Multi-Body Interactions in Molecular Docking Program Devised with Key Water Molecules in Protein Binding Sites


Abstract
1. Introduction
2. Results and Discussion
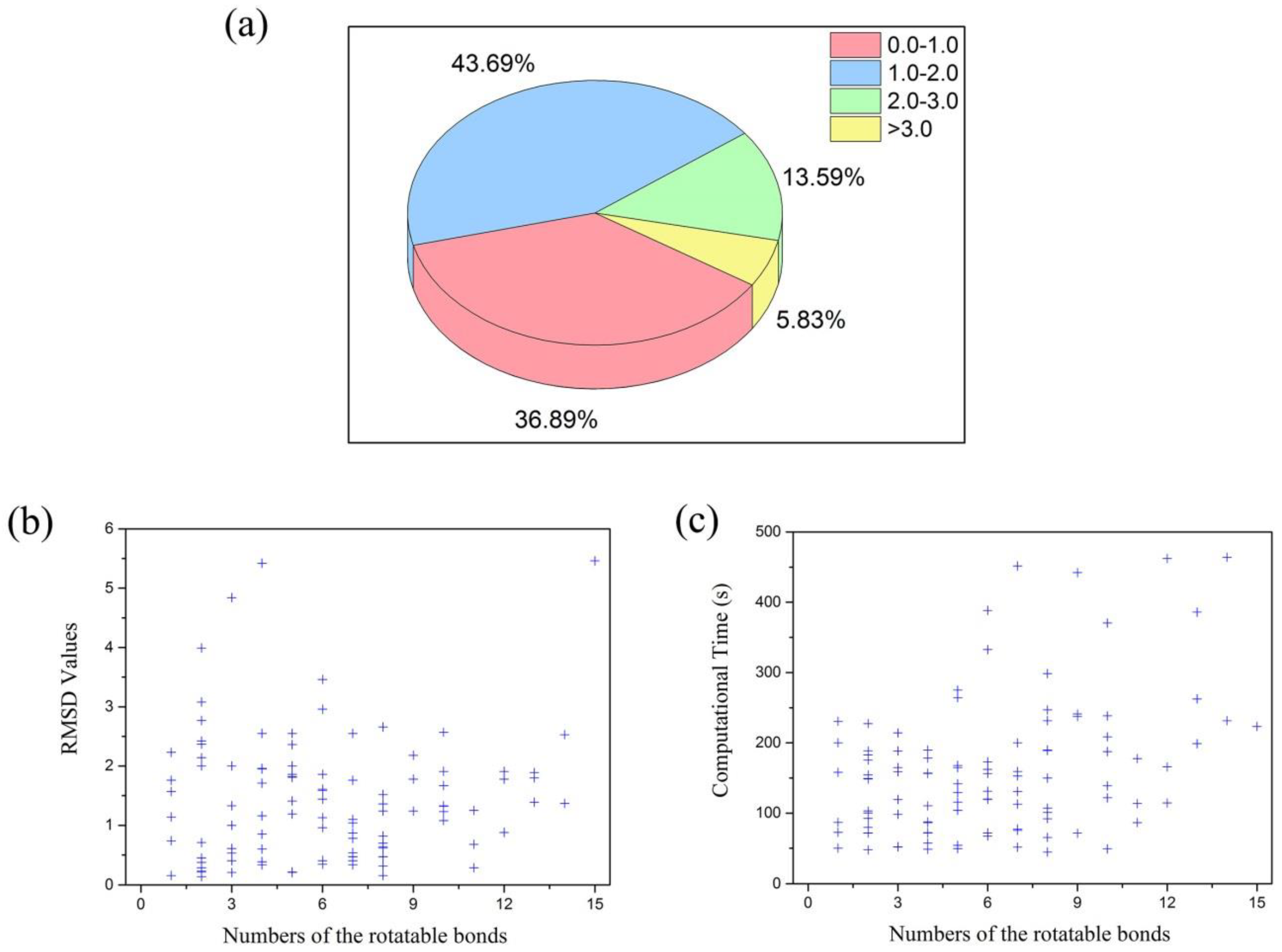
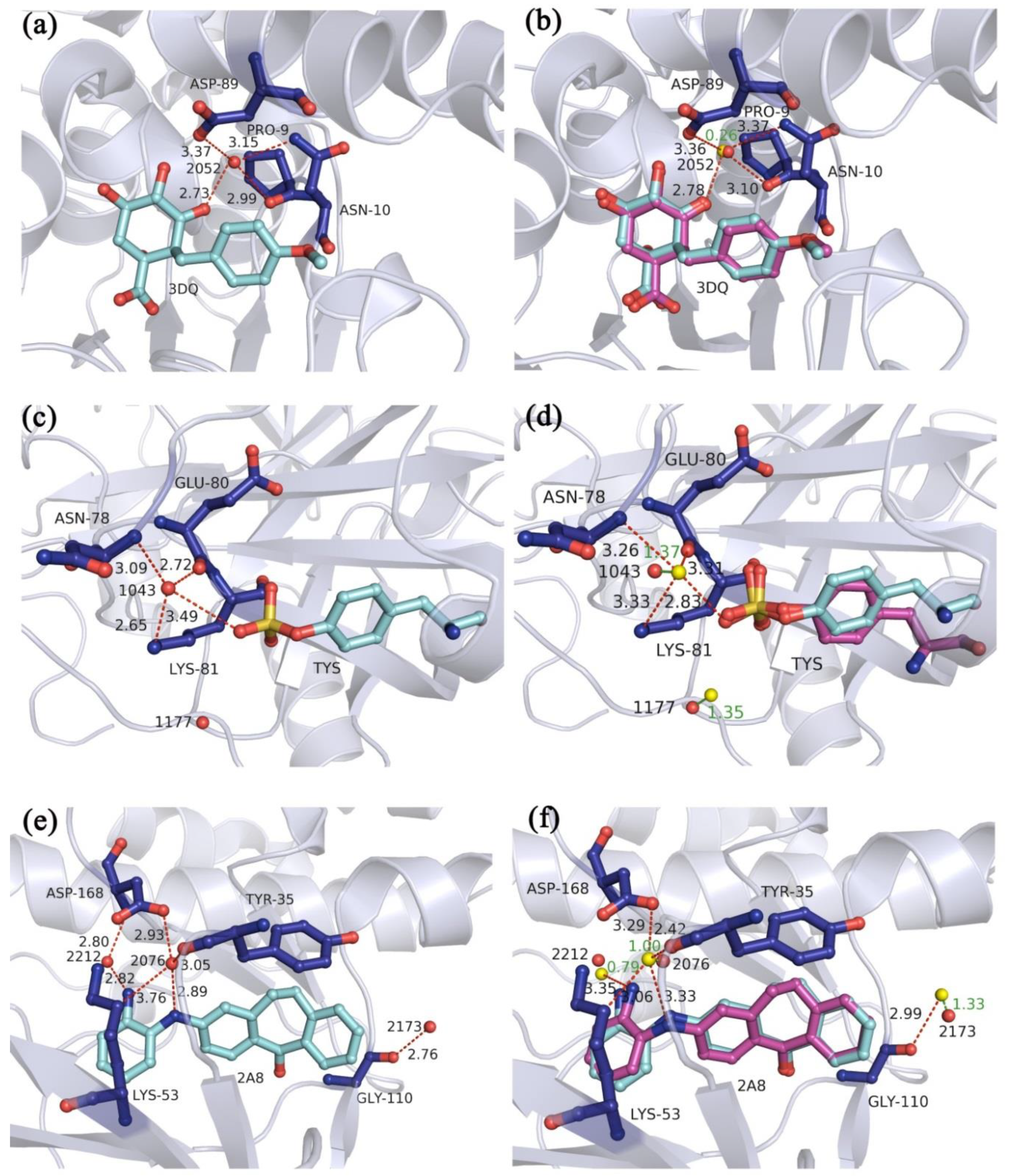
2.1. Results of Multi-Body Docking Considering the Key Water Molecules as Fixed-Length Optimization Variables
2.2. Validation of Multi-Body Docking Considering the Key Water Molecules as Fixed-Length Optimization Variables
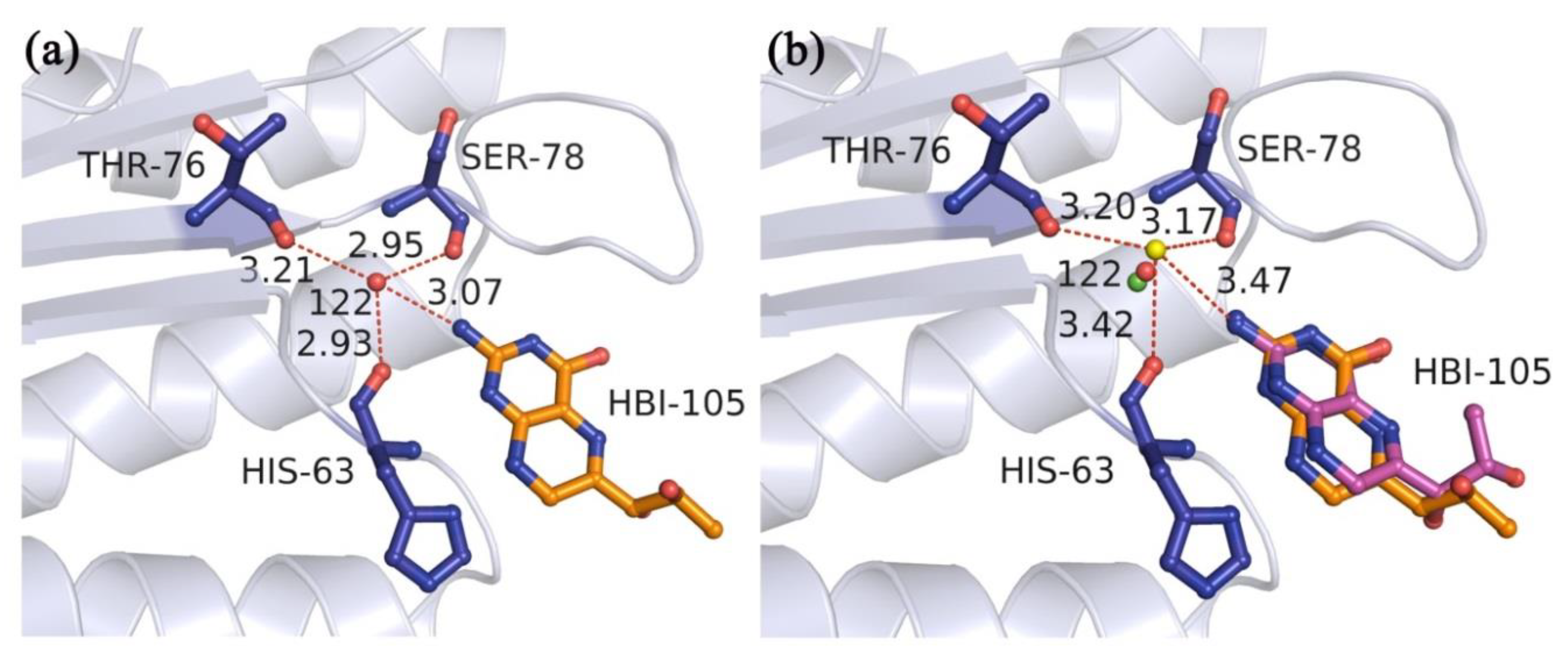
2.2.1. Docking Accuracy
2.2.2. Cross-Docking Accuracy
2.2.3. Screening Efficiency
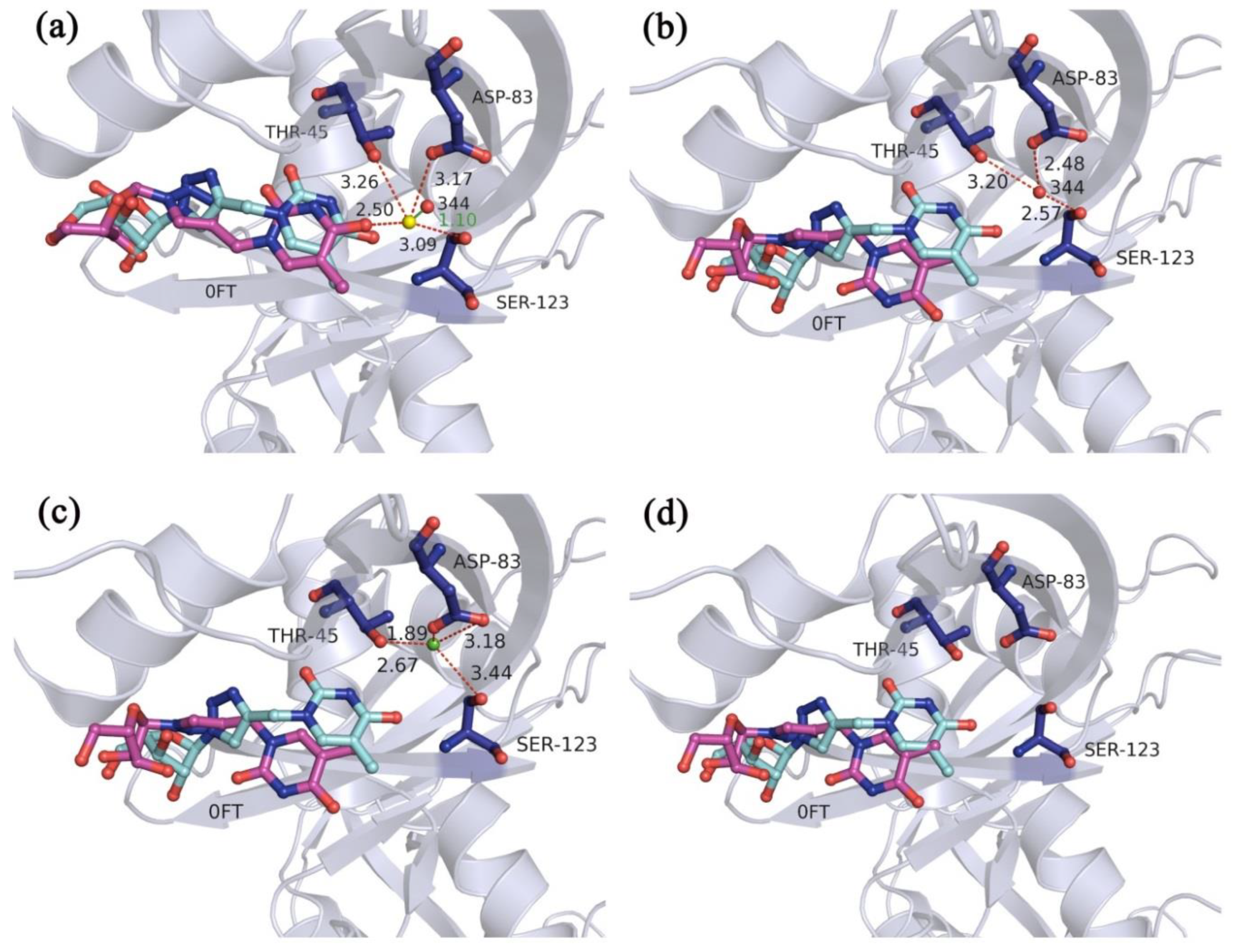
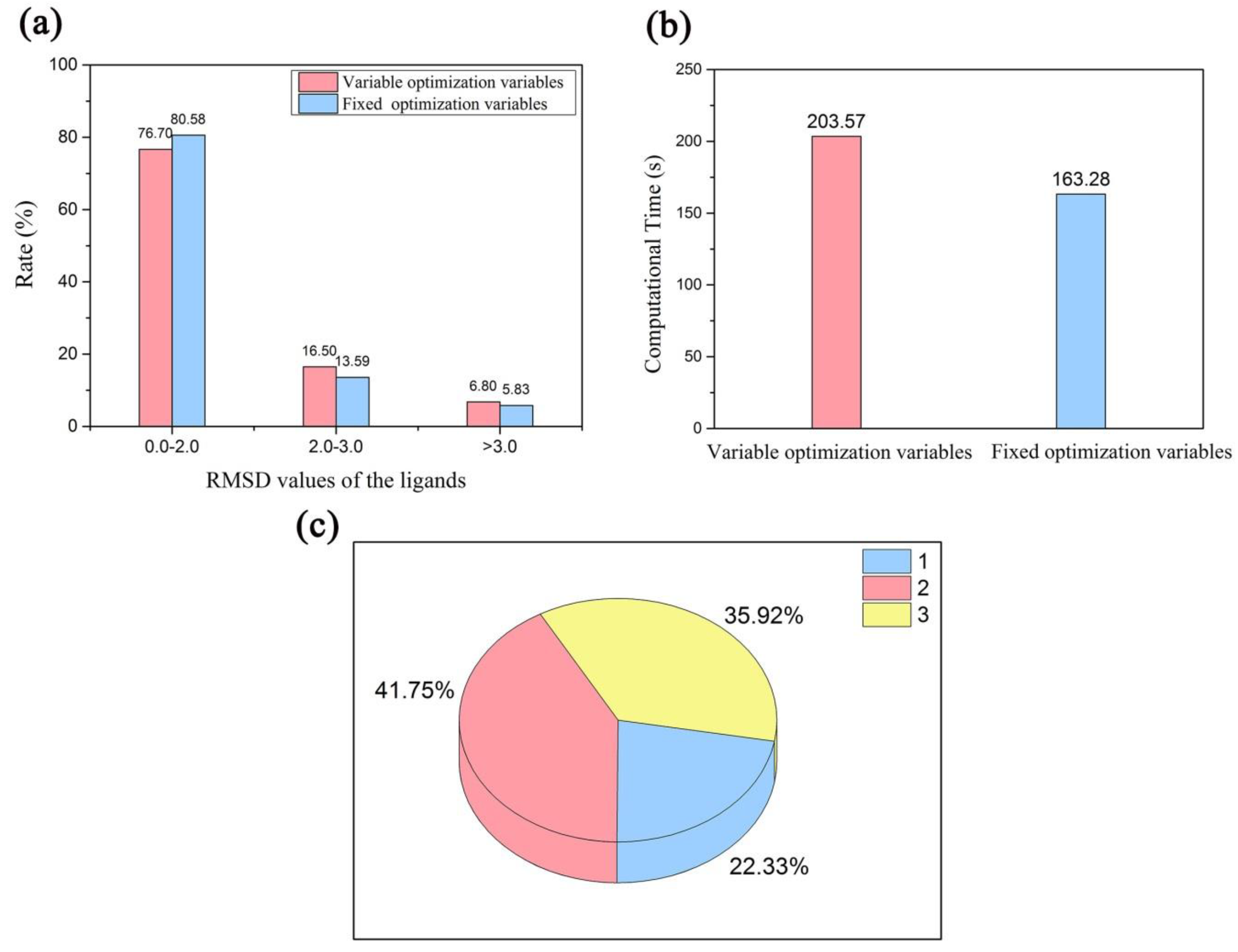
2.3. Validation of Multi-Body Docking Considering the Key Water Molecules as Variable-Length Optimization Variables
2.3.1. Docking Accuracy
2.3.2. Cross-Docking Accuracy
2.3.3. Screening Efficiency
3. Methods
3.1. Model of the Multi-Body Interaction
3.2. Multi-Body Interaction Considering the Key Water Molecules as Variable-Length Optimization Variables
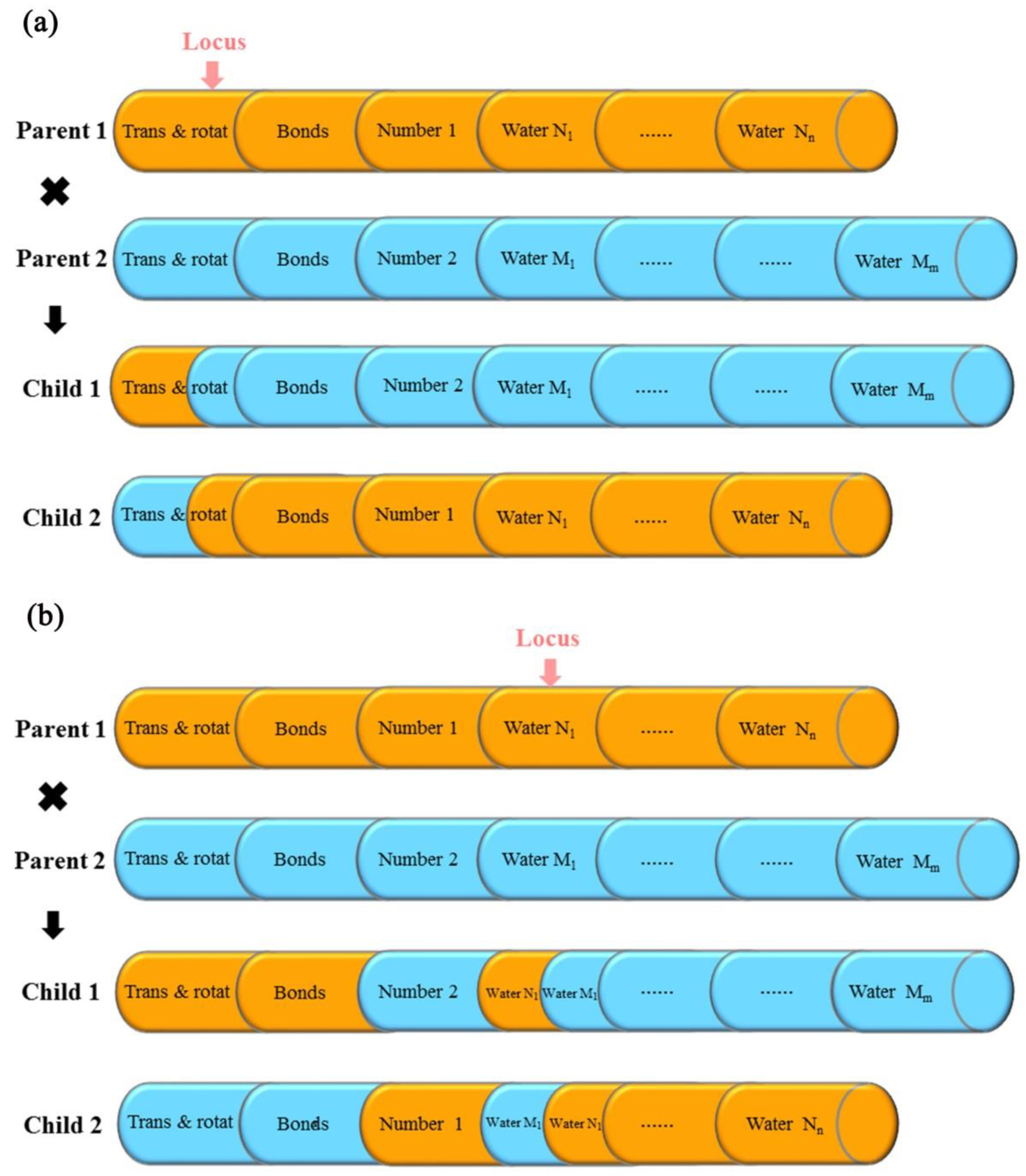
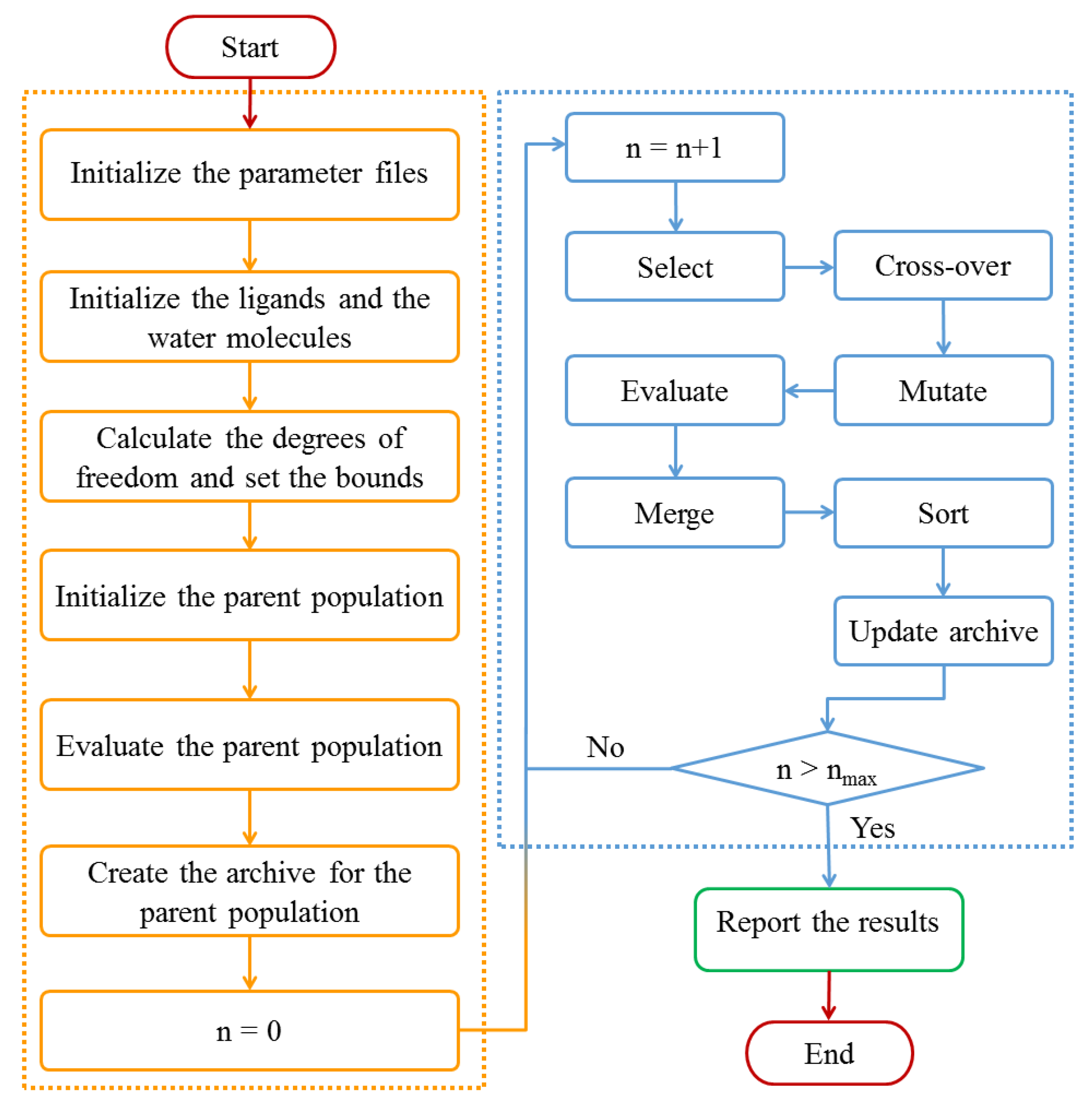
3.3. Design of Multi-Body Docking
3.3.1. Multi-Objective Optimization Model and Algorithm for Multi-Body Docking
3.3.2. Scoring Function Designed for Multi-Body Docking.
3.4. Properties of Multi-Body Docking
3.5. Preparation of the Data Sets
3.6. Validation of Multi-Body Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Protein-ligand docking: Current status and future challenges. Proteins 2006, 65, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Skolnick, J. Assessment of programs for ligand binding affinity prediction. J. Comput. Chem. 2008, 29, 1316–1331. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; He, Z.; Sun, M.; Li, S.; Li, H. Statistical Analysis, Investigation, and Prediction of the Water Positions in the Binding Sites of Proteins. J. Chem. Inf. Model. 2017, 57, 1517–1528. [Google Scholar] [CrossRef] [PubMed]
