
Effects of Population Dynamics on Establishment of a Restriction-Modification System in a Bacterial Host


Abstract
1. Introduction
2. Methods
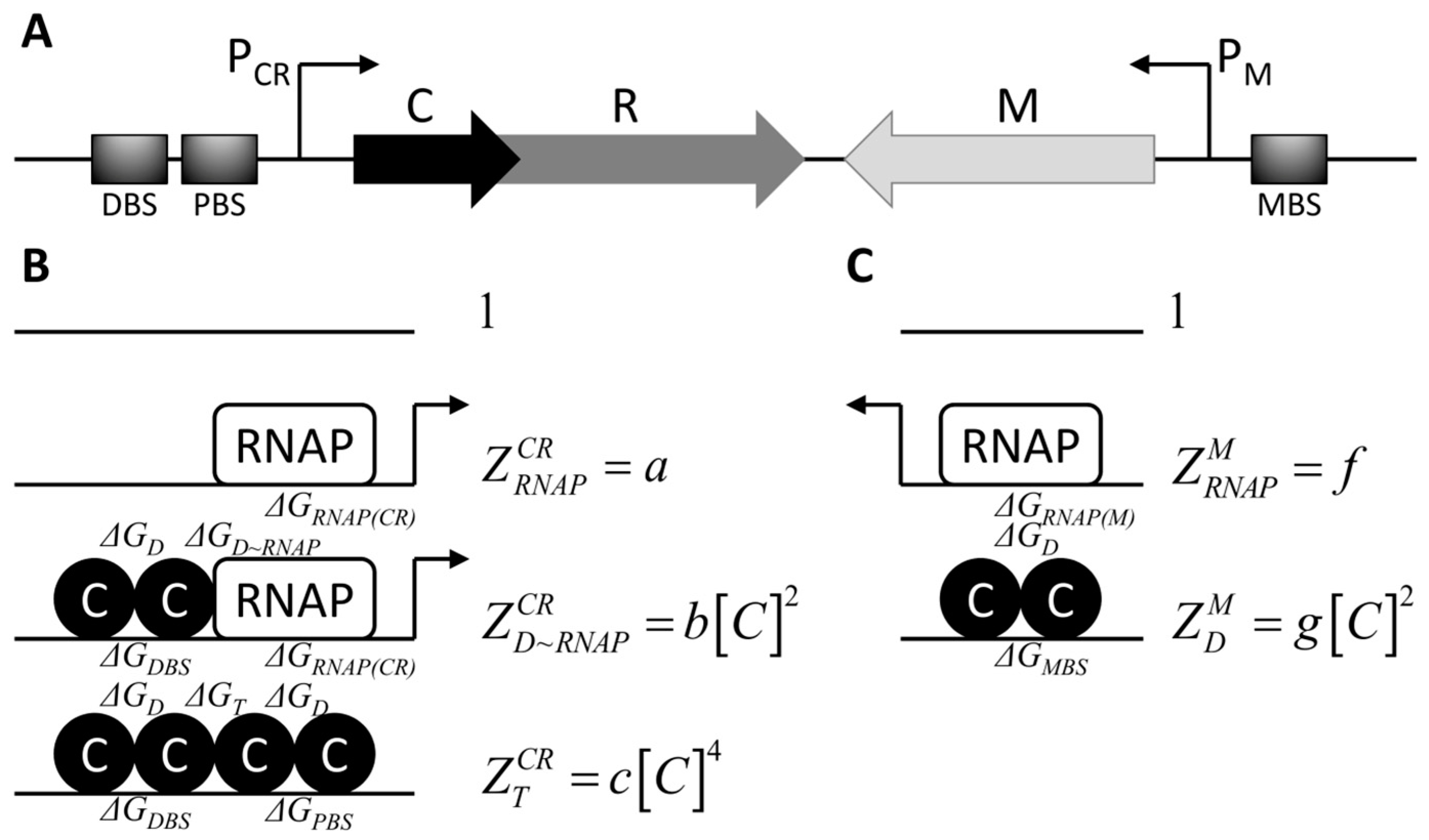
2.1. Modeling cr Operon and m Gene Transcription Activities
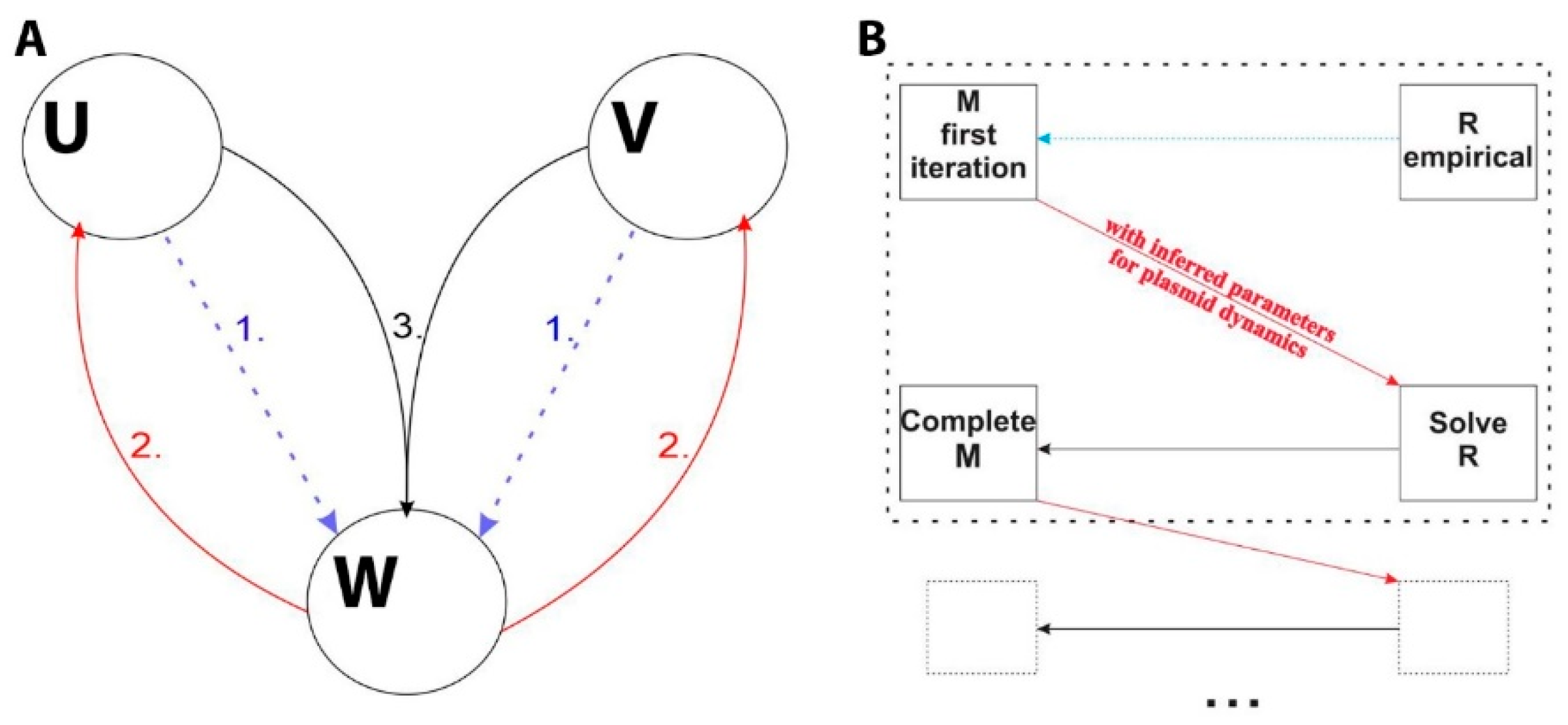
2.2. Introducing the Interplay of Cell and Plasmid Division Rates
2.3. Numerically Solving and Fitting the Model Equations
3. Results and Discussion
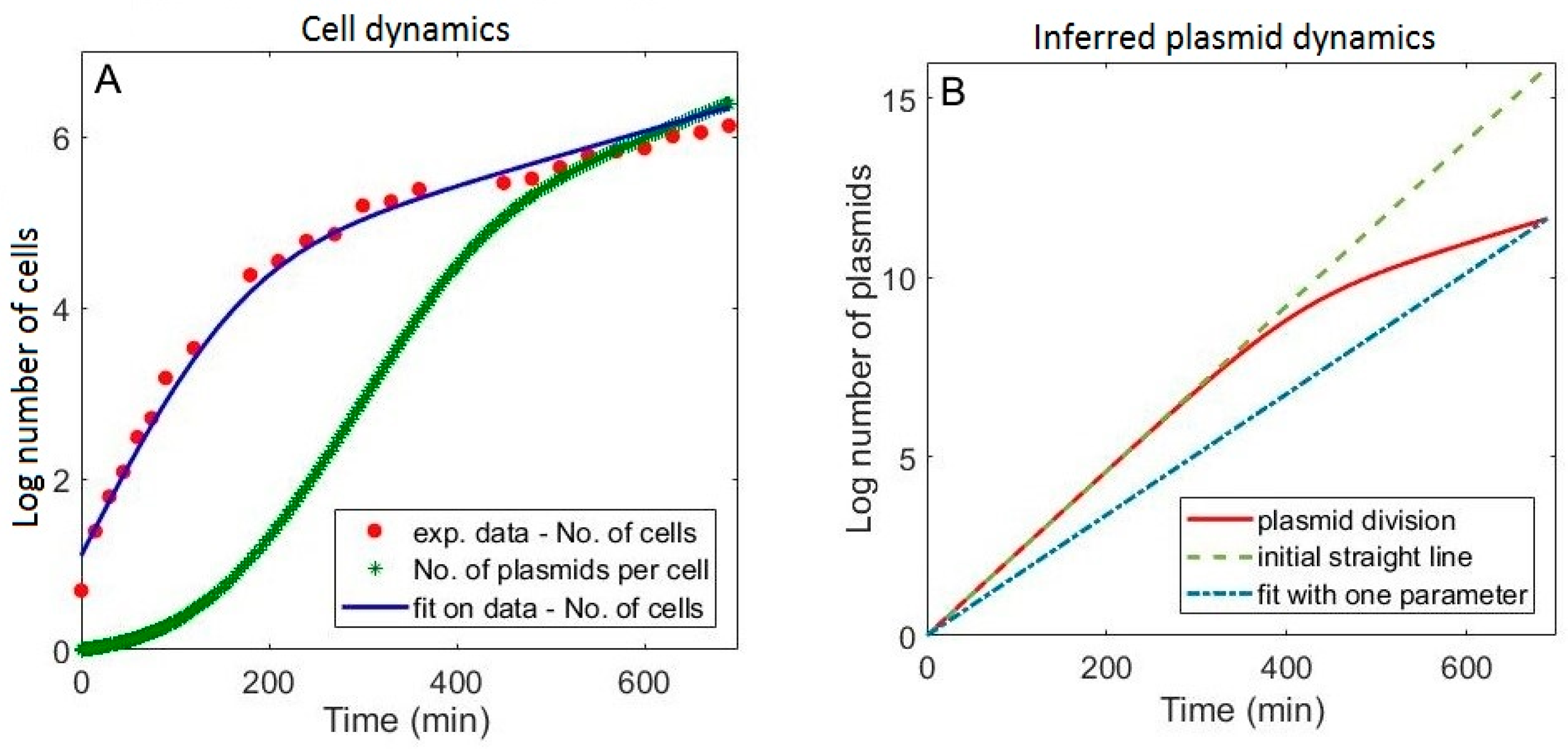
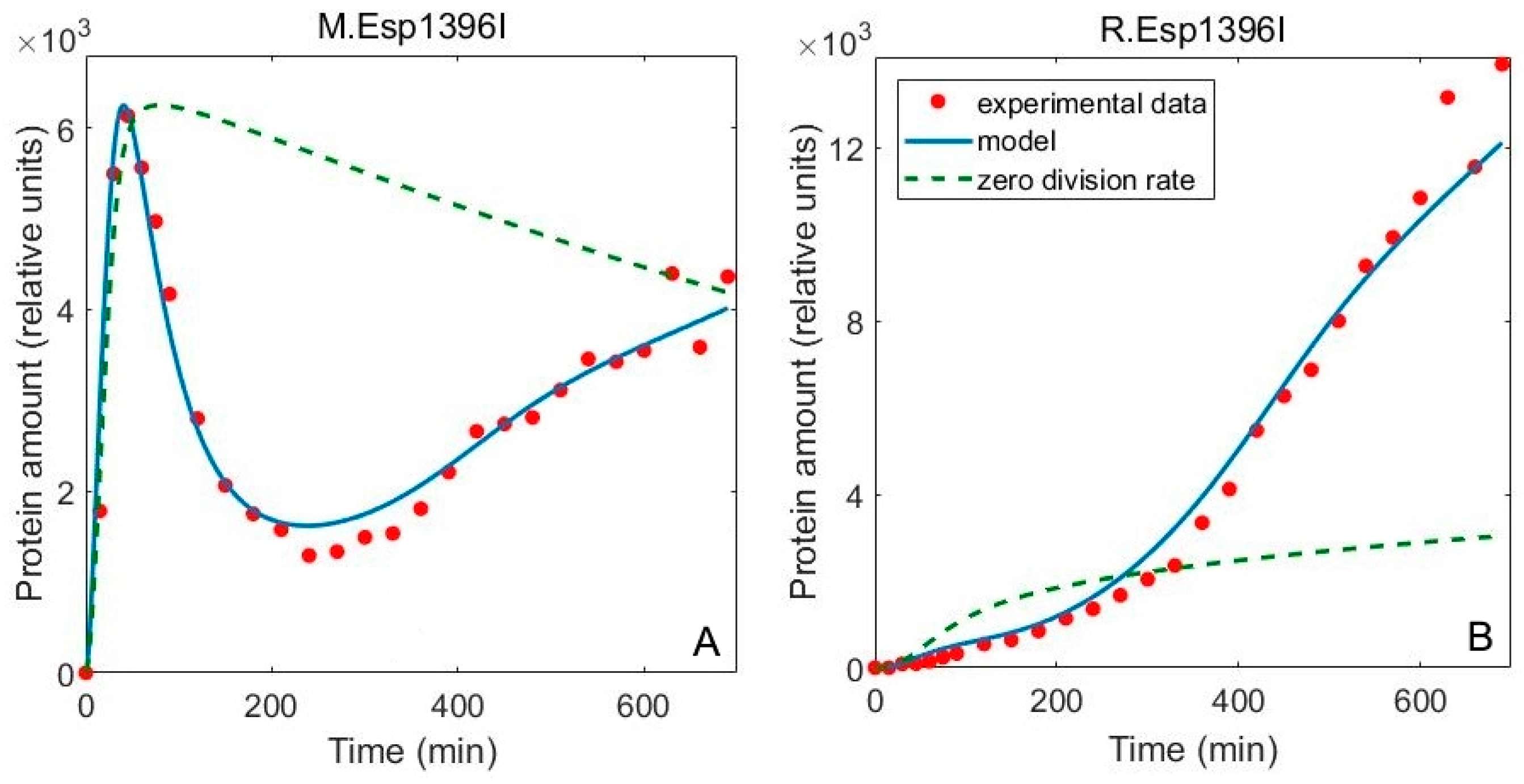
3.1. Including the Population Dynamics Effects
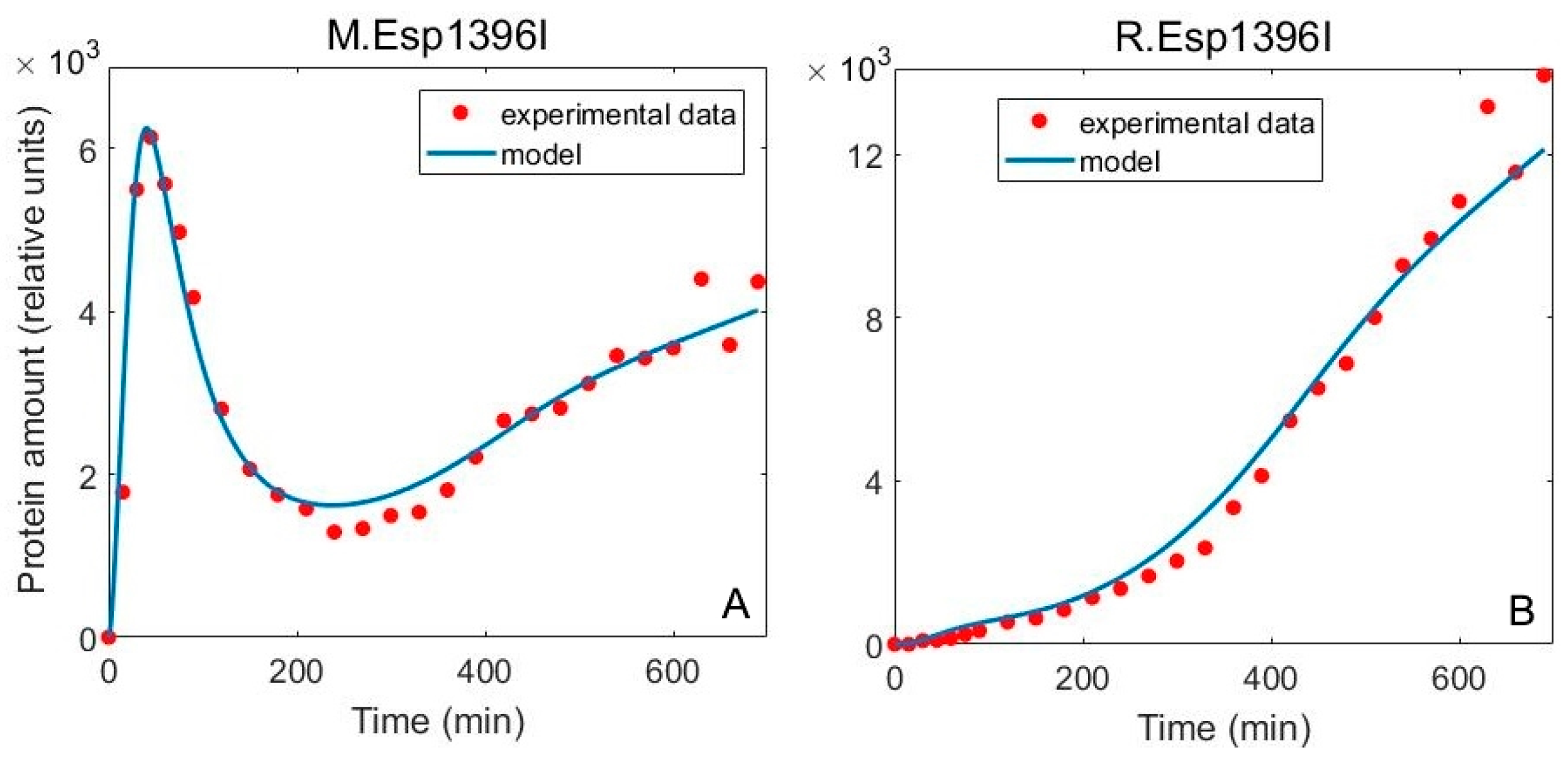
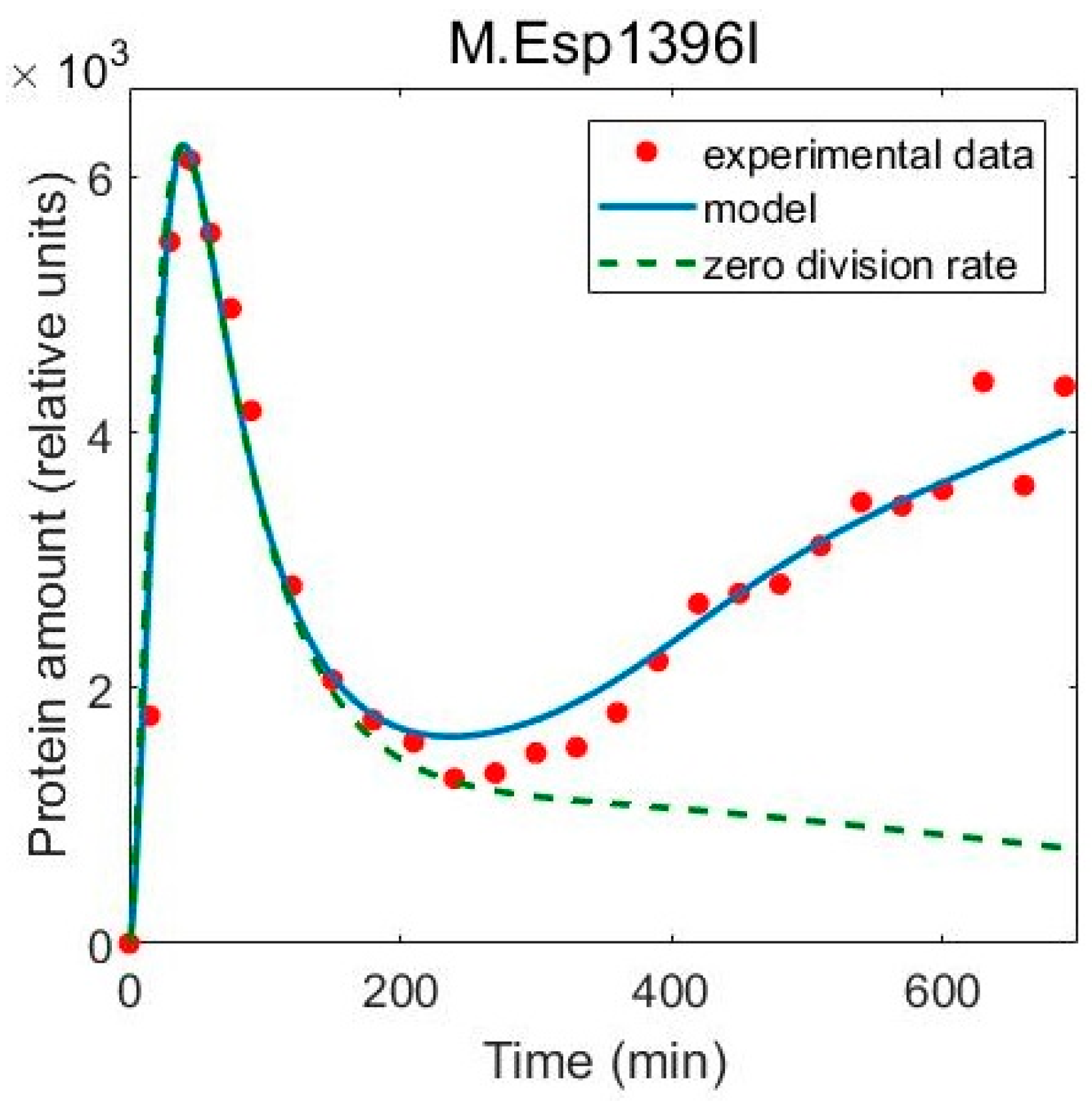
3.2. Including Population Dynamics Improves Agreement with the Expression Measurements
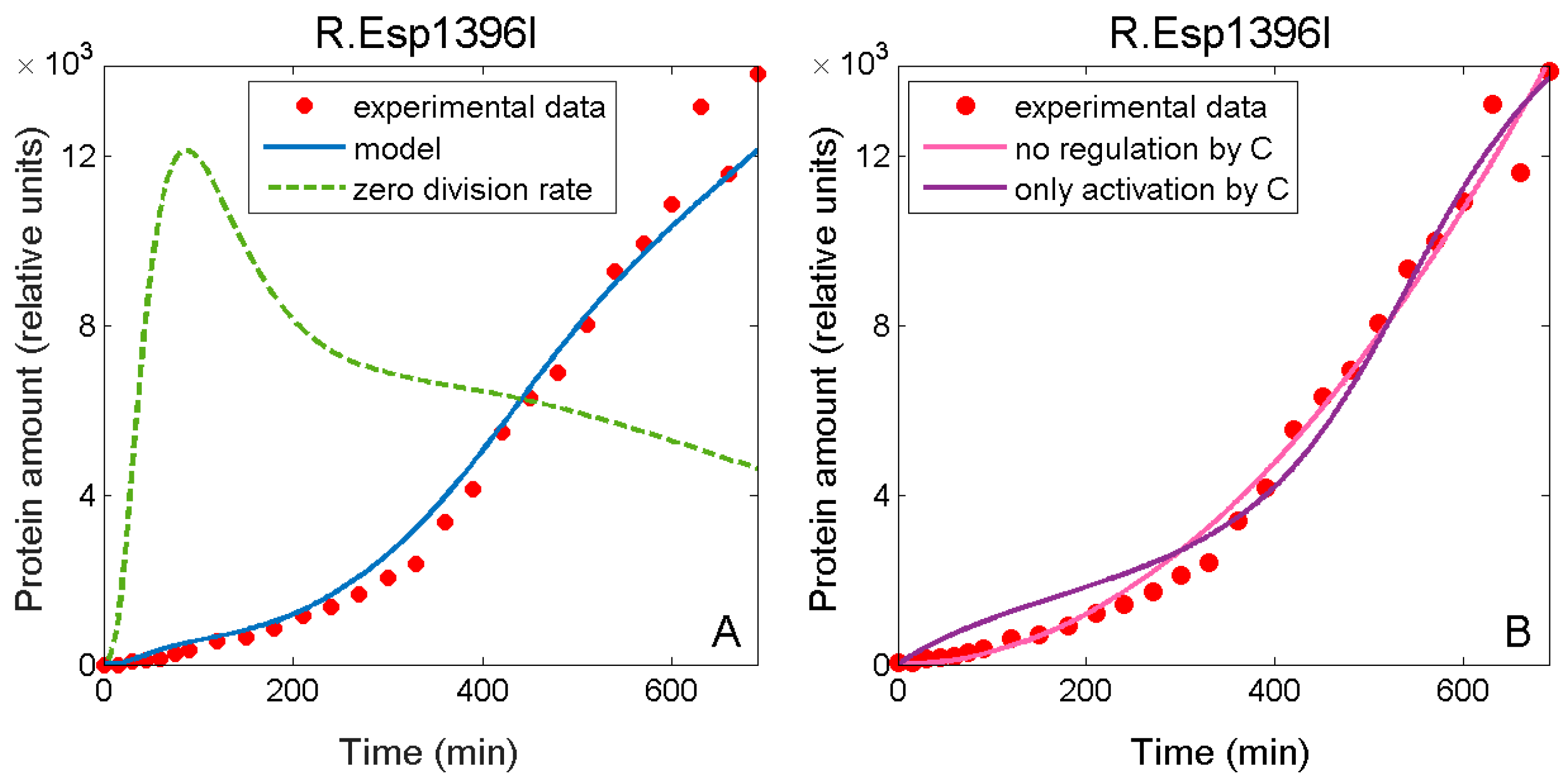
3.3. Falsely Identifying Regulation when Plasmid Dynamics is not Included
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Longo, D.; Hasty, J. Dynamics of single-cell gene expression. Mol. Syst. Biol. 2006, 2, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Ohno, M.; Karagiannis, P.; Taniguchi, Y. Protein expression analyses at the single cell level. Molecules 2014, 19, 13932–13947. [Google Scholar] [CrossRef]
