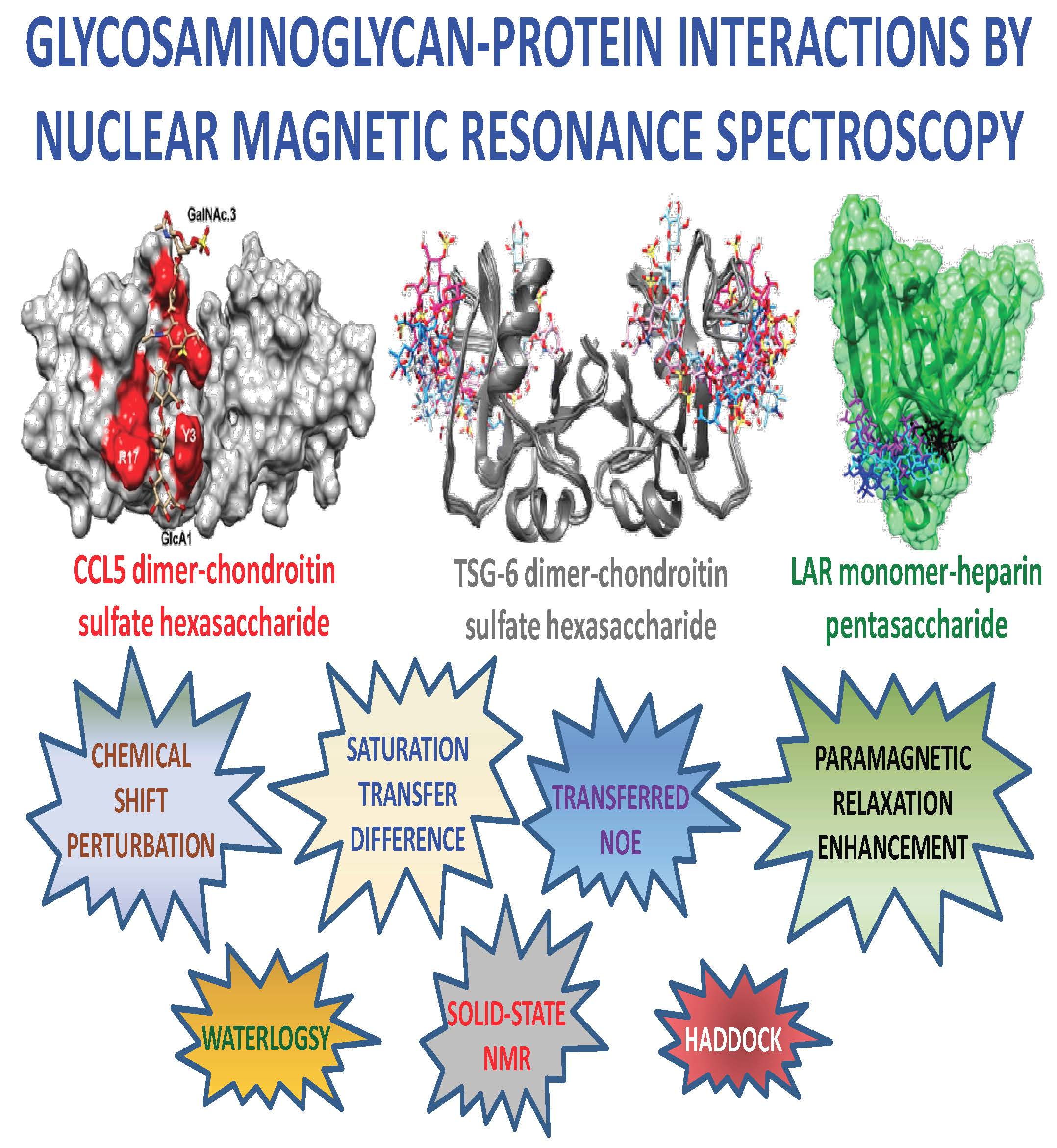
Glycosaminoglycan-Protein Interactions by Nuclear Magnetic Resonance (NMR) Spectroscopy



Abstract

1. Introduction
2. Common NMR Experiments
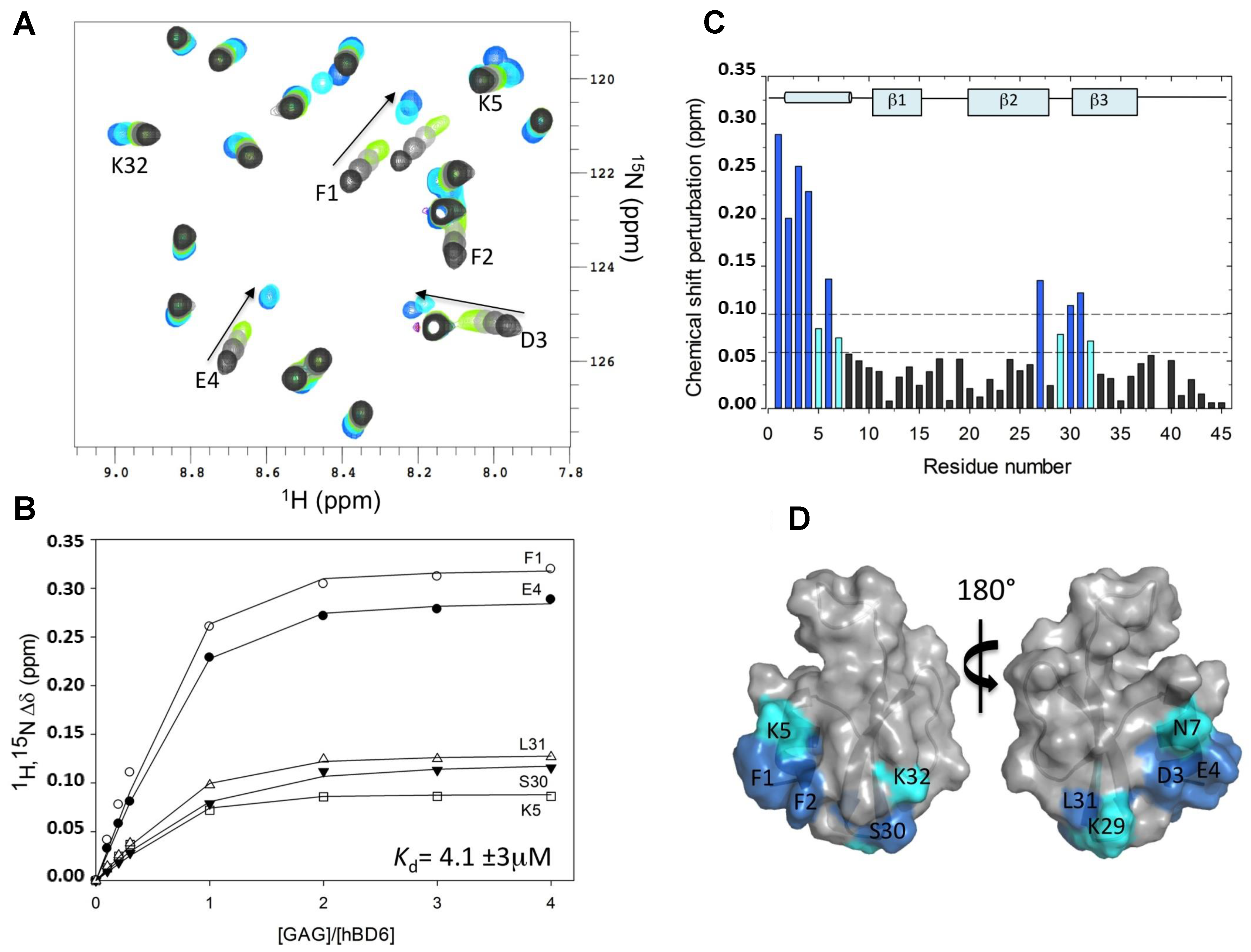
2.1. Chemical Shift Perturbation
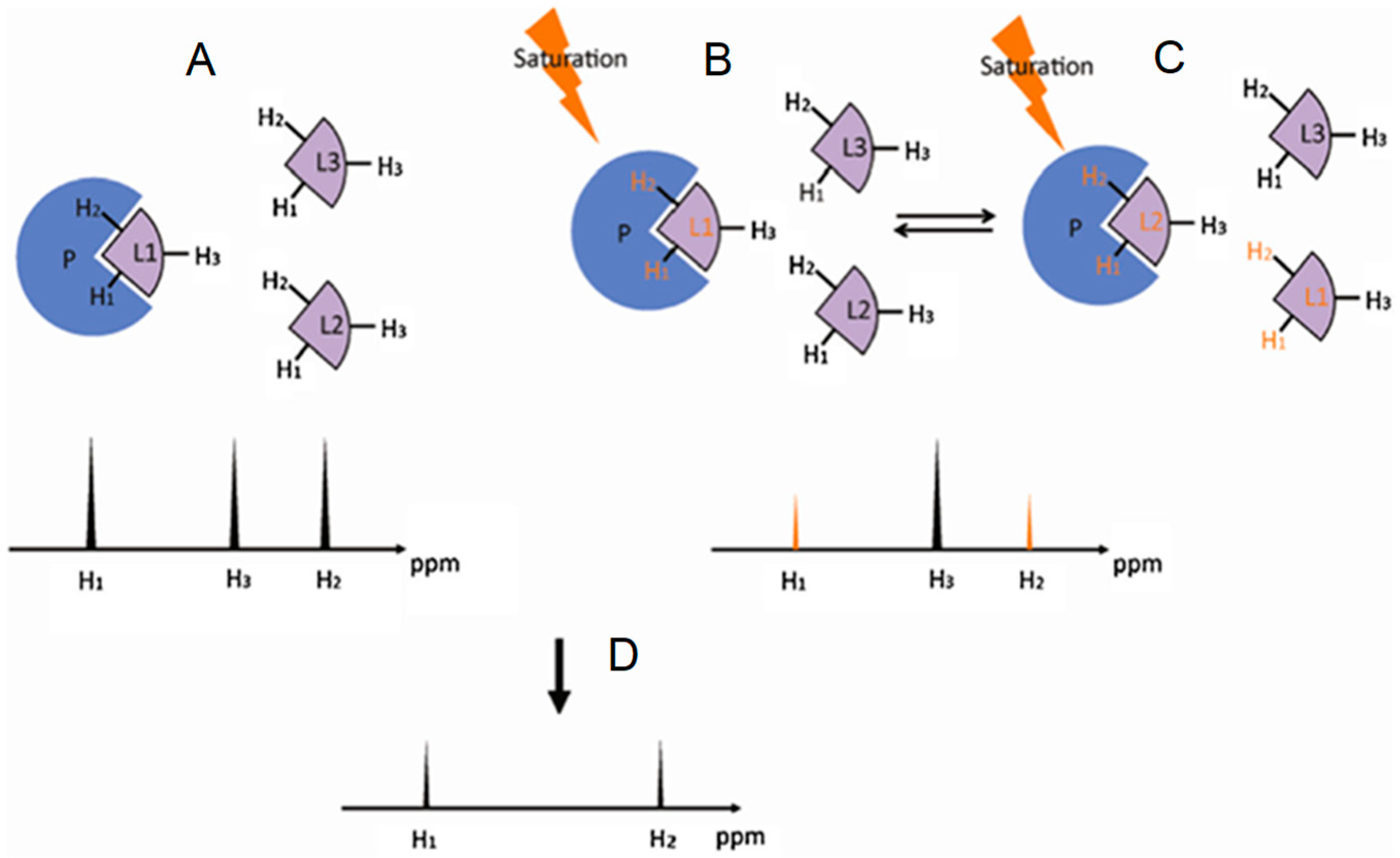
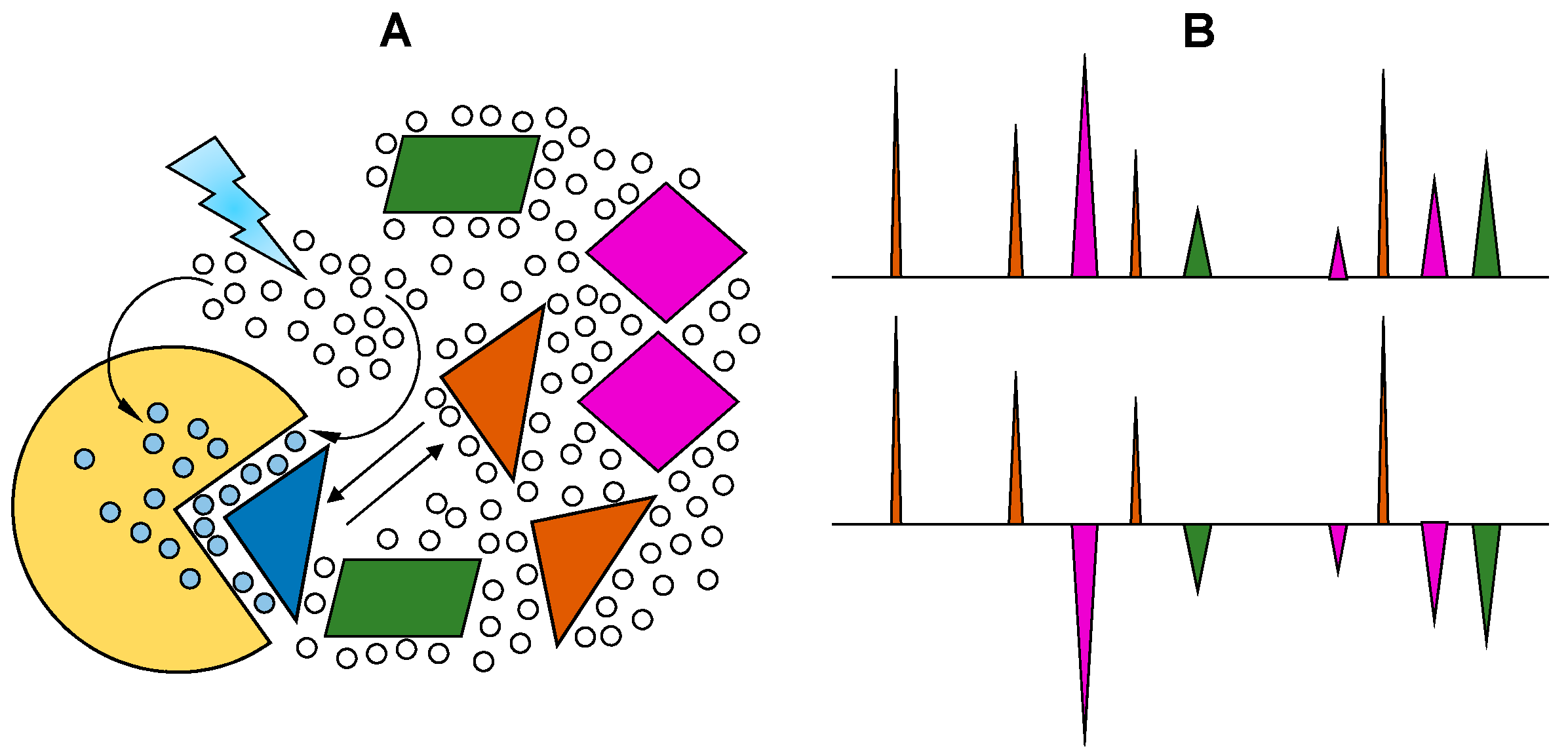
2.2. Saturation Transfer Difference
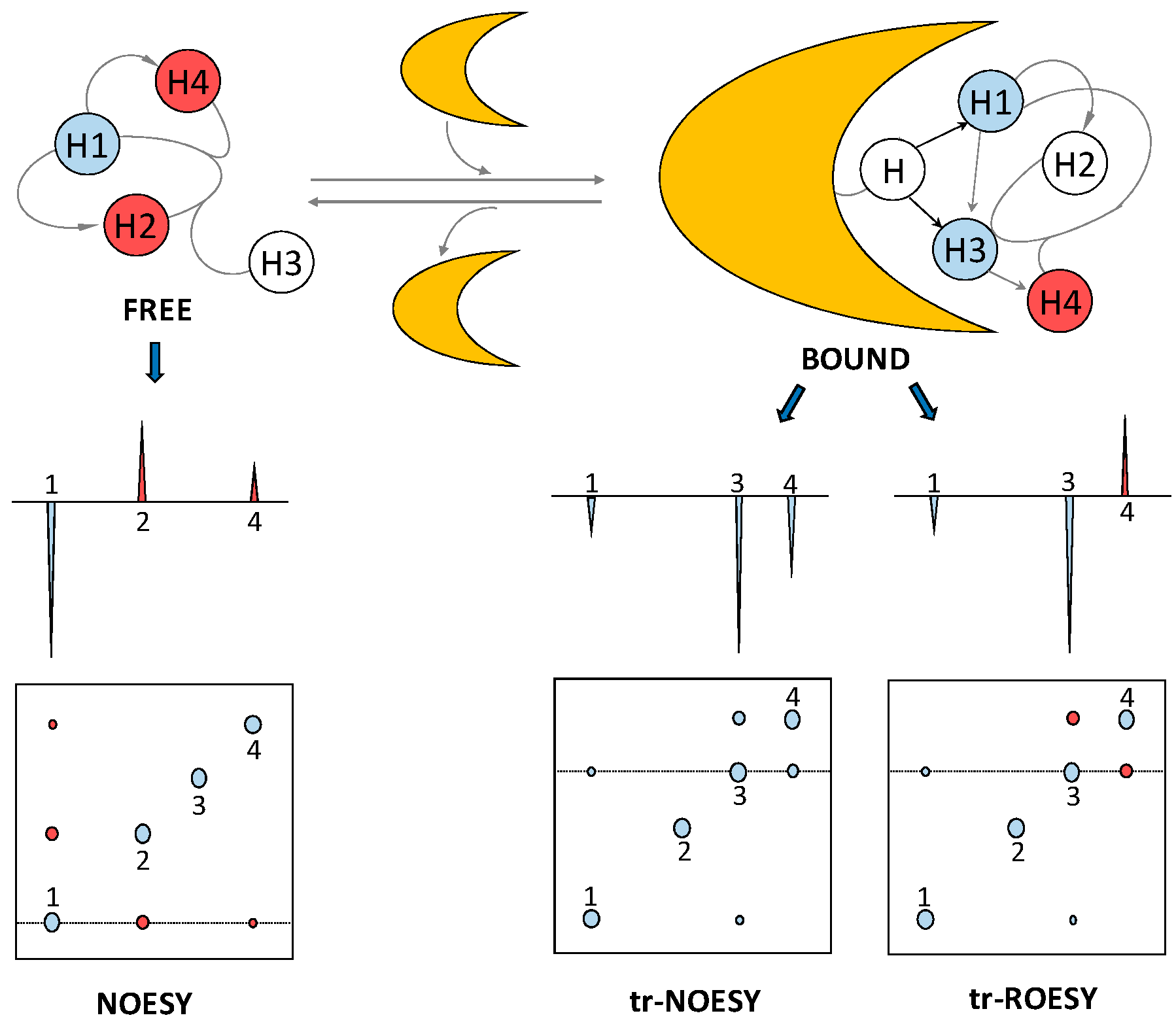
2.3. Transferred Nuclear Overhauser Effect
2.4. WaterLOGSY
3. Production of GAG Ligands for NMR
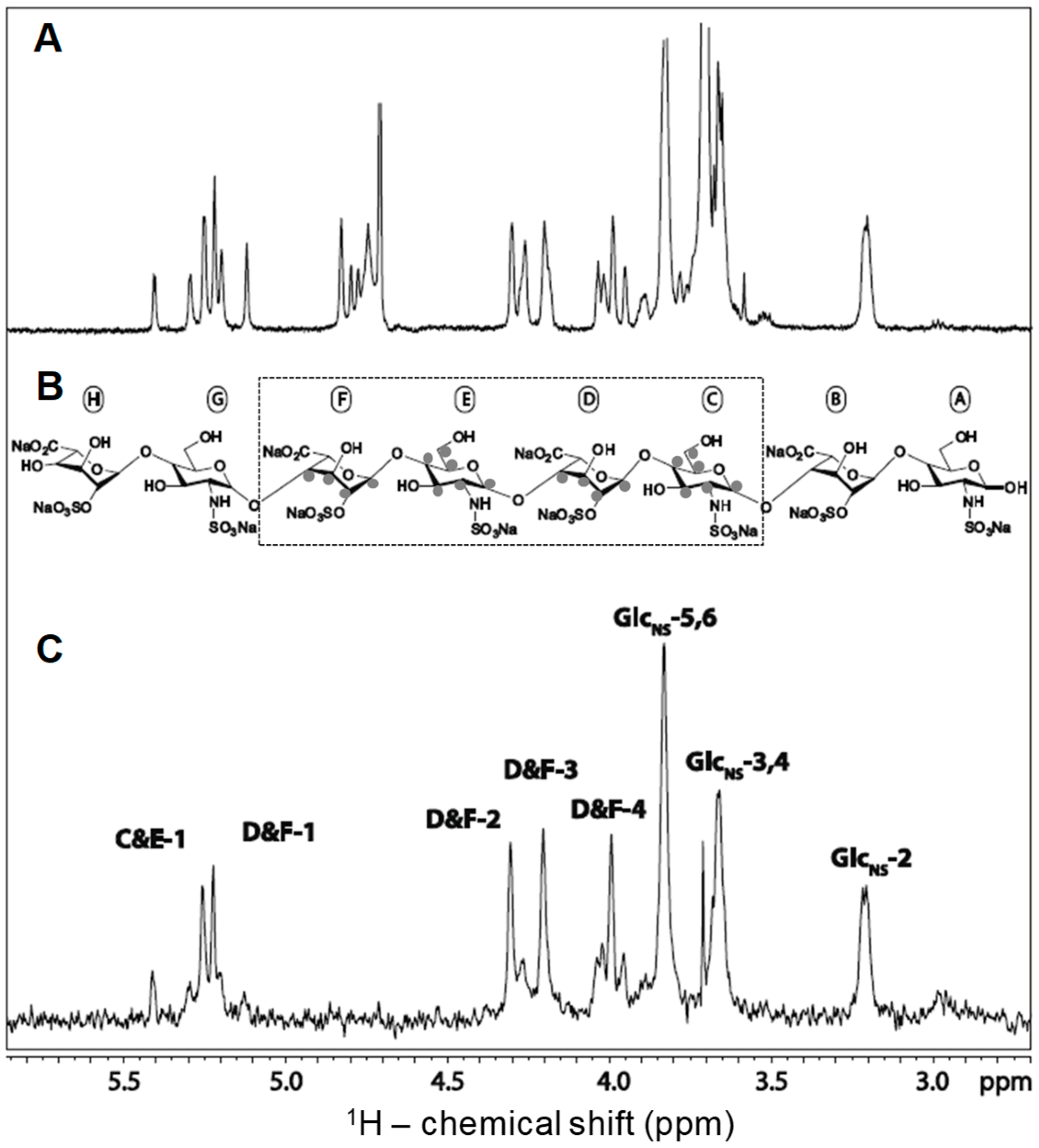
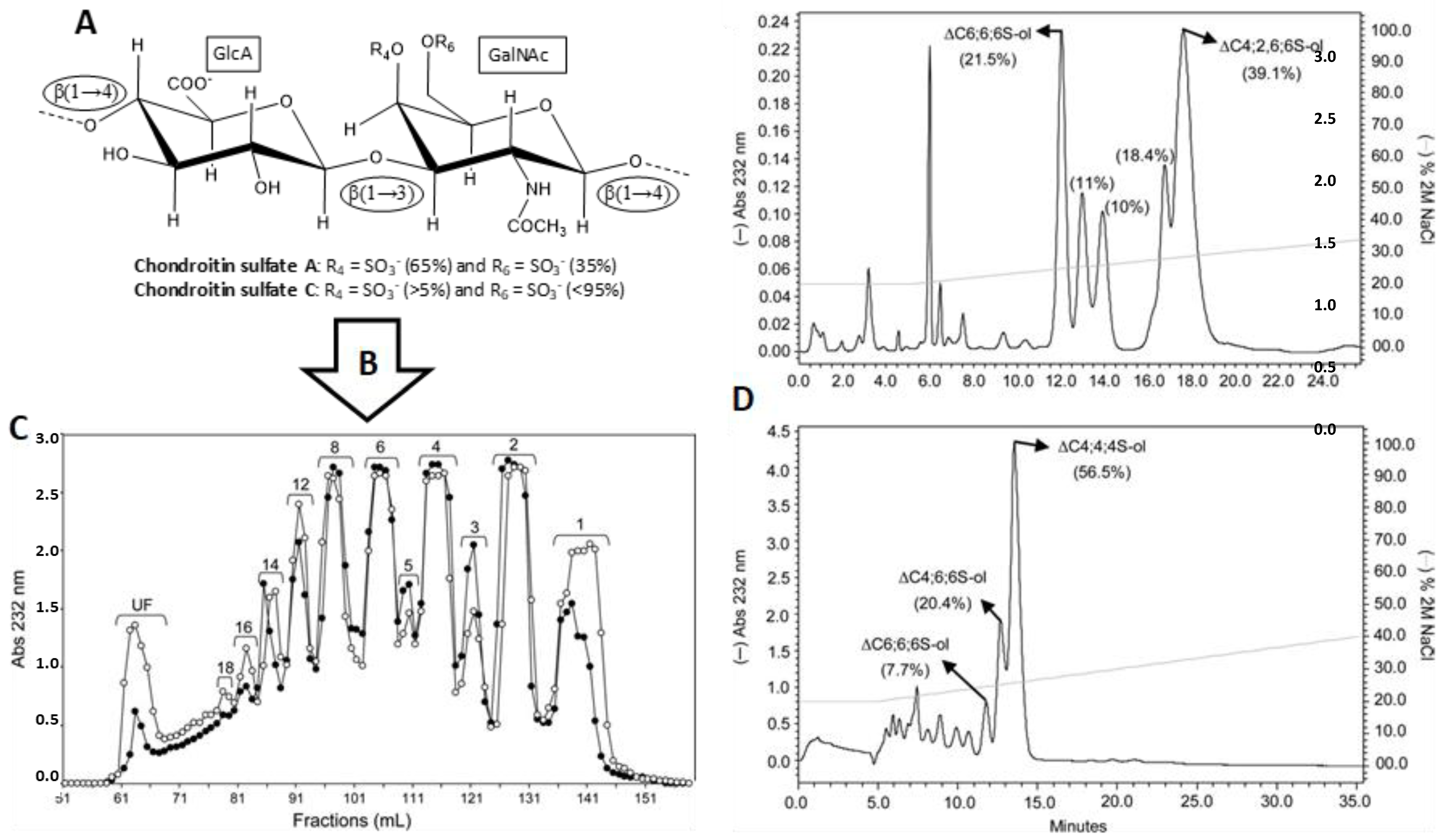
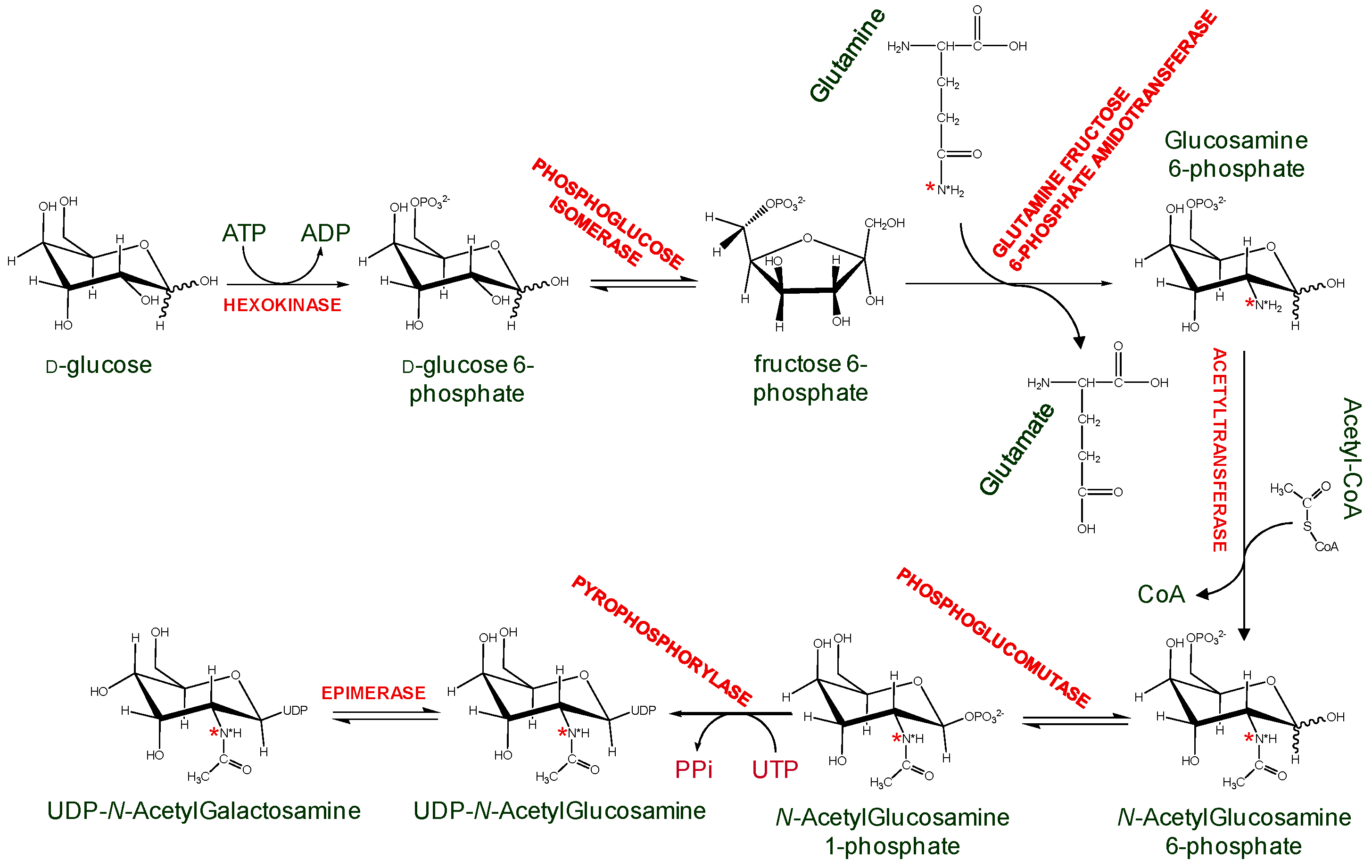
3.1. Unlabeled Oligosaccharides
3.2. Isotopically Labeled Oligosaccharides
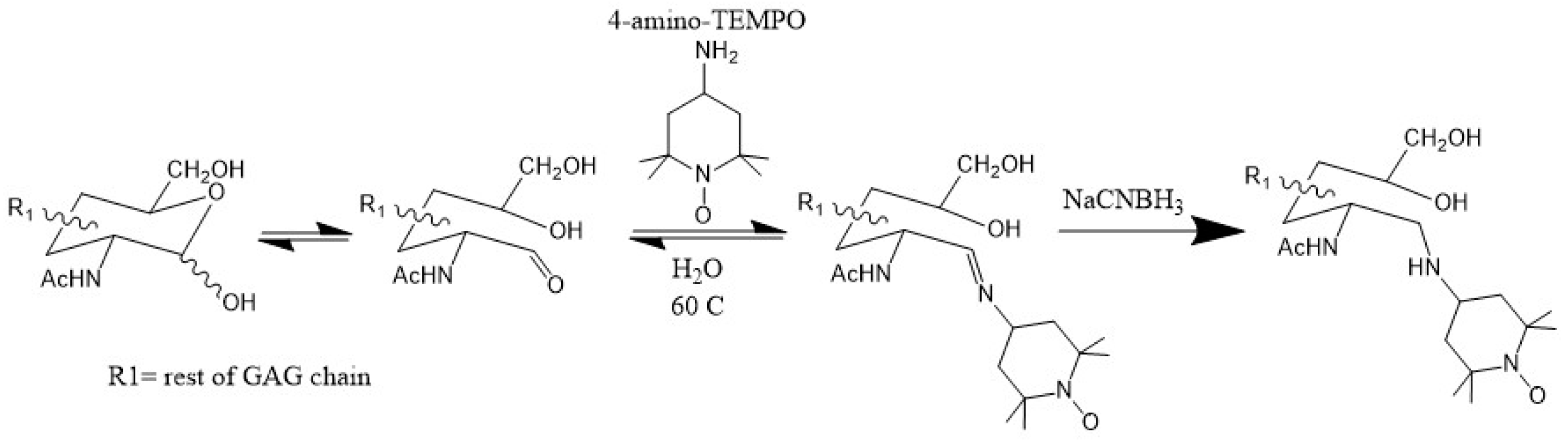
3.3. Paramagnetically Labeled Oligosaccharides
4. Characterizing Lysine and Arginine Residues in GAG-Binding Sites
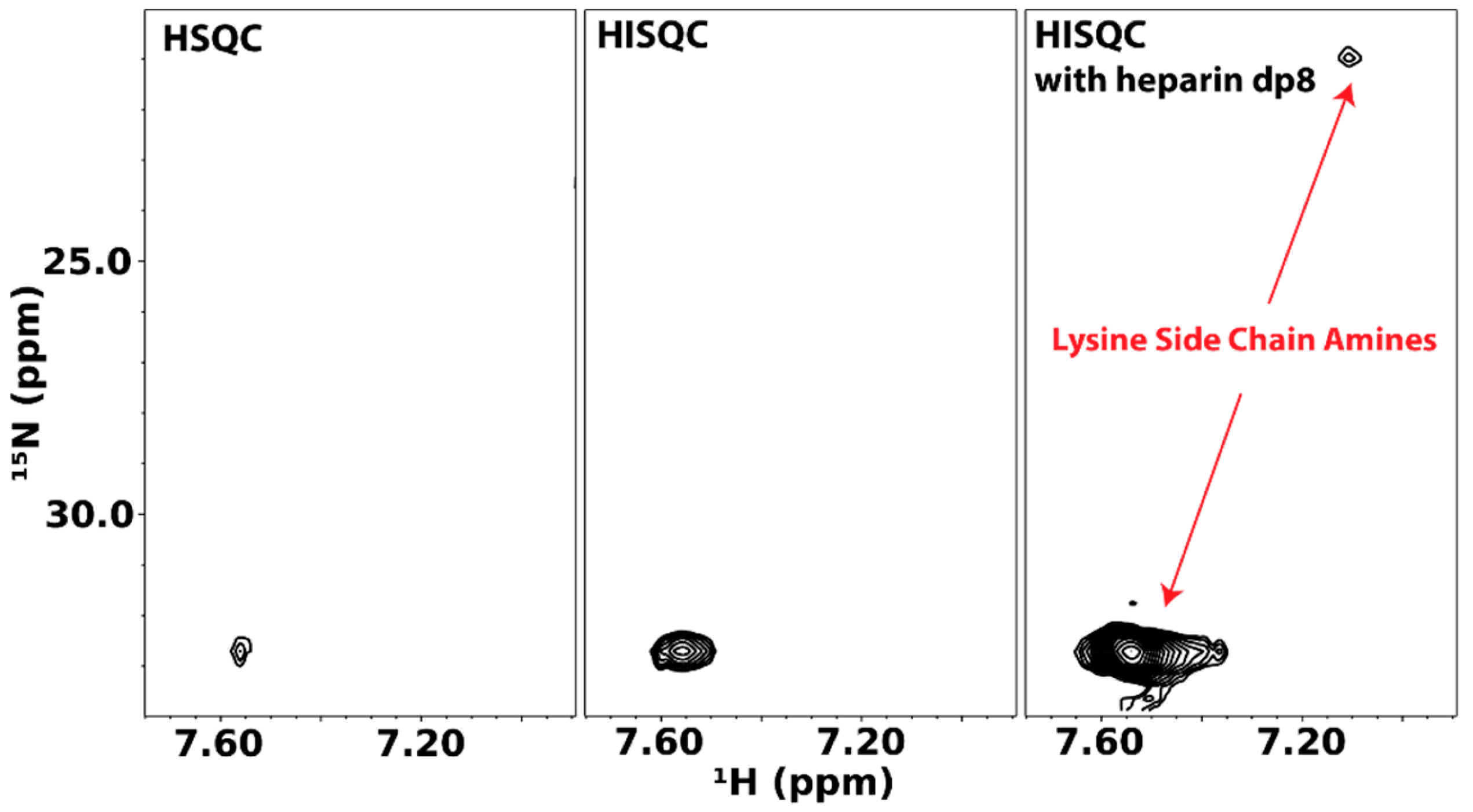
4.1. Direct Observation of Lysine Amine Signals
4.2. Specific Labeling of Lysine and Arginine Side Chains
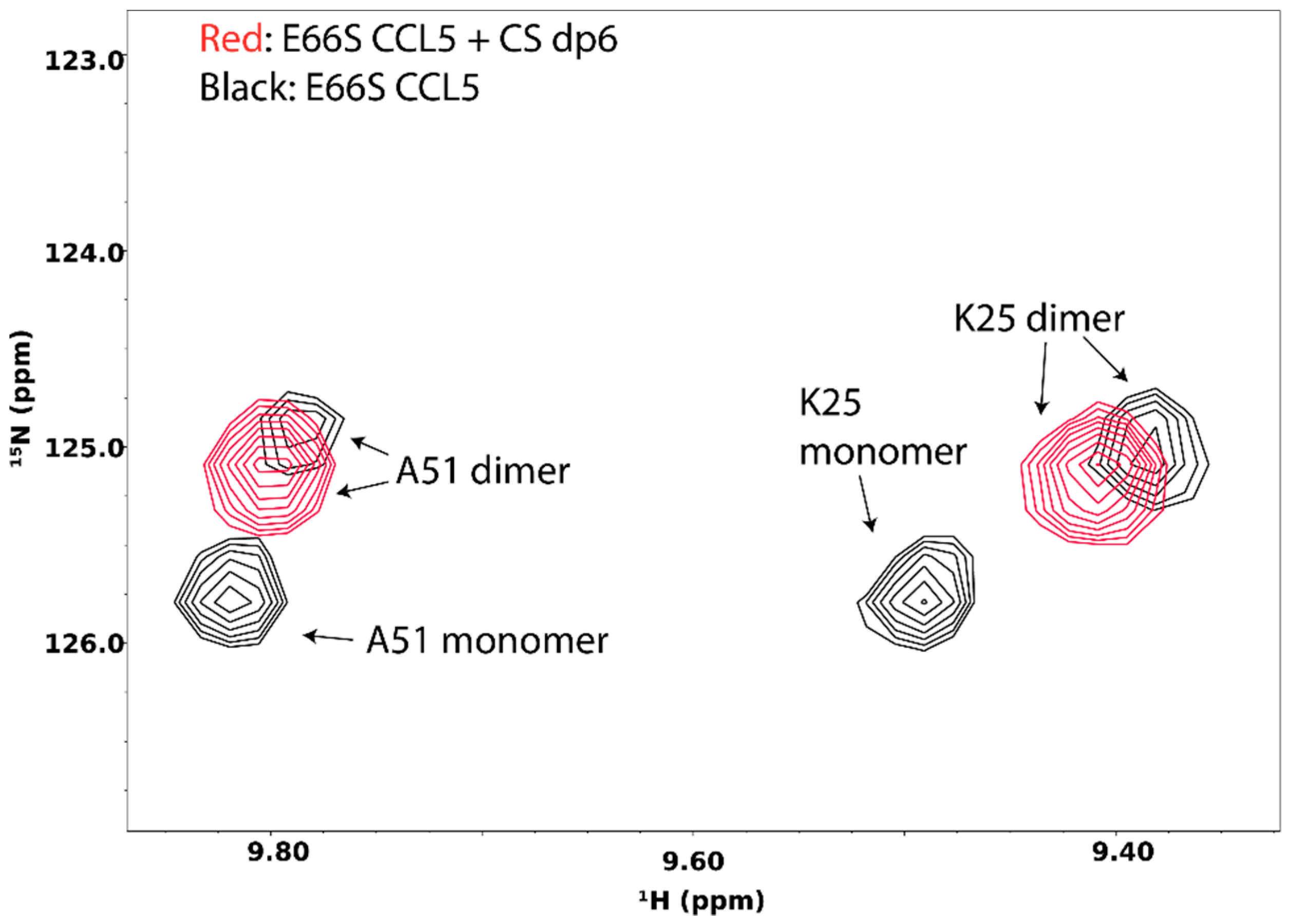
5. Probing GAG-Induced Changes in Protein Dynamics and Oligomerization
6. Solid-State NMR Studies of GAG-Protein Interactions
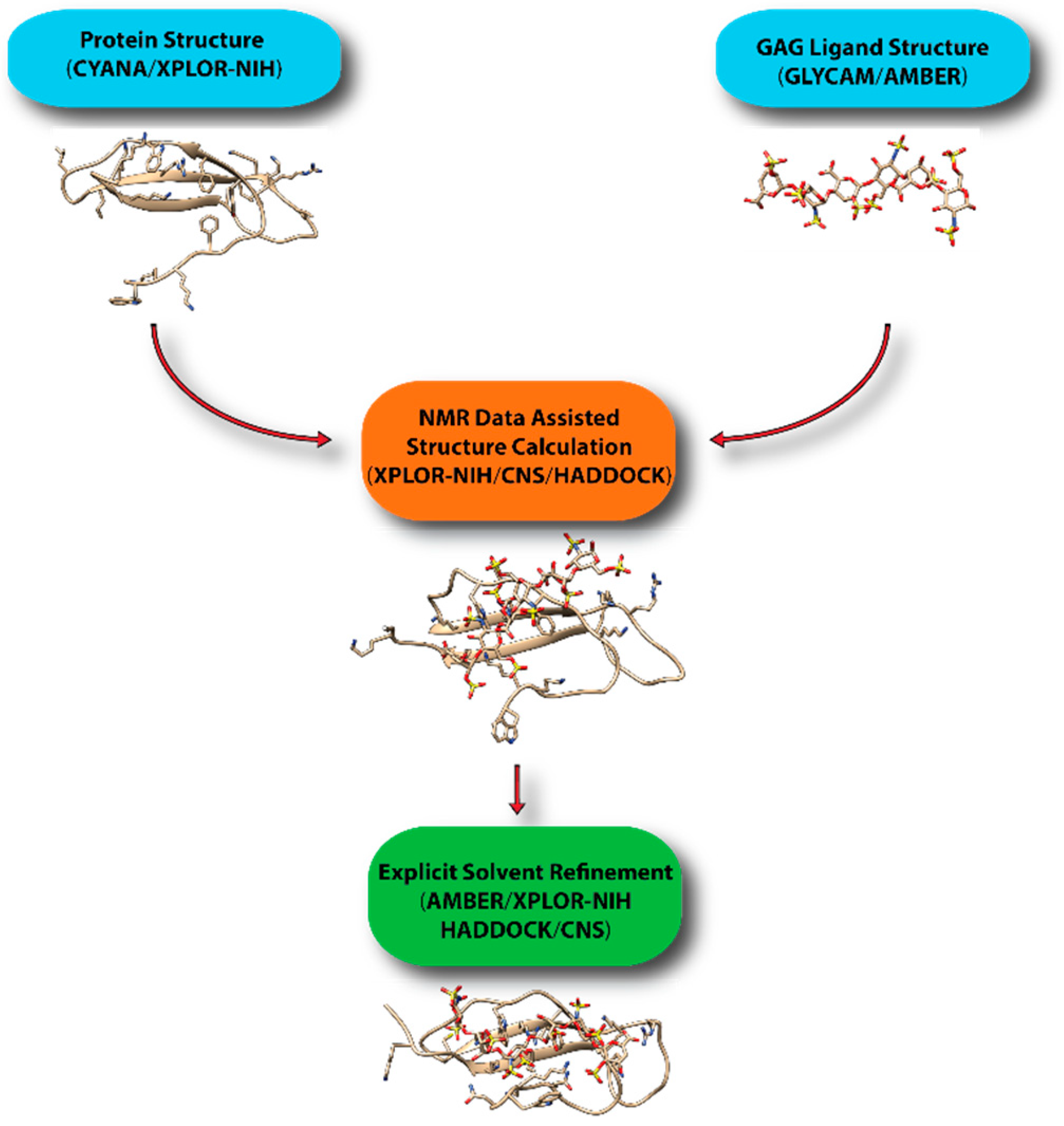
7. Computational Modeling of Protein-GAG Complexes Using Sparse Constraints
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pomin, V.H.; Mulloy, B. Current structural biology of the heparin interactome. Curr. Opin. Struct. Biol. 2015, 34, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.S.; Mancera, R.L. The structure of glycosaminoglycans and their interactions with proteins. Chem. Biol. Drug Des. 2008, 72, 455–482. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.E.I.; Johnson, Z.; Bonvin, P.; Handel, T.M. Glycosaminoglycan Interactions with Chemokines Add Complexity to a Complex System. Pharmaceuticals (Basel) 2017, 10, 70. [Google Scholar] [CrossRef] [PubMed]
- Imberty, A.; Lortat-Jacob, H.; Pérez, S. Structural view of glycosaminoglycan-protein interactions. Carbohydr. Res. 2007, 342, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Canales, A.; Angulo, J.; Ojeda, R.; Bruix, M.; Fayos, R.; Lozano, R.; Giménez-Gallego, G.; Martín-Lomas, M.; Nieto, P.M.; Jiménez-Barbero, J. Conformational flexibility of a synthetic glycosylaminoglycan bound to a fibroblast growth factor. FGF-1 recognizes both the (1)C(4) and (2)S(O) conformations of a bioactive heparin-like hexasaccharide. J. Am. Chem. Soc. 2005, 127, 5778–5779. [Google Scholar] [CrossRef] [PubMed]
- Pomin, V.H. Heparin-Binding Proteins (Chemokines and Defensins) and their complexes with glycosaminoglycans from the solution NMR perspective. Curr. Protein Pept. Sci. 2014, 15, 738–744. [Google Scholar] [CrossRef] [PubMed]