- Mancera, R.L. De novo ligand design with explicit water molecules: An application to bacterial neuraminidase. J. Comput. Aided Mol. Des. 2002, 16, 479–499. [Google Scholar] [CrossRef] [PubMed]
- Breiten, B.; Lockett, M.R.; Sherman, W.; Fujita, S.; Al-Sayah, M.; Lange, H.; Bowers, C.M.; Heroux, A.; Krilov, G.; Whitesides, G.M. Water networks contribute to enthalpy/entropy compensation in protein-ligand binding. J. Am. Chem. Soc. 2013, 135, 15579–15584. [Google Scholar] [CrossRef] [PubMed]
- De Beer, S.; Vermeulen, N.P.; Oostenbrink, C. The role of water molecules in computational drug design. Curr. Top. Med. Chem. 2010, 10, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Shoichet, B.K. Exploiting ordered waters in molecular docking. J. Med. Chem. 2008, 51, 4862–4865. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.C.; Mancera, R.L. Ligand-protein docking with water molecules. J. Chem. Inf. Model. 2008, 48, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Melville, J.L.; Hirst, J.D. Molecular docking and QSAR of aplyronine A and analogues: Potent inhibitors of actin. J. Comput. Aided Mol. Des. 2010, 24, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Chessari, G.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Nissink, J.W.M.; Taylor, R.D.; Taylor, R. Modeling water molecules in protein-ligand docking using GOLD. J. Med. Chem. 2005, 48, 6504–6515. [Google Scholar] [CrossRef] [PubMed]
- Thilagavathi, R.; Mancera, R.L. Ligand-protein cross-docking with water molecules. J. Chem. Inf. Model. 2010, 50, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, D.G.; Garcia-Sosa, A.T.; Alberts, I.L.; Todorov, N.P.; Mancera, R.L. The effect of tightly bound water molecules on the structural interpretation of ligand-derived pharmacophore models. J. Comput. Aided Mol. Des. 2004, 18, 89–100. [Google Scholar] [CrossRef] [PubMed]
