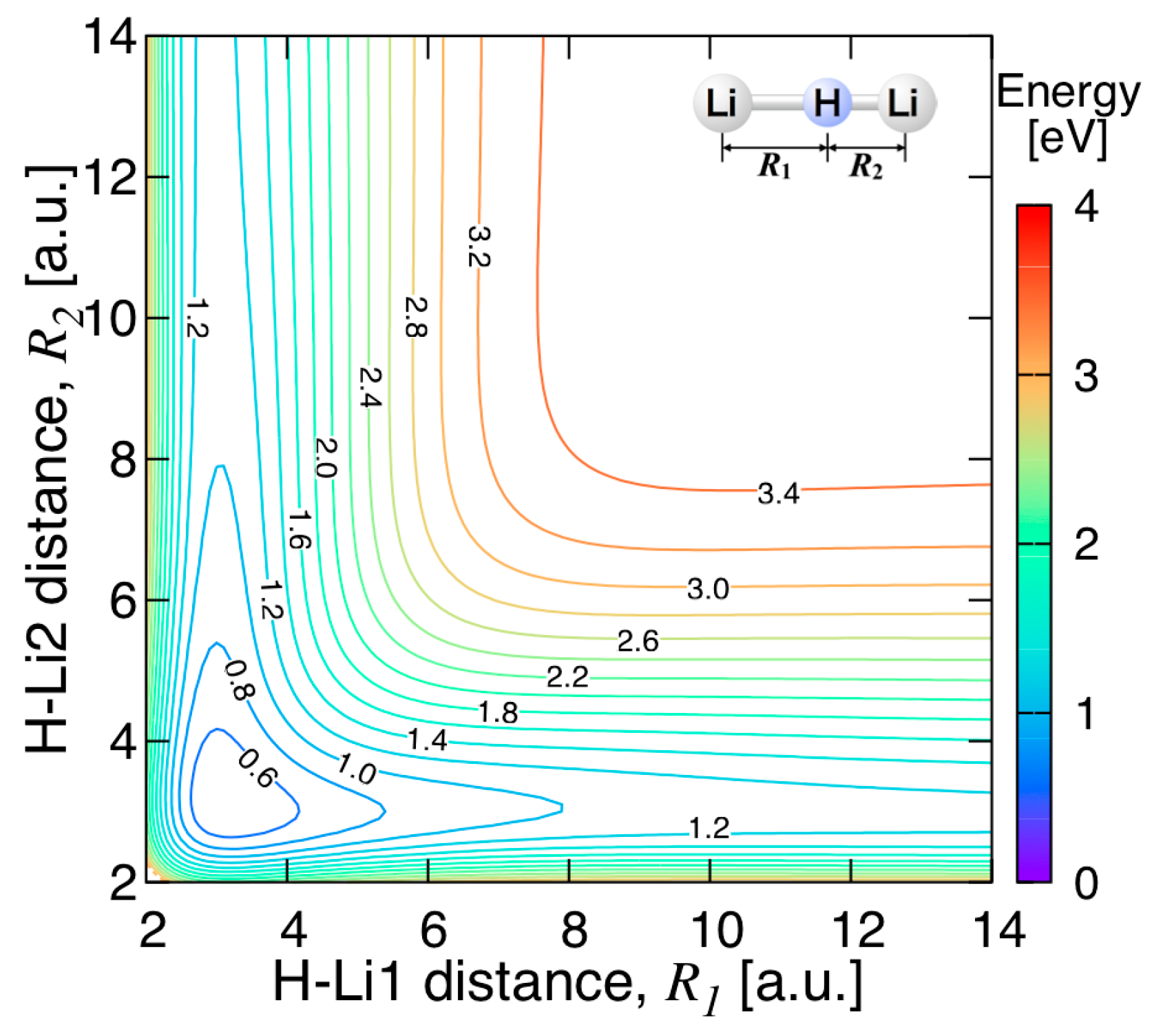
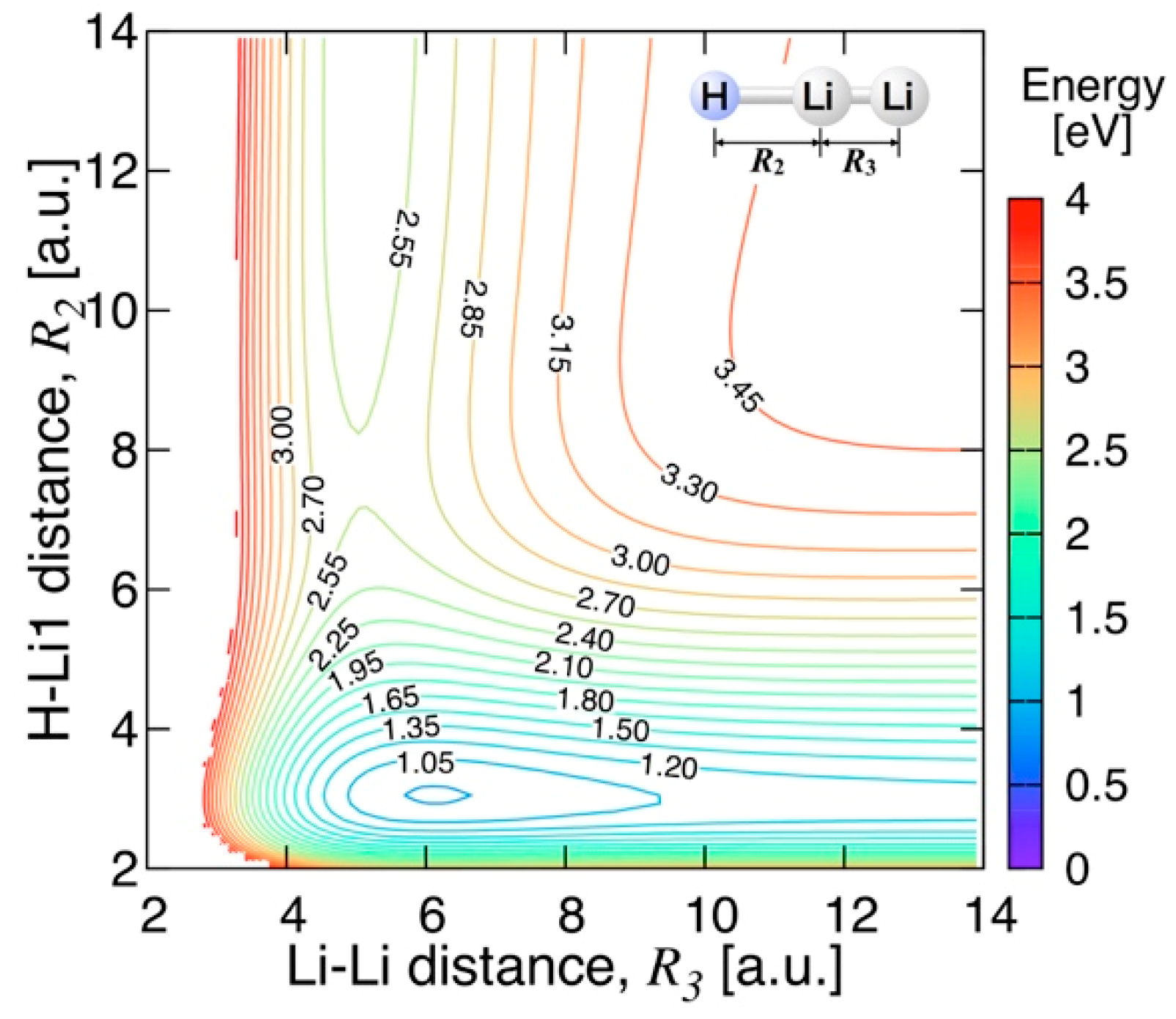
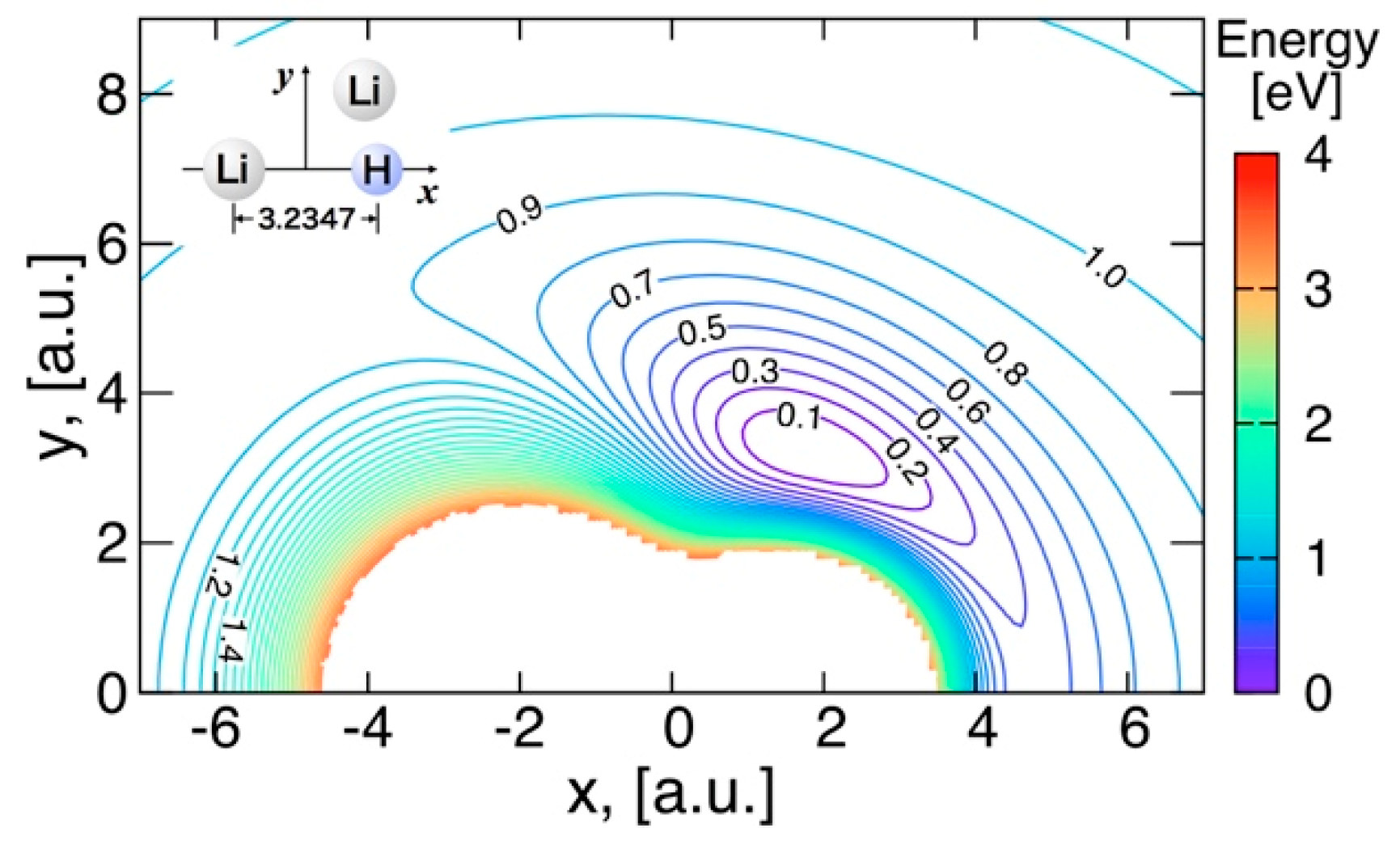
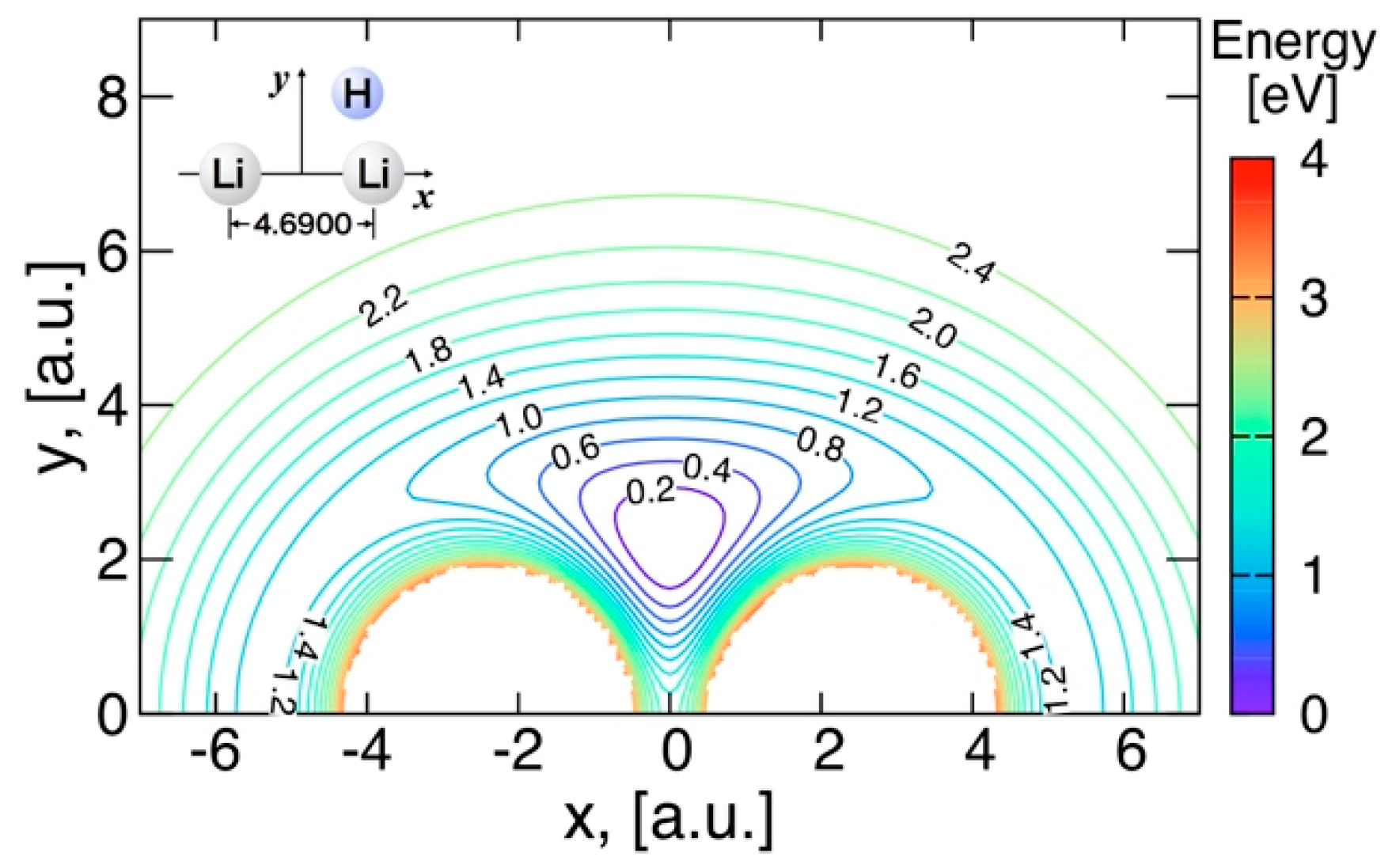
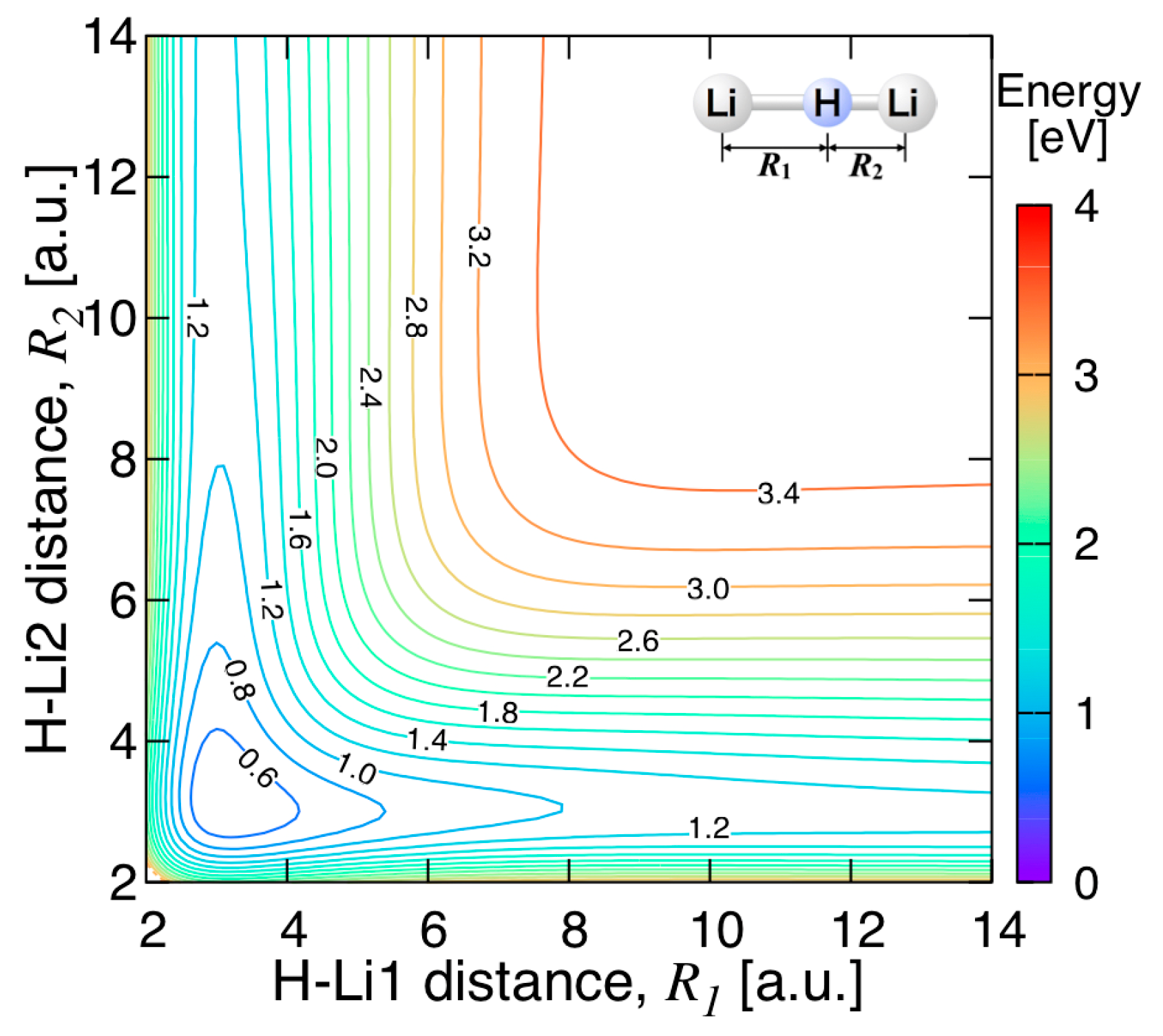
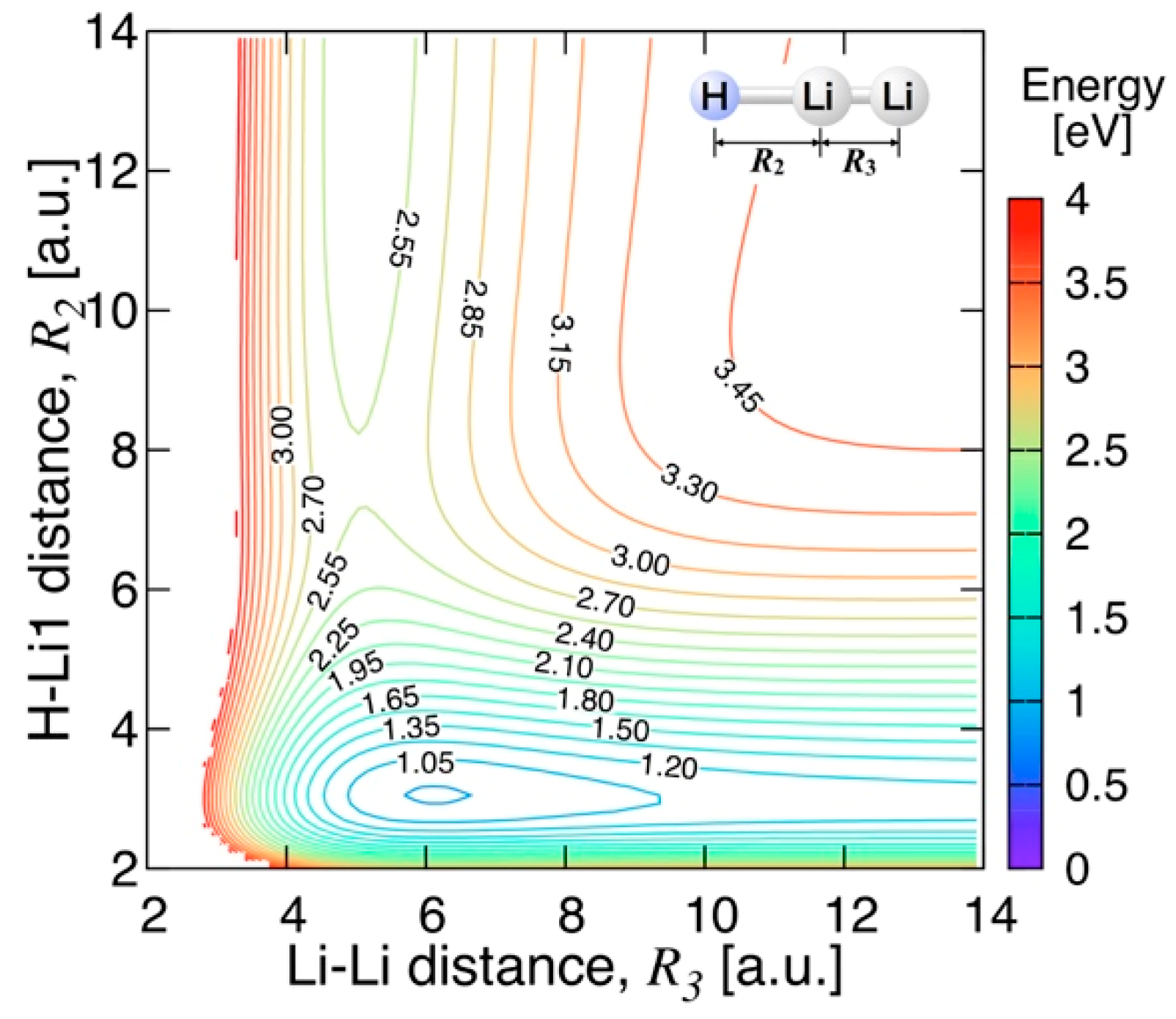
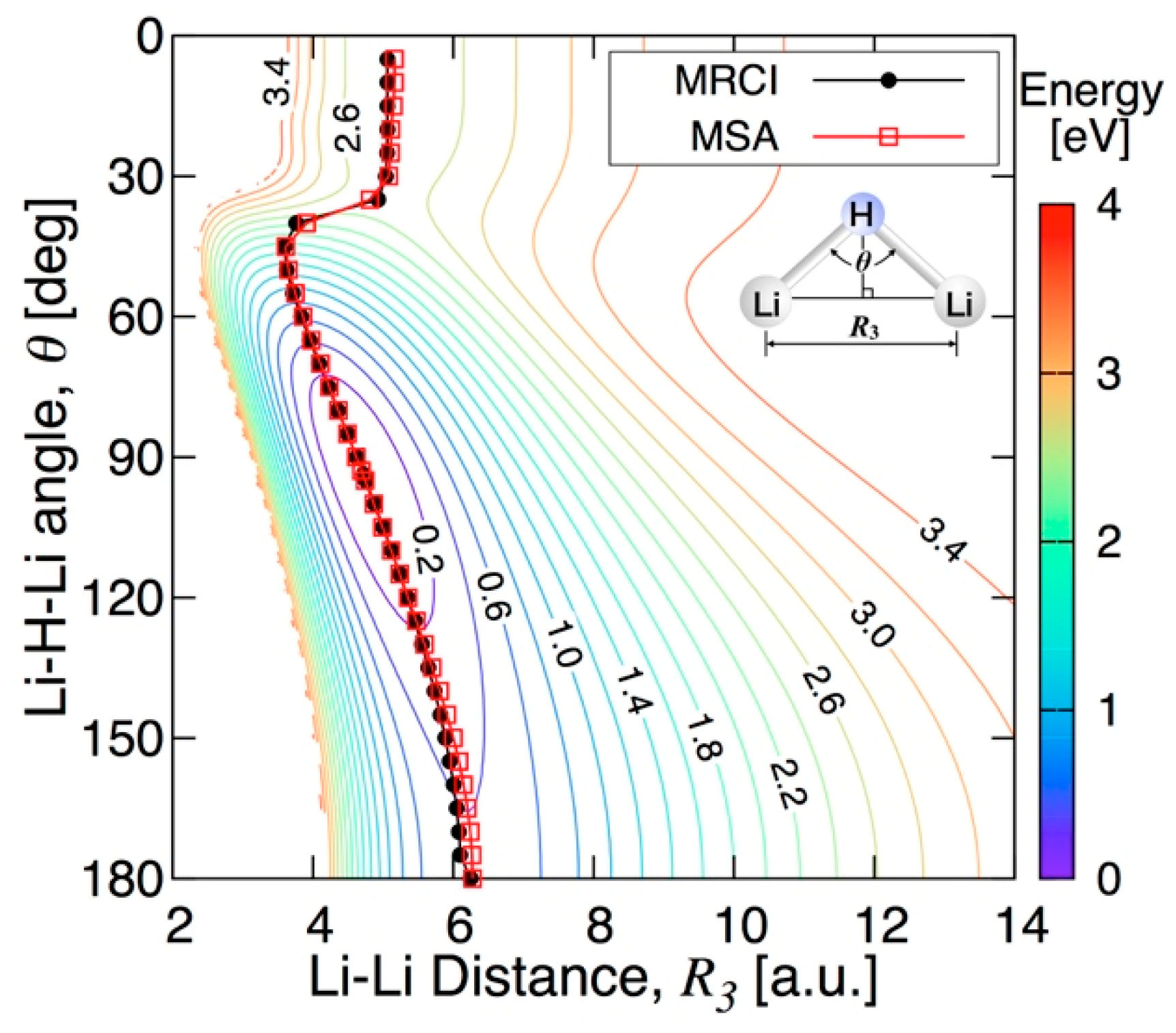
3.3.2. Features of the Present Potential Energy Surface
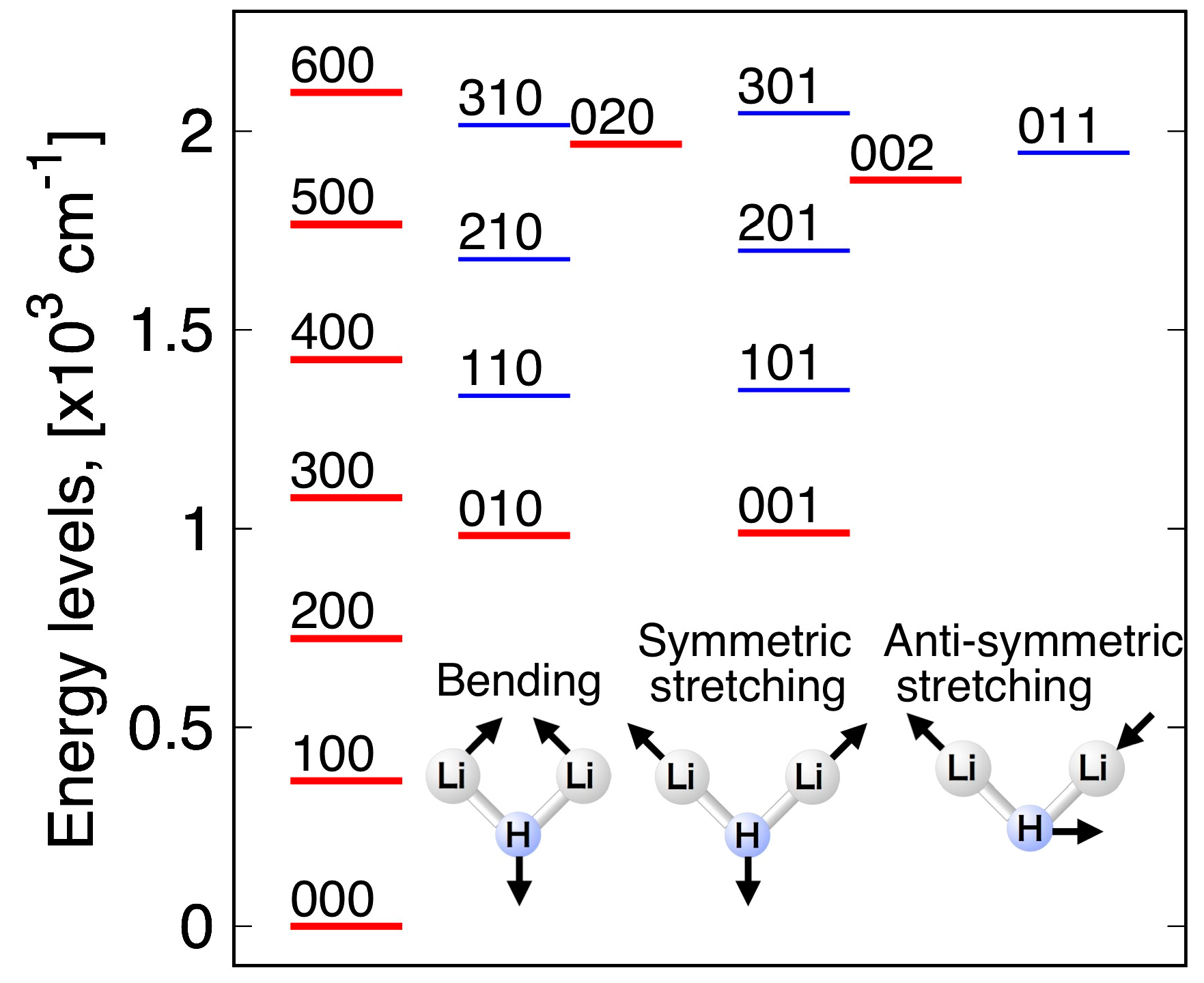
3.4. Vibrational Levels
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jeung, G.-H. Multiple ionic-covalent couplings in molecules and clusters. Chin. J. Chem. Phys. 2009, 22, 187. [Google Scholar] [CrossRef]
- Pekeris, C.L. 1 1S, 2 1S, and 2 3S states of H- and of He. Phys. Rev. 1962, 126, 1470–1476. [Google Scholar] [CrossRef]
- Sakintuna, B.; Lamari-Darkrim, F.; Hirscher, M. Metal hydride materials for solid hydrogen storage: A review. Int. J. Hydrogen Energy 2007, 32, 1121–1140. [Google Scholar] [CrossRef]
- Siegbahn, P.; Schaefer, H.F. Potential energy surfaces for H + Li2 → LiH + Li ground state surface from large scale configuration interaction. J. Chem. Phys. 1975, 62, 3488–3495. [Google Scholar] [CrossRef]