- Mulloy, B.; Forster, M.J.; Jones, C.; Davies, D.B. N.m.r. and molecular-modelling studies of the solution conformation of heparin. Biochem. J. 1993, 293, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Shaya, D.; Zhao, W.; Garron, M.L.; Xiao, Z.; Cui, Q.; Zhang, Z.; Sulea, T.; Linhardt, R.J.; Cygler, M. Catalytic mechanism of heparinase II investigated by site-directed mutagenesis and the crystal structure with its substrate. J. Biol. Chem. 2010, 285, 20051–20061. [Google Scholar] [CrossRef] [PubMed]
- Michel, G.; Pojasek, K.; Li, Y.; Sulea, T.; Linhardt, R.; Raman, R.; Prabhakar, V.; Sasisekharan, R.; Cygler, M. The Structure of Chondroitin B Lyase Complexed with Glycosaminoglycan Oligosaccharides Unravels a Calcium-Dependent Catalytic Machinery. J. Biol. Chem. 2004, 279, 32882–32896. [Google Scholar] [CrossRef] [PubMed]
- Arnott, S.; Gus, J.M.; Hukins, D.W.; Dea, I.C.; Rees, D.A. Conformation of keratan sulphate. J. Mol. Biol. 1974, 88, 175–184. [Google Scholar] [CrossRef]
- Almond, A.; Deangelis, P.L.; Blundell, C.D. Hyaluronan: The Local Solution Conformation Determined by NMR and Computer Modeling is Close to a Contracted Left-Handed 4-Fold Helix. J. Mol. Biol. 2006, 358, 1256–1269. [Google Scholar] [CrossRef] [PubMed]
- Pomin, V.H.; Mulloy, B. Glycosaminoglycans and Proteoglycans. Pharmaceuticals (Basel) 2018, 11, 27. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.G.; Triandafillou, C.G.; Huang, T.Y.; Zulueta, M.M.; Banerjee, S.; Dinner, A.R.; Hung, S.C.; Tang, W.J. Structural basis for oligomerization and glycosaminoglycan binding of CCL5 and CCL3. Proc. Natl. Acad. Sci. USA 2016, 113, 5000–5005. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Yarovoi, S.V.; Zhu, Z.; Rauova, L.; Hayes, V.; Lebedeva, T.; Liu, Q.; Poncz, M.; Arepally, G.; Cines, D.B.; et al. Atomic description of the immune complex involved in heparin-induced thrombocytopenia. Nat. Commun. 2015, 6, 8277. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Viola, C.M.; Brzozowski, A.M.; Davies, G.J. Structural characterization of human heparanase reveals insights into substrate recognition. Nat. Struct. Mol. Biol. 2015, 22, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Ke, J.; Gu, X.; Fang, J.; Wang, W.; Cong, Q.; Li, J.; Tan, J.; Brunzelle, J.S.; Zhang, C.; et al. Structural and Functional Study of d-Glucuronyl C5-epimerase. J. Biol. Chem. 2015, 290, 4620–4630. [Google Scholar] [CrossRef] [PubMed]
- Aguda, A.H.; Panwar, P.; Du, X.; Nguyen, N.T.; Brayer, G.D.; Bromme, D. Structural basis of collagen fiber degradation by cathepsin K. Proc. Natl. Acad. Sci. USA 2014, 111, 17474–17479. [Google Scholar] [CrossRef] [PubMed]
- Whalen, D.M.; Malinauskas, T.; Gilbert, R.J.C.; Siebold, C. Structural Insights into Proteoglycan-Shaped Hedgehog Signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 16420–16425. [Google Scholar] [CrossRef] [PubMed]