- García-Sosa, A.T.; Mancera, R.L. The effect of a tightly bound water molecule on scaffold diversity in the computer-aided de novo ligand design of CDK2 inhibitors. J. Mol. Model. 2006, 12, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Rarey, M.; Kramer, B.; Lengauer, T. The particle concept: Placing discrete water molecules during protein-ligand docking predictions. Proteins 1999, 34, 17–28. [Google Scholar] [CrossRef]
- Osterberg, F.; Morris, G.M.; Sanner, M.F.; Olson, A.J.; Goodsell, D.S. Automated docking to multiple target structures: Incorporation of protein mobility and structural water heterogeneity in AutoDock. Proteins 2002, 46, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Corbeil, C.R.; Moitessier, N. Docking ligands into flexible and solvated macromolecules. 3. Impact of input ligand conformation, protein flexibility, and water molecules on the accuracy of docking programs. J. Chem. Inf. Model. 2009, 49, 997–1009. [Google Scholar] [CrossRef] [PubMed]
- Lie, M.A.; Thomsen, R.; Pedersen, C.N.; Schiott, B.; Christensen, M.H. Molecular docking with ligand attached water molecules. J. Chem. Inf. Model. 2011, 51, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Goodsell, D.S.; Olson, A.J. Automated docking of substrates to proteins by simulated annealing. Proteins Struct. Funct. Bioinf. 1990, 8, 195–202. [Google Scholar] [CrossRef] [PubMed]
- García-Sosa, A.T.; Firth-Clark, S.; Mancera, R.L. Including tightly-bound water molecules in de novo drug design. Exemplification through the in silico generation of poly (ADP-ribose) polymerase ligands. J. Chem. Inf. Model. 2005, 45, 624–633. [Google Scholar] [CrossRef] [PubMed]
- García-Sosa, A.T.; Mancera, R.L. Free Energy Calculations of Mutations Involving a Tightly Bound Water Molecule and Ligand Substitutions in a Ligand-Protein Complex. Mol. Inform. 2010, 29, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, J.F.; Cvitkovic, M.W.; Bettens, R.P. Trouble with the many-body expansion. J. Chem. Theory Comput. 