- Elowitz, M.B.; Leibler, S. A synthetic oscillatory network of transcriptional regulators. Nature 2000, 403, 335–338. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, N.; Young, J.W.; Alon, U.; Swain, P.S.; Elowitz, M.B. Gene regulation at the single-cell level. Science 2005, 307, 1962–1965. [Google Scholar] [CrossRef] [PubMed]
- Young, J.W.; Locke, J.C.; Altinok, A.; Rosenfeld, N.; Bacarian, T.; Swain, P.S.; Mjolsness, E.; Elowitz, M.B. Measuring single-cell gene expression dynamics in bacteria using fluorescence time-lapse microscopy. Nat. Protoc. 2012, 7, 80–97. [Google Scholar] [CrossRef] [PubMed]
- Morozova, N.; Sabantsev, A.; Bogdanova, E.; Fedorova, Y.; Maikova, A.; Vedyaykin, A.; Rodic, A.; Djordjevic, M.; Khodorkovskii, M.; Severinov, K. Temporal dynamics of methyltransferase and restriction endonuclease accumulation in individual cells after introducing a restriction-modification system. Nucleic Acids Res. 2016, 44, 790–800. [Google Scholar] [CrossRef]
- Kobayashi, I. Behavior of restriction–modification systems as selfish mobile elements and their impact on genome evolution. Nucleic Acids Res. 2001, 29, 3742–3756. [Google Scholar] [CrossRef]
- Mruk, I.; Kobayashi, I. To be or not to be: Regulation of restriction–modification systems and other toxin–antitoxin systems. Nucleic Acids Res. 2013, 42, 70–86. [Google Scholar] [CrossRef]
- Nagornykh, M.; Bogdanova, E.; Protsenko, A.; Solonin, A.; Zakharova, M.; Severinov, K. Regulation of gene expression in a type II restriction-modification system. Russ. J. Genet. 2008, 44, 523–532. [Google Scholar] [CrossRef]