- Pomin, V.H. Biological findings from the recent NMR-based studies of glycosaminoglycan-protein interactions. Glycobiology 2014, 24, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Farmer, B.T., 2nd; Constantine, K.L.; Goldfarb, V.; Friedrichs, M.S.; Wittekind, M.; Yanchunas, J., Jr.; Robertson, J.G.; Mueller, L. Localizing the NADP+ binding site on the MurB enzyme by NMR. Nat. Struct. Biol. 1996, 3, 995–997. [Google Scholar] [CrossRef] [PubMed]
- De Paula, V.S.; Pomin, V.H.; Valente, A.P. Unique Properties of Human β-Defensin 6 (hBD6) and Glycosaminoglycan Complex: Sandwich-like dimerization and competition with the chemokine receptor 2 (CCR2) binding site. J. Biol. Chem. 2014, 289, 22969–22979. [Google Scholar] [CrossRef] [PubMed]
- Bhunia, A.; Bhattacharjya, S.; Chatterjee, S. Applications of saturation transfer difference NMR in biological systems. Drug Discov. Today 2012, 17, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Cala, O.; Guillière, F.; Krimm, I. NMR-based analysis of protein-ligand interactions. Anal. Bioanal. Chem. 2014, 406, 943–956. [Google Scholar] [CrossRef] [PubMed]
- Haselhorst, T.; Lamerz, A.-C.; von Itzstein, M. Saturation transfer difference NMR spectroscopy as a technique to investigate protein-carbohydrate interactions in solution. Methods Mol. Biol. 2009, 534, 375–386. [Google Scholar] [PubMed]
- Yu, F.; Roy, S.; Arevalo, E.; Schaeck, J.; Wang, J.; Holte, K.; Gunay, NS.; Capila, I.; Kaundinya, G.V. Characterization of heparin-protein interaction by saturation transfer difference (STD) NMR. Anal. Bioanal. Chem. 2014, 406, 3079–3089. [Google Scholar] [CrossRef] [PubMed]
- Post, C.B. Exchange-transferred NOE spectroscopy and bound ligand structure determination. Curr. Opin. Struct. Biol. 2003, 13, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Unione, L.; Galante, S.; Diaz, D.; Cañada, J.; Jiménez-Barbero, J. NMR and molecular recognition. The application of ligand-based NMR methods to monitor molecular interactions. Med. Chem. Commun. 2014, 5, 1280–1289. [Google Scholar] [CrossRef]
- Kogelberg, H.; Solís, D.; Jiménez-Barbero, J. New structural insights into carbohydrate-protein interactions from NMR spectroscopy. Curr. Opin. Struct. Biol. 2003, 13, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Bax, A.; Sklenar, V.; Summers, M.F. Direct identification of relayed nuclear Overhauser effects. J. Magn. Reson. 1986, 70, 327–331. [Google Scholar] [CrossRef]
- Künze, G.; Gehrcke, J.-P.; Pisabarro, M.T.; Huster, D. NMR characterization of the binding properties and conformation of glycosaminoglycans interacting with interleukin-10. Glycobiology 2014, 24, 1036–1049. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Yang, J.