2014, 10, 3699–3707. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, J.F.; Bettens, R.P. Many-body basis set superposition effect. J. Chem. Theory Comput. 2015, 11, 5132–5143. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein–ligand docking using GOLD. Proteins Struct. Funct. Bioinf. 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Therrien, E.; Weill, N.; Tomberg, A.; Corbeil, C.R.; Lee, D.; Moitessier, N. Docking ligands into flexible and solvated macromolecules. 7. Impact of protein flexibility and water molecules on docking-based virtual screening accuracy. J. Chem. Inf. Model. 2014, 54, 3198–3210. [Google Scholar] [CrossRef] [PubMed]
- Deb, K.; Pratap, A.; Agarwal, S.; Meyarivan, T. A fast and elitist multiobjective genetic algorithm: NSGA-II. IEEE Trans. Evol. Comput. 2002, 6, 182–197. [Google Scholar] [CrossRef]
- Bai, F.; Liao, S.; Gu, J.; Jiang, H.; Wang, X.; Li, H. An accurate metalloprotein-specific scoring function and molecular docking program devised by a dynamic sampling and iteration optimization strategy. J. Chem. Inf. Model. 2015, 55, 833–847. [Google Scholar] [CrossRef] [PubMed]
- Forli, S.; Olson, A.J. A force field with discrete displaceable waters and desolvation entropy for hydrated ligand docking. J. Med. Chem. 2012, 55, 623–638. [Google Scholar] [CrossRef] [PubMed]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins Struct. Funct. Bioinf. 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, R.C.; Aynechi, T.; Case, D.A.; Kuntz, I.D. Estimation of absolute free energies of hydration using continuum methods: Accuracy of partial charge models and optimization of nonpolar contributions. J. Chem. Theory Comput. 2006, 2, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Huey, R.; Morris, G.M.; Olson, A.J.; Goodsell, D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Babakhani, A.; Talley, T.T.; Taylor, P.; McCammon, J.A. A virtual screening study of the acetylcholine binding protein using a relaxed-complex approach. Comput. Biol. Chem. 2009, 33, 160–170. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nrot a | Ncom b | Min RMSD (Å) | Max RMSD (Å) | Avg RMSD (Å) | Min Time (s) | max Time (s) | Avg Time (s) |
|---|---|---|---|---|---|---|---|
| 1–5 | 49 | 0.14 | 5.42 | 1.51 | 47.70 | 275.06 | 129.01 |
| 6–10 | 42 | 0.16 | 3.46 | 1.28 | 44.70 | 451.77 | 181.18 |
| 11–15 | 12 | 0.29 | 5.46 | 1.77 | 86.54 | 464.05 | 240.54 |
| all | 103 | 0.14 | 5.46 | 1.47 | 44.70 | 464.05 | 166.19 |
| Nrot a (Ncom b) | 1–5 | 6–10 | 11–15 | All Ligands | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RMSD (Å) | (49 Cases) | (42 Cases) | (12 Cases) | (103 Cases) | ||||||||||
| Method | min | max | avg | min | max | avg | min | max | avg | min | max | avg | ||
| Multi-body docking | 0.14 | 5.42 | 1.51 | 0.16 | 3.46 | 1.28 | 0.29 | 5.46 | 1.77 | 0.14 | 5.46 | 1.47 | ||
| AutoDock | 0.14 | 3.68 | 1.27 | 0.39 | 6.93 | 1.46 | 0.68 | 2.96 | 1.65 | 0.14 | 6.93 | 1.39 | ||
| AutoDock Vina | 0.22 | 7.03 | 1.88 | 0.48 | 5.84 | 1.86 | 1.19 | 3.27 | 2.16 | 0.22 | 7.03 | 1.90 | ||
| Gold (ChemScore) | 0.14 | 11.36 | 1.31 | 0.17 | 4.99 | 1.14 | 0.20 | 9.14 | 1.80 | 0.14 | 11.36 | 1.30 | ||
| Gold (GoldScore) | 0.22 | 4.75 | 0.89 | 0.35 | 3.41 | 0.96 | 0.35 | 4.68 | 1.20 | 0.22 | 4.75 | 0.86 | ||
| Protein Target | L a | 1B8N | 1B8O | 2AI1 | 2AI2 | 3FUC | ||
| V b | ||||||||
| R c | ||||||||
| PNP | 1B8N | 0.19 (5.71) | 0.30 (0.62) | 1.72 (6.40) | 1.26 (6.46) | 4.88 (4.69) | ||
| 1B8O | 0.28 (0.35) | 0.16 (0.30) | 1.17 (1.25) | 1.10 (1.51) | 4.83 (4.55) | |||
| 2AI1 | 1.09 (0.98) | 0.65 (5.12) | 0.56 (6.21) | 0.43 (6.22) | 5.46 (6.36) | |||
| 2AI2 | 2.62 (5.70) | 0.88 (5.78) | 3.61 (7.01) | 0.55 (0.45) | 5.00 (7.80) | |||
| 3FUC | 5.14 (5.07) | 5.17 (5.12) | 3.48 (7.66) | 4.62 (3.88) | 4.62 (7.29) | |||
| COX-1 | 1EQG | 1IGZ | 1Q4G | 2AYL | 2OYE | |||
