New Coordination Complexes Based on the 2,6-bis[1-(Phenylimino)ethyl] Pyridine Ligand: Effective Catalysts for the Synthesis of Propylene Carbonates from Carbon Dioxide and Epoxides


Abstract
:1. Introduction
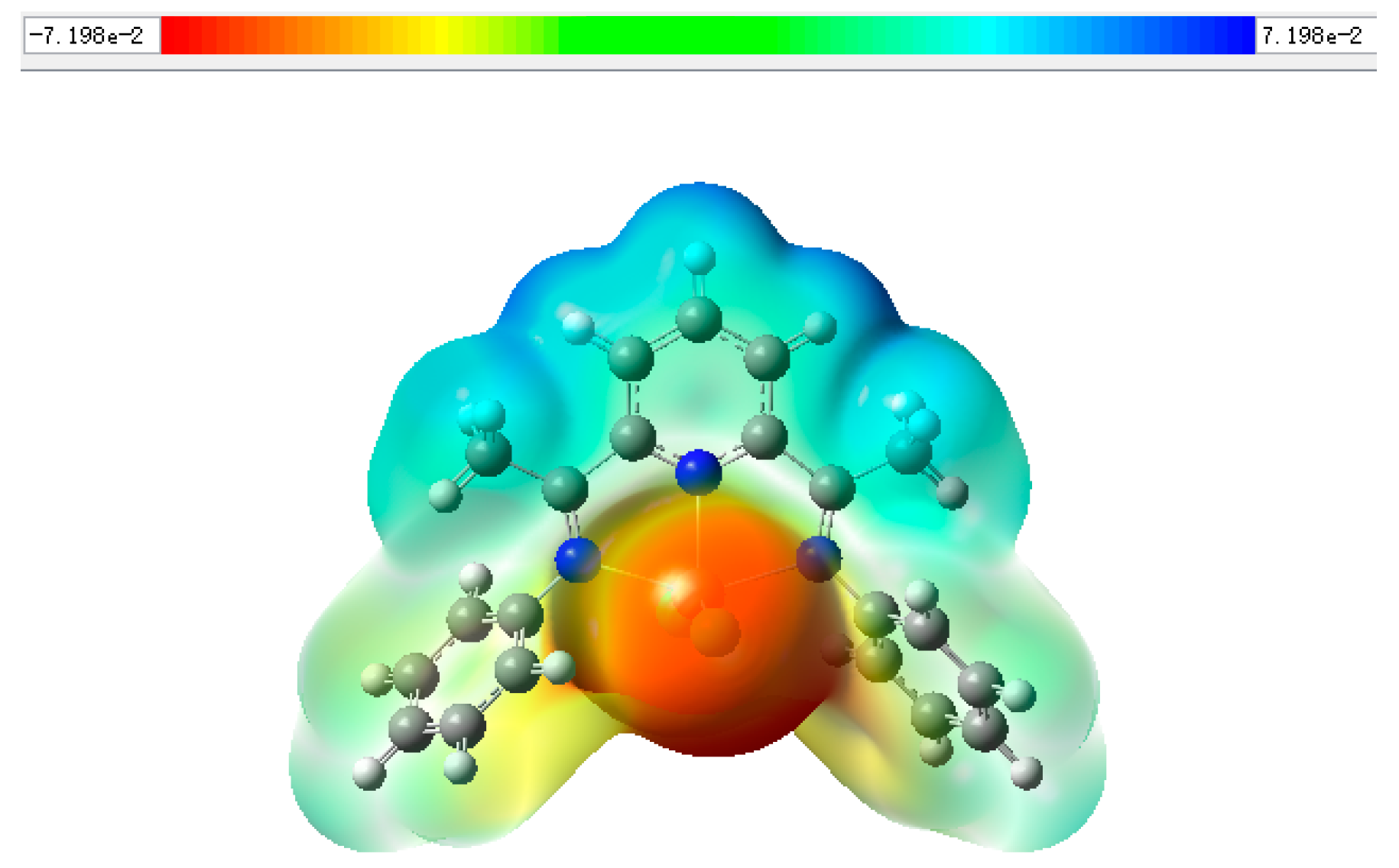