-Y.; Moremen, K.W.; Flanagan, J.G.; Prestegard, J.H. Structural characterization of a heparin sulfate pentamer interacting with LAR-Ig1-2. Biochemistry 2018, 57, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Dalvit, C.; Fogliatto, G.; Stewart, A.; Veronesi, M.; Stockman, B. WaterLOGSY as a method for primary NMR screening: Practical aspects and range of applicability. J. Biomol. NMR 2001, 21, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Mandaliti, W.; Nepravishta, R.; Pica, F.; Vallebona, P.S.; Garaci, E.; Paci, M. Thymosin alpha1 Interacts with Hyaluronic Acid Electrostatically by Its Terminal Sequence LKEKK. Molecules 2017, 22, 1843. [Google Scholar] [CrossRef] [PubMed]
- Geist, L.; Mayer, M.; Cockcroft, X.L.; Wolkerstorfer, B.; Kessler, D.; Engelhardt, H.; McConnell, D.B.; Konrat, R. Direct NMR Probing of Hydration Shells of Protein Ligand Interfaces and Its Application to Drug Design. J. Med. Chem. 2017, 60, 8708–8715. [Google Scholar] [CrossRef] [PubMed]
- Pomin, V.H.; Wang, X. Synthetic Oligosaccharide Libraries and Microarray Technology: A Powerful Combination for the Success of Current Glycosaminoglycan Interactomics. Chem. Med. Chem. 2018, 13, 648–661. [Google Scholar] [CrossRef] [PubMed]
- Wang, X. NMR studies of protein-glycosaminoglycan interactions. In NMR in Glycoscience and Glycotechnology, 1st ed.; Kato, K., Peters, T., Eds.; Royal Society of Chemistry: Croydon, UK, 2017; Chapter 11. [Google Scholar]
- Pomin, V.H.; Park, Y.; Huang, R.; Heiss, C.; Sharp, J.S.; Azadi, P.; Prestegard, J.H. Exploiting enzyme specificities in digestions of chondroitin sulfates A and C: Production of well-defined hexasaccharides. Glycobiology 2012, 22, 826–838. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Bhaskar, U.; Li, G.; Fu, L.; Li, L.; Zhang, F.; Dordick, J.S.; Linhardt, R.J. Immobilized enzymes to convert N-sulfo, N-acetyl heparosan to a critical intermediate in the production of bioengineered heparin. J. Biotechnol. 2013, 167, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; McCallum, S.A.; Xie, J.; Nieto, L.; Corzana, F.; Jiménez-Barbero, J.; Chen, M.; Liu, J.; Linhardt, R.J. Solution structures of chemoenzymatically synthesized heparin and its precursors. J. Am. Chem. Soc. 2008, 130, 12998–13007. [Google Scholar] [CrossRef] [PubMed]
- Laguri, C.; Sapay, N.; Simorre, J.P.; Brutscher, B.; Imberty, A.; Gans, P.; Lortat-Jacob, H. 13C-labeled heparan sulfate analogue as a tool to study protein/heparan sulfate interactions by NMR spectroscopy: Application to the CXCL12α chemokine. J. Am. Chem. Soc. 2011, 133, 9642–9645. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Fu, L.; Li, G.; Andrew Jones, J.; Linhardt, R.J.; Koffas, M. Production of chondroitin in metabolically engineered E. coli. Metab. Eng. 2015, 27, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Pomin, V.H.; Sharp, J.S.; Li, X.; Wang, L.; Prestegard, J.H. Characterization of glycosaminoglycans by 15N NMR spectroscopy and in vivo isotopic labeling. Anal. Chem. 2010, 82, 4078–4088. [Google Scholar] [CrossRef] [PubMed]