- England, W.B.; Sabelli, N.H.; Wahl, A.C. A theoretical study of Li2H. I. Basis set and computational survey of excited states and possible reaction paths. J. Chem. Phys. 1975, 63, 4596–4605. [Google Scholar] [CrossRef]
- Talbi, D.; Saxon, R.P. Low-lying states of Li3H: Is there an ion-pair minimum? Chem. Phys. Lett. 1989, 157, 419–425. [Google Scholar] [CrossRef]
- Rao, B.K.; Khanna, S.N.; Jena, P. Energetics and electronic structures of hydrogenated metal clusters. Phys. Rev. B 1991, 43, 1416–1421. [Google Scholar] [CrossRef]
- Fang, W.; You, X.; Yin, Z. A theoretical study on the low-lying electronic states of the Li2H molecule. Chem. Phys. Lett. 1995, 233, 237–242. [Google Scholar] [CrossRef]
- Allouche, A.R.; Aubert-Frecon, M.; Spiegelmann, F. Theoretical study of the electronic structure of the low-lying states of the Li2H molecule. Can. J. Phys. 1996, 74, 194–201. [Google Scholar] [CrossRef]
- Antoine, R.; Dugourd, P.; Rayane, D.; Allouche, A.-R.; Aubert-Frécon, M.; Broyer, M. On the optical absorption spectrum of Li2H. Chem. Phys. Lett. 1996, 261, 670–676. [Google Scholar] [CrossRef]
- Vezin, B.; Dugourd, P.; Rayane, D.; Labastie, P.; Broyer, M. First observation of an excited state of Li2H. Chem. Phys. Lett. 1993, 202, 209–215. [Google Scholar] [CrossRef]
- Maniero, A.M.; Acioli, P.H.; e Silva, G.M.; Gargano, R. Theoretical calculations of a new potential energy surface for the H + Li2 reaction. Chem. Phys. Lett. 2010, 490, 123–126. [Google Scholar] [CrossRef]
- Song, Y.-Z.; Li, Y.-Q.; Gao, S.-B.; Meng, Q.-T. Accurate ab initio-based DMBE potential energy surface for HLi2(X2A′) via scaling of the external correlation. Eur. Phys. J. D 2014, 68, 3. [Google Scholar] [CrossRef]
- Yuan, M.; Li, W.; Chen, M. Global X2A′ potential energy surface of Li2H and quantum dynamics of H + Li2 (X1Σg+) → Li + LiH (X1Σ+) reaction. Int. J. Quantum Chem. 2017, 117, e25380. [Google Scholar] [CrossRef]