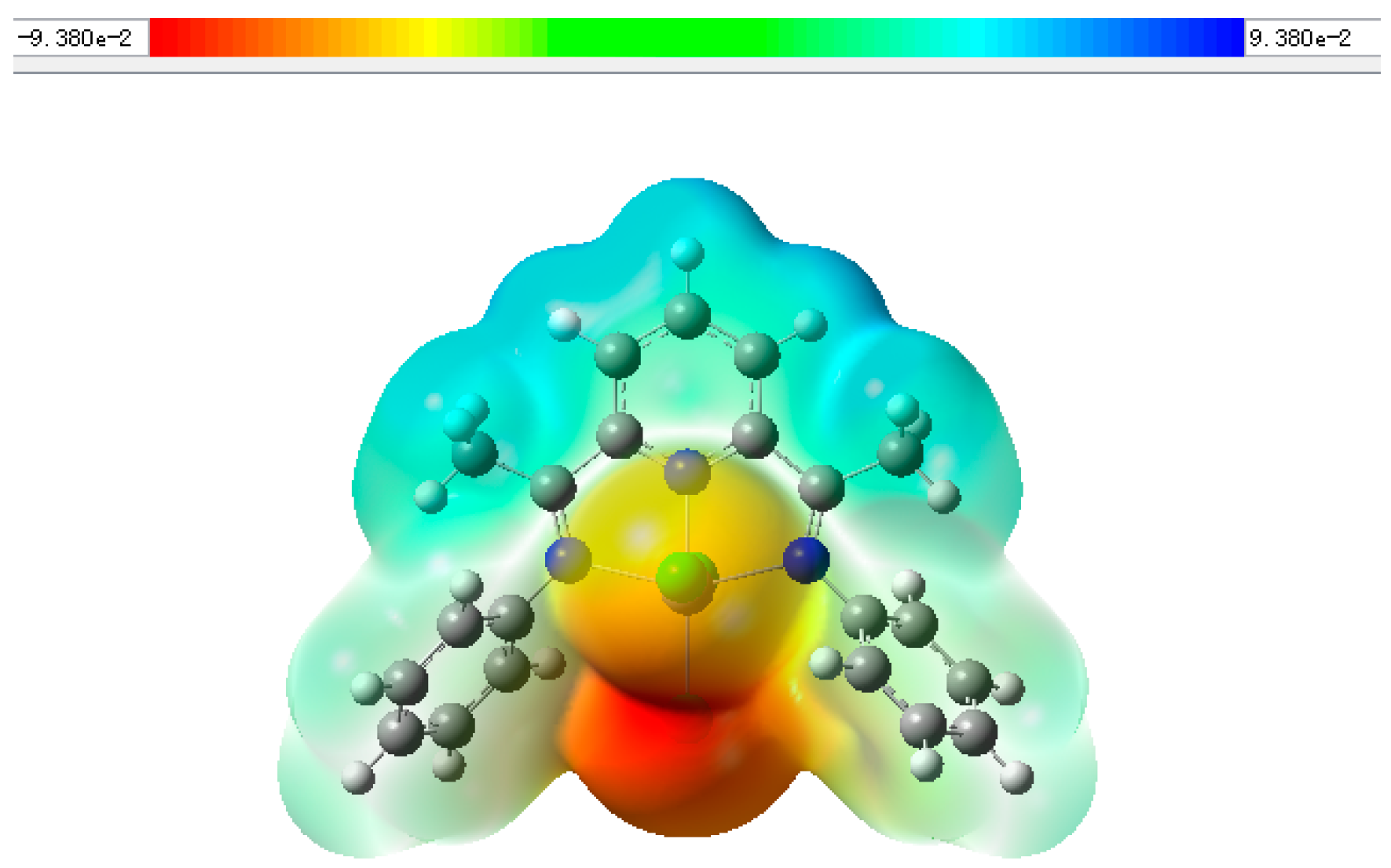
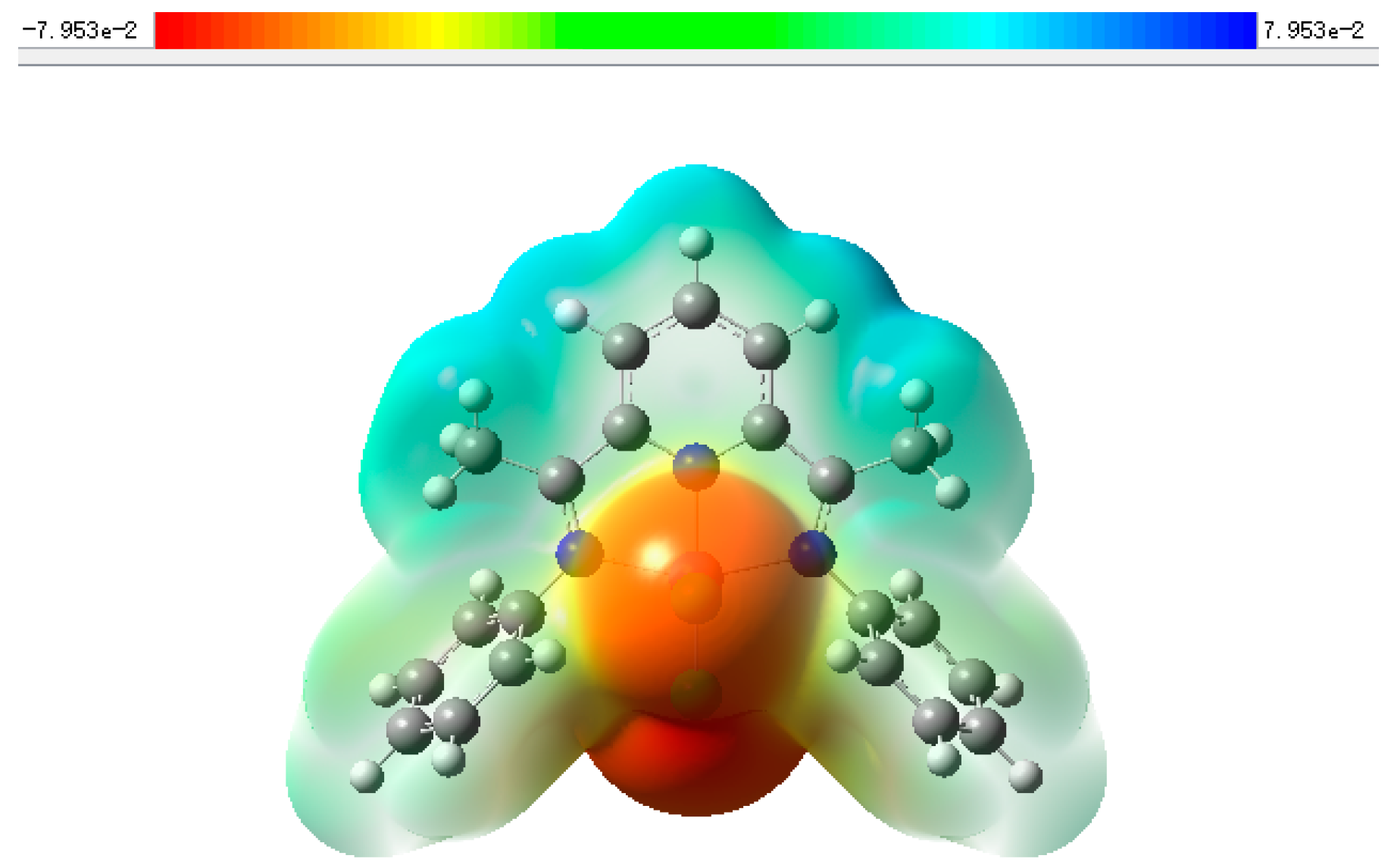
2. Results and Discussion