- Pomin, V.H. Advances in glycosaminoglycanomics by 15N-NMR spectroscopy. Anal. Bioanal. Chem. 2013, 405, 3035–3048. [Google Scholar] [CrossRef] [PubMed]
- Baik, J.Y.; Gasimli, L.; Yang, B.; Datta, P.; Zhang, F.; Glass, C.A.; Esko, J.D.; Linhardt, R.J.; Sharfstein, S.T. Metabolic engineering of Chinese hamster ovary cells: Towards a bioengineered heparin. Metab. Eng. 2012, 14, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Deshauer, C.; Morgan, A.M.; Ryan, E.O.; Handel, T.M.; Prestegard, J.H.; Wang, X. Interactions of the Chemokine CCL5/RANTES with Medium-Sized Chondroitin Sulfate Ligands. Structure 2015, 23, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Wang, X. Structure of decorin binding protein B from Borrelia burgdorferi and its interactions with glycosaminoglycans. Biochim. Biophys. Acta 2015, 1854, 1823–1832. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.; Sepuru, K.M.; Feng, W.; Rajarathnam, K.; Wang, X. Flexible Linker Modulates Glycosaminoglycan Affinity of Decorin Binding Protein, A. Biochemistry 2015, 54, 5113–5119. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.M.; Wang, X. Structural mechanisms underlying sequence-dependent variations in GAG affinities of decorin binding protein A, a Borrelia burgdorferi adhesin. Biochem. J. 2015, 467, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Jowitt, T.A.; Day, A.J.; Prestegard, J.H. Nuclear Magnetic Resonance Insight into the Multiple Glycosaminoglycan Binding Modes of the Link Module from Human TSG-6. Biochemistry 2016, 55, 262–276. [Google Scholar] [CrossRef] [PubMed]
- Koehler, J.; Meiler, J. Expanding the utility of NMR restraints with paramagnetic compounds: Background and practical aspects. Prog. Nucl. Magn. Reson. Spectrosc. 2011, 59, 360–389. [Google Scholar] [CrossRef] [PubMed]
- Iwahara, J.; Tang, C.; Marius Clore, G. Practical aspects of (1) H transverse paramagnetic relaxation enhancement measurements on macromolecules. J. Magn. Reson. 2007, 184, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Calle, L.P.; Cañada, F.J.; Jiménez-Barbero, J. Application of NMR methods to the study of the interaction of natural products with biomolecular receptors. Nat. Prod. Rep. 2011, 28, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.; Wang, X. The novel heparin-binding motif in decorin-binding protein A from strain B31 of Borrelia burgdorferi explains the higher binding affinity. Biochemistry 2013, 52, 8237–8245. [Google Scholar] [CrossRef] [PubMed]
- Iwahara, J.; Jung, Y.S.; Clore, G.M. Heteronuclear NMR spectroscopy for lysine NH (3) groups in proteins: Unique effect of water exchange on (15) N transverse relaxation. J. Am. Chem. Soc. 2007, 129, 2971–2980. [Google Scholar] [CrossRef] [PubMed]