- Gao, S.; Zhang, J.; Song, Y.; Meng, Q.-T. Cross sections for vibrational inhibition at low collision energies for the reaction H + Li2(1XΣg+) → Li + LiH (X1Σg+). Eur. Phys. J. D 2015, 69, 111. [Google Scholar] [CrossRef]
- Zhu, Z.; Dong, B.; Li, W. Quantum wave packet dynamics of the H + Li2 reaction at state-to-state level of theory. Comput. Theor. Chem. 2017, 1115, 248–252. [Google Scholar] [CrossRef]
- Gao, S.; Zhang, L.; Song, Y.; Meng, Q. Coriolis coupling effects in the H + Li2(1XΣg+) → LiH (X1Σg+) + Li reaction: A time-dependent wave packet investigation. Chem. Phys. Lett. 2016, 651, 233–237. [Google Scholar] [CrossRef]
- Vila, H.V.R.; Ribeiro, L.A.; de Macedo, L.G.M.; Gargano, R. On the angular distribution of the H + Li2 cross sections: A converged time-independent quantum scattering study. Sci. Rep. 2018, 8, 1044. [Google Scholar] [CrossRef]
- Wu, C.H.; Ihle, H.R. Binding energies of Li2H and Li2H+ and the ionization potential of Li2H. J. Chem. Phys. 1977, 66, 4356–4359. [Google Scholar] [CrossRef]