| 1EQG | 0.93 (0.40) | 3.61 (5.01) | 0.25 (0.30) | 2.19 (2.88) | 2.87 (4.13) | |||
| 1IGZ | 3.15 (3.77) | 3.48 (4.70) | 3.01 (3.10) | 3.39 (3.85) | 3.71 (3.84) | |||
| 1Q4G | 0.43 (0.48) | 5.41 (4.70) | 0.20 (0.48) | 2.45 (5.22) | 3.81 (2.78) | |||
| 2AYL | 1.00 (0.54) | 4.89 (5.08) | 0.24 (0.26) | 2.18 (4.92) | 2.67 (3.23) | |||
| 2OYE | 2.00 (1.91) | 4.84 (5.33) | 1.83 (2.29) | 5.66 (2.28) | 1.33 (1.32) | |||
| HIVRT | 1C1B | 1RT1 | 1RTH | 1VRT | 1VRU | |||
| 1C1B | 2.34 (5.04) | 1.79 (4.69) | 1.94 (1.95) | 1.37 (1.38) | 1.47 (1.92) | |||
| 1RT1 | 0.99 (2.00) | 0.90 (3.37) | 1.81 (1.72) | 1.42 (1.71) | 1.99 (5.63) | |||
| 1RTH | 3.14 (4.93) | 2.61 (3.42) | 0.32 (0.36) | 0.38 (0.38) | 2.29 (5.11) | |||
| 1VRT | 2.98 (3.79) | 1.86 (3.79) | 1.59 (1.66) | 0.30 (0.31) | 2.11 (4.70) | |||
| 1VRU | 2.42 (3.67) | 3.81 (3.87) | 1.20 (1.19) | 0.84 (0.91) | 2.00 (2.07) | |||
| ER agonist | 1GWQ | 1GWR | 1L2I | 1XPC | 2IOG | |||
| 1GWQ | 0.58 (0.78) | 0.51 (0.86) | 0.59 (6.70) | 3.90 (2.30) | 4.52 (5.18) | |||
| 1GWR | 0.72 (0.75) | 0.18 (0.30) | 0.51 (0.59) | 3.29 (5.75) | 3.18 (2.77) | |||
| 1L2I | 0.44 (0.59) | 0.52 (0.59) | 0.55 (0.45) | 3.34 (6.01) | 4.98 (4.83) | |||
| 1XPC | 3.24 (5.41) | 2.63 (4.99) | 4.29 (5.08) | 1.88 (1.76) | 3.74 (4.80) | |||
| 2IOG | 2.69(2.64) | 3.53 (4.07) | 4.11 (4.39) | 2.26 (2.56) | 3.32 (5.67) | |||
| Protein Targets | Programs | True Hits Rate in the Top 200 Scorers | EF in the Top 5% Scorers | AUC Values |
|---|---|---|---|---|
| PNP | Multi-body docking | 42.00% | 3.62% | 0.68 |
| AutoDock | 26.00% | 3.62% | 0.61 | |
| AutoDock Vina | 16.00% | 0.00% | 0.44 | |
| Gold (ChemScore) | 10.00% | 0.80% | 0.33 | |
| Gold (GoldScore) | 14.00% | 2.41% | 0.38 | |
| COX-1 | Multi-body docking | 64.00% | 5.70% | 0.77 |
| AutoDock | 4.00% | 0.81% | 0.21 | |
| AutoDock Vina | 64.00% | 9.77% | 0.68 | |
| Gold (ChemScore) | 32.00% | 4.88% | 0.53 | |
| Gold (GoldScore) | 12.00% | 0.00% | 0.45 | |