- Esadze, A.; Chen, C.; Zandarashvili, L.; Roy, S.; Pettitt, B.M.; Iwahara, J. Changes in conformational dynamics of basic side chains upon protein-DNA association. Nucleic Acids Res. 2016, 44, 6961–6970. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Hoffpauir, Z.A.; Iwahara, J. Internal Motions of Basic Side Chains of the Antennapedia Homeodomain in the Free and DNA-Bound States. Biochemistry 2017, 56, 5866–5869. [Google Scholar] [CrossRef] [PubMed]
- Sepuru, K.M.; Iwahara, J.; Rajarathnam, K. Direct detection of lysine side chain NH3+ in protein–heparin complexes using NMR spectroscopy. Analyst 2018, 143, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Blaum, B.S.; Deakin, J.A.; Johansson, C.M.; Herbert, A.P.; Barlow, P.N.; Lyon, M.; Uhrin, D. Lysine and arginine side chains in glycosaminoglycan-protein complexes investigated by NMR, cross-linking, and mass spectrometry: A case study of the factor H-heparin interaction. J. Am. Chem. Soc. 2010, 132, 6374–6381. [Google Scholar] [CrossRef] [PubMed]
- Koharudin, L.M.; Bonvin, A.M.; Kaptein, R.; Boelens, R. Use of very long-distance NOEs in a fully deuterated protein: An approach for rapid protein fold determination. J. Magn. Reson. 2003, 163, 228–235. [Google Scholar] [CrossRef]
- Kay, L.E.; Torchia, D.A.; Bax, A. Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: Application to staphylococcal nuclease. Biochemistry 1989, 28, 8972–8979. [Google Scholar] [CrossRef] [PubMed]
- Jeong, K.-W.; Jeong, M.-C.; Jin, B.; Kim, Y. Relationship between Structural Flexibility and Function in the C-Terminal Region of the Heparin-Binding Domain of VEGF165. Biochemistry 2013, 52, 8823–8832. [Google Scholar] [CrossRef] [PubMed]
- Abedini, A.; Tracz, S.M.; Cho, J.H.; Raleigh, D.P. Characterization of the heparin binding site in the N-terminus of human pro-islet amyloid polypeptide: Implications for amyloid formation. Biochemistry 2006, 45, 9228–9237. [Google Scholar] [CrossRef] [PubMed]
- Cohlberg, J.A.; Li, J.; Uversky, V.N.; Fink, A.L. Heparin and other glycosaminoglycans stimulate the formation of amyloid fibrils from alpha-synuclein in vitro. Biochemistry 2002, 41, 1502–1511. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Crowther, R.A.; Jakes, R.; Goedert, M. Alzheimer-like changes in microtubule-associated protein Tau induced by sulfated glycosaminoglycans. Inhibition of microtubule binding, stimulation of phosphorylation, and filament assembly depend on the degree of sulfation. J. Biol. Chem. 1997, 272, 33118–33124. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.; Patil, S.M.; Gibson, J.; Nelson, C.E.; Alder, N.N.; Alexandrescu, A.T. Mechanism of amylin fibrillization enhancement by heparin. J. Biol. Chem. 2011, 286, 22894–22904. [Google Scholar] [CrossRef] [PubMed]
- Malmos, K.G.; Bjerring, M.; Jessen, C.M.; Nielsen, E.H.; Poulsen, E.T.; Christiansen, G.; Vosegaard, T.; Skrydstrup, T.; Enghild, J.J.; Pedersen, J.S.; et al. How Glycosaminoglycans Promote Fibrillation of Salmon Calcitonin. J. Biol. Chem. 2016, 291, 16849–16862. [Google Scholar] [CrossRef] [PubMed]