- Bogdanova, E.; Djordjevic, M.; Papapanagiotou, I.; Heyduk, T.; Kneale, G.; Severinov, K. Transcription regulation of the type II restriction-modification system AhdI. Nucleic Acids Res. 2008, 36, 1429–1442. [Google Scholar] [CrossRef]
- Rodic, A.; Blagojevic, B.; Zdobnov, E.; Djordjevic, M. Understanding key features of bacterial restriction-modification systems through quantitative modeling. BMC Syst. Biol. 2017, 11, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Klimuk, E.; Bogdanova, E.; Nagornykh, M.; Rodic, A.; Djordjevic, M.; Medvedeva, S.; Pavlova, O.; Severinov, K. Controller protein of restriction–modification system Kpn2I affects transcription of its gene by acting as a transcription elongation roadblock. Nucleic Acids Res. 2018, 46, 10810–10826. [Google Scholar] [CrossRef] [PubMed]
- Stefan, D.; Pinel, C.; Pinhal, S.; Cinquemani, E.; Geiselmann, J.; De Jong, H. Inference of quantitative models of bacterial promoters from time-series reporter gene data. PLoS Comput. Biol. 2015, 11, e1004028. [Google Scholar] [CrossRef] [PubMed]
- Klumpp, S.; Zhang, Z.; Hwa, T. Growth rate-dependent global effects on gene expression in bacteria. Cell 2009, 139, 1366–1375. [Google Scholar] [CrossRef] [PubMed]