- Crooks, J.B.; Way, K.R.; Yang, S.-C.; Wu, C.-Y.R.; Stwalley, W.C. Photon and positive ion production from collisions of superthermal hydrogen atoms with lithium atoms and molecules. J. Chem. Phys. 1978, 69, 490–491. [Google Scholar] [CrossRef]
- Xie, Z.; Bowman, J.M. Permutationally invariant polynomial basis for molecular energy surface fitting via monomial symmetrization. J. Chem. Theory Comput. 2010, 6, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Bowman, J.M.; Carter, S.; Huang, X. MULTIMODE: A code to calculate rovibrational energies of polyatomic molecules. Int. Rev. Phys. Chem. 2003, 22, 533–549. [Google Scholar] [CrossRef]
- Rosmus, P.; Meyer, W. Spectroscopic constants and the dipole moment functions for the 1Σ+ ground state of NaLi. J. Chem. Phys. 1976, 65, 492–493. [Google Scholar] [CrossRef]
- Jeung, G.H.; Malrieu, J.P.; Daudey, J.P. Inclusion of core-valence correlation effects in pseudopotential calculations. I. Alkali atoms and diatoms. J. Chem. Phys. 1982, 77, 3571–3577. [Google Scholar] [CrossRef]
- Jeung, G.H.; Daudey, J.P.; Malrieu, J.P. Inclusion of core-valence correlation effects in pseudopotential calculations. II. K2 and KH lowest Σ+ potential curves from valence-correlated wavefunctions. J. Phys. B At. Mol. Phys. 1983, 16, 699. [Google Scholar] [CrossRef]
- Yiannopoulou, A.; Jeung, G.-H.; Park, S.J.; Lee, H.S.; Lee, Y.S. Undulations of the potential-energy curves for highly excited electronic states in diatomic molecules related to the atomic orbital undulations. Phys. Rev. A 1999, 59, 1178–1186. [Google Scholar] [CrossRef]
- Prascher, B.P.; Woon, D.E.; Peterson, K.A.; Dunning, T.H.; Wilson, A.K. Gaussian basis sets for use in correlated molecular calculations. VII. Valence, core-valence, and scalar relativistic basis sets for Li, Be, Na, and Mg. Theor. Chem. Acc. 2011, 128, 69–82. [Google Scholar] [CrossRef]
- Werner, H.-J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M.; Celani, P.; Korona, T.; Lindh, R.; Mitrushenkov, A.; Rauhut, G.; et al. MOLPRO, a Package of ab Initio Programs, Version 2012.1. 2012. Available online: http://www.molpro.net/ (accessed on 21 November 2018).