- Madine, J.; Clayton, J.C.; Yates, E.A.; Middleton, D.A. Exploiting a (13) C-labelled heparin analogue for in situ solid-state NMR investigations of peptide-glycan interactions within amyloid fibrils. Org. Biomol. Chem. 2009, 7, 2414–2420. [Google Scholar] [CrossRef] [PubMed]
- Madine, J.; Pandya, M.J.; Hicks, M.R.; Rodger, A.; Yates, E.A.; Radford, S.E.; Middleton, D.A. Site-specific identification of an abeta fibril-heparin interaction site by using solid-state NMR spectroscopy. Angew. Chem. Int. Ed. 2012, 51, 13140–13143. [Google Scholar] [CrossRef] [PubMed]
- Stewart, K.L.; Hughes, E.; Yates, E.A.; Akien, G.R.; Huang, T.Y.; Lima, M.A.; Rudd, T.R.; Guerrini, M.; Hung, S.C.; Radford, S.E.; et al. Atomic Details of the Interactions of Glycosaminoglycans with Amyloid-beta Fibrils. J. Am. Chem. Soc. 2016, 138, 8328–8331. [Google Scholar] [CrossRef] [PubMed]
- Stewart, K.L.; Hughes, E.; Yates, E.A.; Middleton, D.A.; Radford, S.E. Molecular Origins of the Compatibility between Glycosaminoglycans and Abeta40 Amyloid Fibrils. J. Mol. Biol. 2017, 429, 2449–2462. [Google Scholar] [CrossRef] [PubMed]
- Schwieters, C.D.; Kuszewski, J.J.; Tjandra, N.; Clore, G.M. The Xplor-NIH NMR molecular structure determination package. J. Magn. Res. 2003, 160, 65–73. [Google Scholar] [CrossRef]
- Brunger, A.T.; Adams, P.D.; Clore, G.M.; DeLano, W.L.; Gros, P.; Grosse-Kunstleve, R.W.; Jiang, J.S.; Kuszewski, J.; Nilges, M.; Pannu, N.S.; et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998, 54, 905–921. [Google Scholar] [PubMed]
- Singh, A.; Tessier, M.B.; Pederson, K.; Wang, X.; Venot, A.P.; Boons, G.J.; Prestegard, J.H.; Woods, R.J. Extension and validation of the GLYCAM force field parameters for modeling glycosaminoglycans. Can. J. Chem. 2016, 94, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.; Boelens, R.; Bonvin, A.M. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Poluri, K.M.; Joseph, P.R.; Sawant, K.V.; Rajarathnam, K. Molecular basis of glycosaminoglycan heparin binding to the chemokine CXCL1 dimer. J. Biol. Chem. 2013, 288, 25143–25153. [Google Scholar] [CrossRef] [PubMed]
- Sepuru, K.M.; Nagarajan, B.; Desai, U.R.; Rajarathnam, K. Molecular Basis of Chemokine CXCL5-Glycosaminoglycan Interactions. J. Biol. Chem. 2016, 291, 20539–20550. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Chen, C.-Y.; Zong, C.; Wang, S.; Ramiah, A.; Prabhakar, P.; Morris, L.C.; Boons, G.-J.; Moremen, K.W.; Prestegard, J.H. Structural Aspects of Heparan Sulfate Binding to Robo1–Ig1–2. ACS Chem. Biol. 2016, 11, 3106–3113. [Google Scholar] [CrossRef] [PubMed]