| HIVRT | Multi-body docking | 32.56% | 11.54% | 0.76 |
| AutoDock | 27.90% | 6.41% | 0.50 | |
| AutoDock Vina | 9.30% | 1.28% | 0.26 | |
| Gold (ChemScore) | 2.33% | 0.00% | 0.32 | |
| Gold (GoldScore) | 39.53% | 14.10% | 0.59 | |
| ER agonist | Multi-body docking | 26.87% | 5.67% | 0.82 |
| AutoDock | 29.85% | 4.17% | 0.75 | |
| AutoDock Vina | 50.75% | 8.35% | 0.80 | |
| Gold (ChemScore) | 0.00% | 0.00% | 0.11 | |
| Gold (GoldScore) | 1.49% | 0.00% | 0.28 |
| Protein Target | L a | 1B8N | 1B8O | 2AI1 | 2AI2 | 3FUC | ||
| V b | ||||||||
| R c | ||||||||
| PNP | 1B8N | 0.44 (5.71) | 0.72 (0.62) | 1.17 (6.40) | 1.21 (6.46) | 4.15 (4.69) | ||
| 1B8O | 0.25 (0.35) | 0.29 (0.30) | 1.09 (1.25) | 0.96 (1.51) | 4.77 (4.55) | |||
| 2AI1 | 0.73 (0.98) | 0.62 (5.12) | 0.38 (6.21) | 0.54 (6.22) | 6.86 (6.36) | |||
| 2AI2 | 0.82 (5.70) | 0.71 (5.78) | 6.85 (7.01) | 0.75 (0.45) | 6.83 (7.80) | |||
| 3FUC | 2.96 (5.07) | 2.98 (5.12) | 4.32 (7.66) | 4.71 (3.88) | 5.68 (7.29) | |||
| COX-1 | 1EQG | 1IGZ | 1Q4G | 2AYL | 2OYE | |||
| 1EQG | 0.22 (0.40) | 4.57 (5.01) | 0.21 (0.30) | 2.34 (2.88) | 4.60 (4.13) | |||
| 1IGZ | 3.53 (3.77) | 4.84 (4.70) | 2.89 (3.10) | 3.74 (3.85) | 4.46 (3.84) | |||
| 1Q4G | 0.44 (0.48) | 4.78 (4.70) | 0.44 (0.48) | 2.42 (5.22) | 2.80 (2.78) | |||
| 2AYL | 1.33 (0.54) | 5.66 (5.08) | 0.85 (0.26) | 2.26 (4.92) | 3.92 (3.23) | |||
| 2OYE | 2.00 (1.91) | 4.24 (5.33) | 1.96 (2.29) | 3.23 (2.28) | 1.49 (1.32) | |||
| HIVRT | 1C1B | 1RT1 | 1RTH | 1VRT | 1VRU | |||
| 1C1B | 1.54 (5.04) | 2.68 (4.69) | 1.83 (1.95) | 1.24 (1.38) | 1.77 (1.92) | |||
| 1RT1 | 1.06 (2.00) | 1.31 (3.37) | 1.82 (1.72) | 0.97 (1.71) | 2.56 (5.63) | |||
| 1RTH | 3.56 (4.93) | 3.11 (3.42) | 0.36 (0.36) | 0.40 (0.38) | 2.79 (5.11) | |||
| 1VRT | 2.73 (3.79) | 1.92 (3.79) | 1.73 (1.66) | 0.30 (0.31) | 2.46 (4.70) | |||
| 1VRU | 2.83 (3.67) | 3.10 (3.87) | 1.20 (1.19) | 0.78 (0.91) | 2.00 (2.07) | |||