- Gerosa, L.; Kochanowski, K.; Heinemann, M.; Sauer, U. Dissecting specific and global transcriptional regulation of bacterial gene expression. Mol. Syst. Biol. 2013, 9, 658–668. [Google Scholar] [CrossRef] [PubMed]
- Berthoumieux, S.; De Jong, H.; Baptist, G.; Pinel, C.; Ranquet, C.; Ropers, D.; Geiselmann, J. Shared control of gene expression in bacteria by transcription factors and global physiology of the cell. Mol. Syst. Biol. 2013, 9, 634–644. [Google Scholar] [CrossRef] [PubMed]
- Savageau, M.A. Design of molecular control mechanisms and the demand for gene expression. Proc. Natl. Acad. Sci. USA 1977, 74, 5647–5651. [Google Scholar] [CrossRef]
- Klumpp, S.; Hwa, T. Bacterial growth: Global effects on gene expression, growth feedback and proteome partition. Curr. Opin. Biotechnol. 2014, 28, 96–102. [Google Scholar] [CrossRef]
- Vilar, J.M.G.; Guet, C.C.; Leibler, S. Modeling network dynamics: The lac operon, a case study. J. Cell Biol. 2003, 161, 471–476. [Google Scholar] [CrossRef]
- De Jong, H.; Geiselmann, J. Fluorescent Reporter Genes and the Analysis of Bacterial Regulatory Networks. In Hybrid Systems Biology; Maler, O., Halász, Á., Dang, T., Piazza, C., Eds.; Springer: Cham, Switzerland, 2015; pp. 27–50. [Google Scholar]
- Wang, L.-Z.; Wu, F.; Flores, K.; Lai, Y.-C.; Wang, X. Build to understand: Synthetic approaches to biology. Integr. Biol. 2015, 8, 394–408. [Google Scholar] [CrossRef]