- Gehrcke, J.P.; Pisabarro, M.T. Identification and characterization of a glycosaminoglycan binding site on interleukin-10 via molecular simulation methods. J. Mol. Gr. Mod. 2015, 62, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Griffith, A.R.; Rogers, C.J.; Miller, G.M.; Abrol, R.; Hsieh-Wilson, L.C.; Goddard, W.A. Predicting glycosaminoglycan surface protein interactions and implications for studying axonal growth. Proc. Natl. Acad. Sci. USA 2017, 114, 13697–13702. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, N.V.; Desai, U.R. Toward a robust computational screening strategy for identifying glycosaminoglycan sequences that display high specificity for target proteins. Glycobiology 2014, 24, 1323–1333. [Google Scholar] [CrossRef] [PubMed]
- Forster, M.; Mulloy, B. Computational approaches to the identification of heparin-binding sites on the surfaces of proteins. Biochem. Soc. Trans. 2006, 34, 431–444. [Google Scholar] [CrossRef] [PubMed]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GAG Type or Sub-Type | Hexosamine * | Uronic Acid or Neutral Sugar * | Sulfation Pattern * |
|---|---|---|---|
| Heparin | 4-linked α-GlcNAc | 4-linked α-IdoA | GlcNS6S, IdoA2S |
| Heparan sulfate | 4-linked α-GlcNAc | 4-linked β-GlcA | non-sulfated |
| Chondroitin sulfate A | 3-linked β-GalNAc | 4-linked β-GlcA | GalNAc4S |
| Chondroitin sulfate B (dermatan sulfate) | 3-linked β-GalNAc | 4-linked α-IdoA | GalNAc4S, IdoA2S |
| Chondroitin sulfate C | 3-linked β-GalNAc | 4-linked β-GlcA | GalNAc6S |
| Chondroitin sulfate D | 3-linked β-GalNAc | 4-linked β-GlcA | GalNAc6S, GlcA2S |
| Chondroitin sulfate E | 3-linked β-GalNAc | 4-linked β-GlcA | GalNAc4S6S |
| Keratan sulfate | 4-linked β-GlcNAc | 3-linked α-Gal | GlcNAc6S, Gal6S |
| Hyaluronan | 3-linked β-GlcNAc | 3-linked β-GlcA | non-sulfated |
| 1H-1H Connection a | Free b | Bound |
|---|---|---|
| AH1-BH4 | 2.62 ± 0.02 c | 2.86 ± 0.21 |
| CH1-DH4 | 2.41 ± 0.02 | 2.65 ± 0.20 |
| CH3-DH1 | 2.43 ± 0.11 | 3.05 ± 0.25 |
| DH1-DH2 | 3.16 ± 0.01 | - |
| DH1-DH3 | 3.27 ± 0.01 | 2.72 ± 0.19 |
| DH1-EH4 | 2.71 ± 0.02 | 3.14 ± 0.25 |
| DH1-EH6 | 2.85 ± 0.02 | 2.91 ± 0.23 |
| DH1-EH6′ | 3.22 ± 0.03 | 2.49 ± 0.19 |
| OMe-EH1 | 2.96 ± 0.02 | 3.22 ± 0.20 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pomin, V.H.; Wang, X. Glycosaminoglycan-Protein Interactions by Nuclear Magnetic Resonance (NMR) Spectroscopy. Molecules 2018, 23, 2314. https://doi.org/10.3390/molecules23092314
Pomin VH, Wang X. Glycosaminoglycan-Protein Interactions by Nuclear Magnetic Resonance (NMR) Spectroscopy. Molecules. 2018; 23(9):2314. https://doi.org/10.3390/molecules23092314
Chicago/Turabian StylePomin, Vitor H., and Xu Wang. 2018. "Glycosaminoglycan-Protein Interactions by Nuclear Magnetic Resonance (NMR) Spectroscopy" Molecules 23, no. 9: 2314. https://doi.org/10.3390/molecules23092314
APA StylePomin, V. H., & Wang, X. (2018). Glycosaminoglycan-Protein Interactions by Nuclear Magnetic Resonance (NMR) Spectroscopy. Molecules, 23(9), 2314. https://doi.org/10.3390/molecules23092314