- Carter, S.; Bowman, J.M. The adiabatic rotation approximation for rovibrational energies of many-mode systems: Description and tests of the method. J. Chem. Phys. 1998, 108, 4397–4404. [Google Scholar] [CrossRef]
- Kim, S.K.; Herschbach, D.R. Angular momentum disposal in atom exchange reactions. Faraday Discuss. Chem. Soc. 1987, 84, 159–169. [Google Scholar] [CrossRef]
- Verma, K.K.; Koch, M.E.; Stwalley, W.C. Observation of levels near dissociation in the X1Σg+ State of 7Li2. J. Chem. Phys. 1983, 78, 3614–3622. [Google Scholar] [CrossRef]
- Stwalley, W.C.; Zemke, W.T. Spectroscopy and structure of the Lithium Hydride diatomic molecules and ions. J. Phys. Chem. Ref. Data 1993, 22, 87–112. [Google Scholar] [CrossRef]
- Neff, M.; Rauhut, G. Toward large scale vibrational configuration interaction calculations. J. Chem. Phys. 2009, 131, 124129. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Li2H (X2A1) | Method | Basis | c.v. a | Re Li–H | ∠LiHLi | Eatom b | Ediss 1 c | Ediss 2 d | Eion e |
|---|---|---|---|---|---|---|---|---|---|
| [a.u.] | [degrees] | [kcal∙mol−1] | [kcal∙mol−1] | [kcal∙mol−1] | [eV] | ||||
| Present | MRCI | J99 | Y | 3.233 | 93.70 | 82.05 | 58.39 | 25.25 | 4.671 |
| MSA fitting | J99 | Y | 3.234 | 92.93 | 82.11 | 56.44 | 26.71 | - | |
| MRCI | CV5Z | Y | 3.227 | 93.60 | 83.04 | 58.98 | 25.34 | 4.683 | |
| MRCI | CVQZ | Y | 3.231 | 93.66 | 82.79 | 58.78 | 25.26 | 4.678 | |
| MRCI | ACVTZ | Y | 3.240 | 93.59 | 82.11 | 58.24 | 25.12 | 4.672 | |
| MRCI | J99 | N | 3.253 | 94.04 | 82.00 | 58.26 | 24.94 | 4.642 | |
| MRCI | CV5Z | N | 3.254 | 93.95 | 82.70 | 58.62 | 25.15 | 4.646 | |
| MRCI | CVQZ | N | 3.256 | 93.88 | 82.52 | 58.48 | 25.10 | 4.647 | |
| MRCI | ACVTZ | N | 3.262 | 93.76 | 81.80 | 57.99 | 24.93 | 4.647 | |
| Yuan g | MRCI-F12 | AVTZ | N | 3.220 | 94.37 | 86.64 | 61.4 | 27.4 | |
| Song h | Fitting | DMBE/SEC PES | N | 3.247 | 94.31 | 87.91 | 65.0 | 31.3 | |
| MRCI | V5Z | N | 3.252 | 94.14 | 84.39 | ||||
| MRCI | VQZ | N | 3.256 | 93.95 | 82.56 | ||||
| Maniero i | Fitting (Full CI) | pseudo-potential 6-311G (2df, 2pd) | N | 3.221 | 94.65 | 86.9 | |||
| Vezin j | Exp. | 3.241 | 94.70 | ||||||
| Wu k | Exp. (linear) | 87.9 ± 3 f | 4.5 ± 0.3 | ||||||
| Exp. (bent) | 82.1 ± 3 | ||||||||
| From Wu f, Verma l and Stwalley m | Exp. | 63.4 ± 3 | 29.8 ± 3 |
| Li2H (12B2) | Method | Re Li–H [a.u.] | ∠LiHLi [degrees] | E–E(X2A1) [cm−1] | |
| Present | MRCI | 3.174 | 180.0 | 3581.7 | |
| Talbi a | MCSCF/SOCI | 3.222 | 180 | 3382 | |
| Allouche b | CASSCF | 3.137 | 180 | 3597 | |
| Fang c | UMP2 | 3.162 | 179.90 | 3458.9 | |
| Li2H (12B1) | Method | Re Li–H [a.u.] | ∠LiHLi [degrees] | E–E(X2A1) [cm−1] | |
| Present | MRCI | 3.035 | 131.6 | 8526.6 | |
| Allouche b | CASSCF | 2.986 | 133 | 8554 | |
| Fang c | UMP2 | 3.012 | 145.0 | 8719.7 | |
| Li2H (12A2) | Method | Re Li–H [a.u.] | ∠LiHLi [degrees] | E–E(X2A1) [cm−1] | |
| Present | MRCI | 3.167 | 180.0 | 17428.3 | |
| Fang c | UMP2 | 3.140 | 179.89 | 16798.6 | |
| Li2H+ (11A1) | Method | Re Li–H [a.u.] | ∠LiHLi [degrees] | E–E(X2A1) [cm−1] | |
| adiabatic | vertical | ||||
| Present | MRCI | 3.104 | 180.0 | 31654.3 | 37673.4 |
| Allouche b | CASSCF | 32584.8 | 38392.0 | ||
| Wu d | Experiment | 36295.0 | |||
| Vibrational Frequency | ||||||
|---|---|---|---|---|---|---|
| Harmonic | Anharmonic | |||||
| ω1 | ω2 | ω3 | ν1 | ν2 | ν3 | |
| [cm−1] | [cm−1] | [cm−1] | [cm−1] | [cm−1] | [cm−1] | |