- Zhang, C.; Tsoi, R.; You, L. Addressing biological uncertainties in engineering gene circuits. Integr. Biol. 2015, 8, 456–464. [Google Scholar] [CrossRef] [PubMed]
- De Jong, H. Modeling and simulation of genetic regulatory systems: A literature review. J. Comput. Biol. 2002, 9, 67–103. [Google Scholar] [CrossRef] [PubMed]
- Bogdanova, E.; Zakharova, M.; Streeter, S.; Taylor, J.; Heyduk, T.; Kneale, G.; Severinov, K. Transcription regulation of restriction-modification system Esp1396I. Nucleic Acids Res. 2009, 37, 3354–3366. [Google Scholar] [CrossRef] [PubMed]
- Sneppen, K.; Zocchi, G. Physics in Molecular Biology; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Rodic, A.; Blagojevic, B.; Djordjevic, M. Systems Biology of Bacterial Immune Systems: Regulation of Restriction-Modification and CRISPR-Cas Systems. In Systems Biology; Rajewsky, N., Jurga, S., Barciszewski, J., Eds.; Springer Nature: Cham, Switzerland, 2018; pp. 37–58. [Google Scholar]
- Shea, M.A.; Ackers, G.K. The OR control system of bacteriophage lambda: A physical-chemical model for gene regulation. J. Mol. Biol. 1985, 181, 211–230. [Google Scholar] [CrossRef]