| ER agonist | 1GWQ | 1GWR | 1L2I | 1XPC | 2IOG | |||
| 1GWQ | 0.38 (0.78) | 0.60 (0.86) | 0.62 (6.70) | 3.28 (2.30) | 5.67 (5.18) | |||
| 1GWR | 0.50 (0.75) | 0.14 (0.30) | 0.57 (0.59) | 3.77 (5.75) | 4.48 (2.77) | |||
| 1L2I | 0.32 (0.59) | 0.56 (0.59) | 0.47 (0.45) | 3.87 (6.01) | 4.34 (4.83) | |||
| 1XPC | 5.28 (5.41) | 5.15 (4.99) | 4.40 (5.08) | 1.20 (1.76) | 3.89 (4.80) | |||
| 2IOG | 2.63 (2.64) | 2.54 (4.07) | 4.02 (4.39) | 1.76 (2.56) | 3.99 (5.67) | |||
| Protein Targets | Fixed/Variable a | True Hits Rate in the Top 200 Scorers | EF in the Top 5% Scorers | AUC Values |
|---|---|---|---|---|
| PNP | Fixed | 42.00% | 3.62% | 0.68 |
| Variable | 56.00% | 7.25% | 0.79 | |
| COX-1 | Fixed | 64.00% | 5.70% | 0.77 |
| Variable | 56.00% | 8.14% | 0.68 | |
| HIVRT | Fixed | 32.56% | 11.54% | 0.76 |
| Variable | 48.84% | 5.12% | 0.75 | |
| ER agonist | Fixed | 26.87% | 5.67% | 0.82 |
| Variable | 25.37% | 5.07% | 0.56 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, W.; Wang, D.; Shen, Z.; Li, S.; Li, H. Multi-Body Interactions in Molecular Docking Program Devised with Key Water Molecules in Protein Binding Sites. Molecules 2018, 23, 2321. https://doi.org/10.3390/molecules23092321
Xiao W, Wang D, Shen Z, Li S, Li H. Multi-Body Interactions in Molecular Docking Program Devised with Key Water Molecules in Protein Binding Sites. Molecules. 2018; 23(9):2321. https://doi.org/10.3390/molecules23092321
Chicago/Turabian StyleXiao, Wei, Disha Wang, Zihao Shen, Shiliang Li, and Honglin Li. 2018. "Multi-Body Interactions in Molecular Docking Program Devised with Key Water Molecules in Protein Binding Sites" Molecules 23, no. 9: 2321. https://doi.org/10.3390/molecules23092321
APA StyleXiao, W., Wang, D., Shen, Z., Li, S., & Li, H. (2018). Multi-Body Interactions in Molecular Docking Program Devised with Key Water Molecules in Protein Binding Sites. Molecules, 23(9), 2321. https://doi.org/10.3390/molecules23092321