| MULTIMODE (Global PES) a | 395.46 | 1039.11 | 1056.73 | 376.92 | 983.87 | 971.79 |
| MULTIMODE (Local PES) b | 379.73 | 1031.62 | 1079.89 | 365.00 | 982.48 | 989.33 |
| Molpro (VCI, MRCI) c | 380.07 | 1034.22 | 1079.41 | 345.29 | 947.18 | 978.44 |
| Molpro (VCI, UCCSD(T)-F12) d | 376.12 | 1023.60 | 1071.50 | 362.22 | 973.49 | 981.20 |
| Levels of A1 Symmetry | Levels of B2 Symmetry | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| i | v1 | v2 | v3 | i | v1 | v2 | v3 | ||
| 1 | 1 | 0 | 0 | 365.00 | 4 | 0 | 0 | 1 | 989.33 |
| 2 | 2 | 0 | 0 | 724.46 | 7 | 1 | 0 | 1 | 1347.74 |
| 3 | 0 | 1 | 0 | 982.48 | 10 | 2 | 0 | 1 | 1699.78 |
| 5 | 3 | 0 | 0 | 1077.97 | 13 | 0 | 1 | 1 | 1945.16 |
| 6 | 1 | 1 | 0 | 1333.73 | 16 | 3 | 0 | 1 | 2044.91 |
| 8 | 4 | 0 | 0 | 1425.01 | Zero point energy: 1222.76 cm−1 | ||||
| 9 | 2 | 1 | 0 | 1678.27 | |||||
| 11 | 5 | 0 | 0 | 1765.03 | |||||
| 12 | 0 | 0 | 2 | 1876.42 | |||||
| 14 | 0 | 2 | 0 | 1966.69 | |||||
| 15 | 3 | 1 | 0 | 2015.30 | |||||
| 17 | 6 | 0 | 0 | 2097.23 | |||||
| Energy | First | Second | Third | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [cm−1] | v1 | v2 | v3 | Coeff. | v1 | v2 | v3 | Coeff. | v1 | v2 | v3 | Coeff. |
| 365.00 | 1 | 0 | 0 | 0.9978 | 1 | 1 | 2 | −0.0515 | 2 | 2 | 0 | 0.0197 |
| 724.46 | 2 | 0 | 0 | 0.9961 | 2 | 1 | 2 | −0.0513 | 0 | 1 | 0 | −0.0335 |
| 982.48 | 0 | 1 | 0 | −0.9746 | 0 | 0 | 2 | 0.1509 | 1 | 1 | 0 | −0.1277 |
| 989.33 | 0 | 0 | 1 | 0.9697 | 0 | 1 | 1 | 0.2111 | 0 | 1 | 3 | −0.1006 |
| 1077.97 | 3 | 0 | 0 | 0.9926 | 1 | 1 | 0 | 0.0586 | 3 | 1 | 2 | −0.0507 |
| 1333.73 | 1 | 1 | 0 | 0.9520 | 2 | 1 | 0 | 0.1861 | 1 | 0 | 2 | −0.1470 |
| 1347.74 | 1 | 0 | 1 | 0.9699 | 1 | 1 | 1 | 0.2024 | 1 | 1 | 3 | −0.1016 |
| 1425.01 | 4 | 0 | 0 | 0.9860 | 2 | 1 | 0 | 0.0819 | 4 | 1 | 0 | −0.0653 |
| 1678.27 | 2 | 1 | 0 | −0.9193 | 3 | 1 | 0 | 0.2344 | 1 | 1 | 0 | 0.1899 |
| 1699.78 | 2 | 0 | 1 | 0.9697 | 2 | 1 | 1 | 0.1915 | 2 | 1 | 3 | −0.1026 |
| 1765.03 | 5 | 0 | 0 | −0.9745 | 3 | 1 | 0 | −0.1018 | 5 | 1 | 0 | 0.0881 |
| 1876.42 | 0 | 0 | 2 | 0.6844 | 0 | 2 | 0 | −0.5207 | 0 | 1 | 2 | 0.4024 |
| 1945.16 | 0 | 1 | 1 | −0.8593 | 0 | 2 | 1 | −0.2739 | 0 | 0 | 3 | 0.2492 |
| 1966.69 | 0 | 2 | 0 | −0.7742 | 0 | 0 | 2 | −0.5558 | 1 | 2 | 0 | −0.2176 |
| 2015.30 | 3 | 1 | 0 | −0.8708 | 4 | 1 | 0 | −0.2767 | 2 | 1 | 0 | −0.2461 |
| 2044.91 | 3 | 0 | 1 | 0.9682 | 3 | 1 | 1 | 0.1777 | 3 | 1 | 3 | −0.1033 |
| 2097.23 | 6 | 0 | 0 | 0.9548 | 3 | 1 | 0 | 0.1158 | 4 | 1 | 0 | −0.1141 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahn Furudate, M.; Hagebaum-Reignier, D.; Jeung, G.-H. Full-Dimensional Ab Initio Potential Energy Surface and Vibrational Energy Levels of Li2H. Molecules 2019, 24, 26. https://doi.org/10.3390/molecules24010026
Ahn Furudate M, Hagebaum-Reignier D, Jeung G-H. Full-Dimensional Ab Initio Potential Energy Surface and Vibrational Energy Levels of Li2H. Molecules. 2019; 24(1):26. https://doi.org/10.3390/molecules24010026
Chicago/Turabian StyleAhn Furudate, Michiko, Denis Hagebaum-Reignier, and Gwang-Hi Jeung. 2019. "Full-Dimensional Ab Initio Potential Energy Surface and Vibrational Energy Levels of Li2H" Molecules 24, no. 1: 26. https://doi.org/10.3390/molecules24010026
APA StyleAhn Furudate, M., Hagebaum-Reignier, D., & Jeung, G.-H. (2019). Full-Dimensional Ab Initio Potential Energy Surface and Vibrational Energy Levels of Li2H. Molecules, 24(1), 26. https://doi.org/10.3390/molecules24010026