- Butcher, J.C. Numerical Methods for Ordinary Differential Equations, 3rd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- Lomax, R.G.; Hahs-Vaughn, D.L. Statistical Concepts: A Second Course, 4th ed.; Routledge: Abingdon, UK, 2013. [Google Scholar]
- Phillips, R.; Kondev, J.; Theriot, J.; Garcia, H. Physical Biology of the Cell; Garland Science: New York, NY, USA, 2012. [Google Scholar]
- Press, W.H.; Teukolsky, S.A.; Vetterling, W.T.; Flannery, B.P. Numerical Recipes: The Art of Scientific Computing, 3rd ed.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- McGeehan, J.; Ball, N.J.; Streeter, S.; Thresh, S.-J.; Kneale, G. Recognition of dual symmetry by the controller protein C. Esp1396I based on the structure of the transcriptional activation complex. Nucleic Acids Res. 2011, 40, 4158–4167. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.N.; McGeehan, J.E.; Ball, N.J.; Streeter, S.D.; Thresh, S.-J.; Kneale, G. Structural analysis of DNA–protein complexes regulating the restriction–modification system Esp1396I. Acta Cryst. 2013, F69, 962–966. [Google Scholar] [CrossRef]
- Barzel, B.; Liu, Y.-Y.; Barabási, A.-L. Constructing minimal models for complex system dynamics. Nat. Commun. 2015, 6, 7186–7193. [Google Scholar] [CrossRef]
- Porreca, R.; Cinquemani, E.; Lygeros, J.; Ferrari-Trecate, G. Structural identification of unate-like genetic network models from time-lapse protein concentration measurements. In Proceedings of the 49th IEEE Conference on Decision and Control, Atlanta, GA, USA, 15–17 December 2010; pp. 2529–2534. [Google Scholar]
Sample Availability: Not applicable. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Notation | Value | Description |
|---|---|---|
| Population Dynamics | ||
| 2.3 × 10−2 | Cell division rate in the first time interval | |
| 3.1 × 10−3 | Cell division rate in the second time interval | |
| 1.5 × 102 | Time of transition between the cell division rates | |
| 2.3 × 10−2 | Plasmid division rate in the first time interval | |
| 7.5 × 10−3 | Plasmid division rate in the second time interval | |
| 4.2 × 102 | Time of transition between the plasmid division rates | |
| Restriction endonuclease dynamics | ||
| 5.3 × 10−1 | Basal transcription activity of the PCR promoter | |
| 2.7 × 10−1 | Constants which absorb the relevant interaction energies and RNA polymerase concentration | |
| 4.3 × 10−3 | ||
| 2.7 × 10−9 | ||
| 5.1 × 10−1 | Translation rate for control protein | |
| Translation rate for restriction endonuclease | ||
| 3.8 × 10−2 | Decay rate for the operon transcript | |
| 1.2 × 10−6 | Decay rate for control protein | |
| Decay rate for restriction endonuclease | ||
| Methyltransferase dynamics | ||
| 4.6 × 101 | Basal transcription activity of the PM promoter | |
| 3.0 × 10−5 | Constants which absorb the relevant interaction energies and RNA polymerase concentration | |
| 1.5 | Translation rate for methyltransferase | |
| 2.0 × 10−1 | Decay rate for the m gene transcript | |
| 8.0 × 10−4 | Decay rate for methyltransferase | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Graovac, S.; Rodic, A.; Djordjevic, M.; Severinov, K.; Djordjevic, M. Effects of Population Dynamics on Establishment of a Restriction-Modification System in a Bacterial Host. Molecules 2019, 24, 198. https://doi.org/10.3390/molecules24010198
Graovac S, Rodic A, Djordjevic M, Severinov K, Djordjevic M. Effects of Population Dynamics on Establishment of a Restriction-Modification System in a Bacterial Host. Molecules. 2019; 24(1):198. https://doi.org/10.3390/molecules24010198
Chicago/Turabian StyleGraovac, Stefan, Andjela Rodic, Magdalena Djordjevic, Konstantin Severinov, and Marko Djordjevic. 2019. "Effects of Population Dynamics on Establishment of a Restriction-Modification System in a Bacterial Host" Molecules 24, no. 1: 198. https://doi.org/10.3390/molecules24010198
APA StyleGraovac, S., Rodic, A., Djordjevic, M., Severinov, K., & Djordjevic, M. (2019). Effects of Population Dynamics on Establishment of a Restriction-Modification System in a Bacterial Host. Molecules, 24(1), 198. https://doi.org/10.3390/molecules24010198