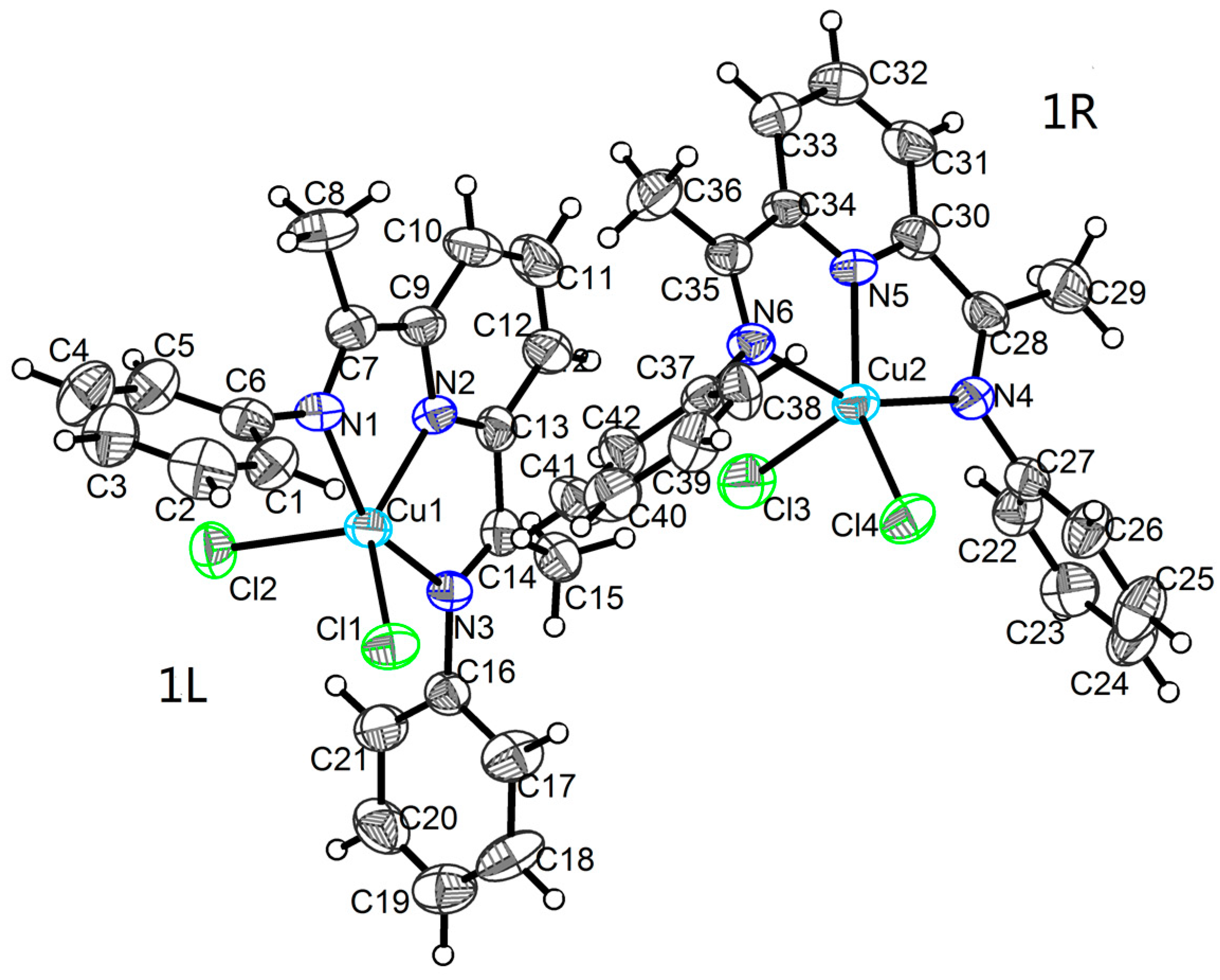
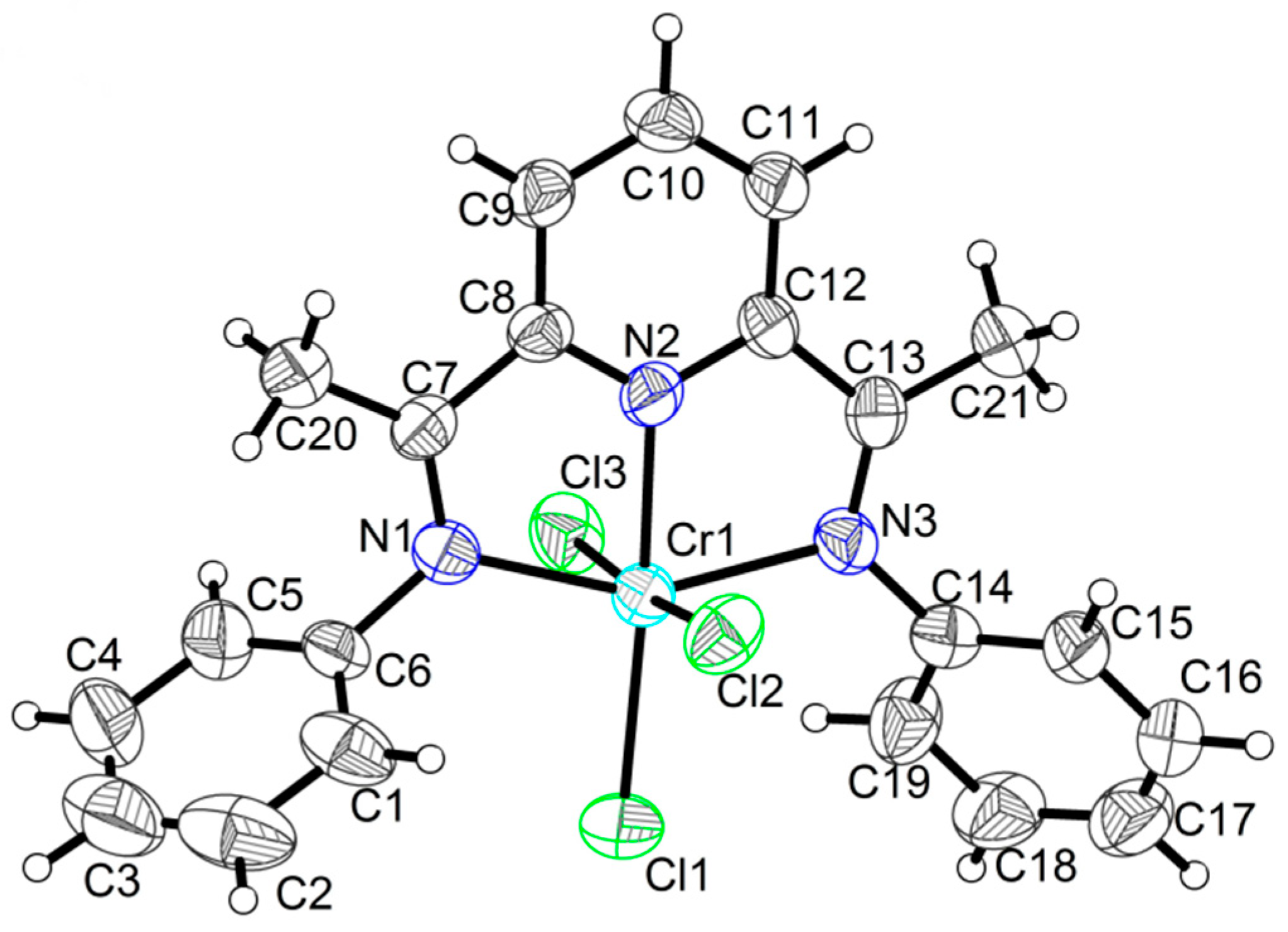
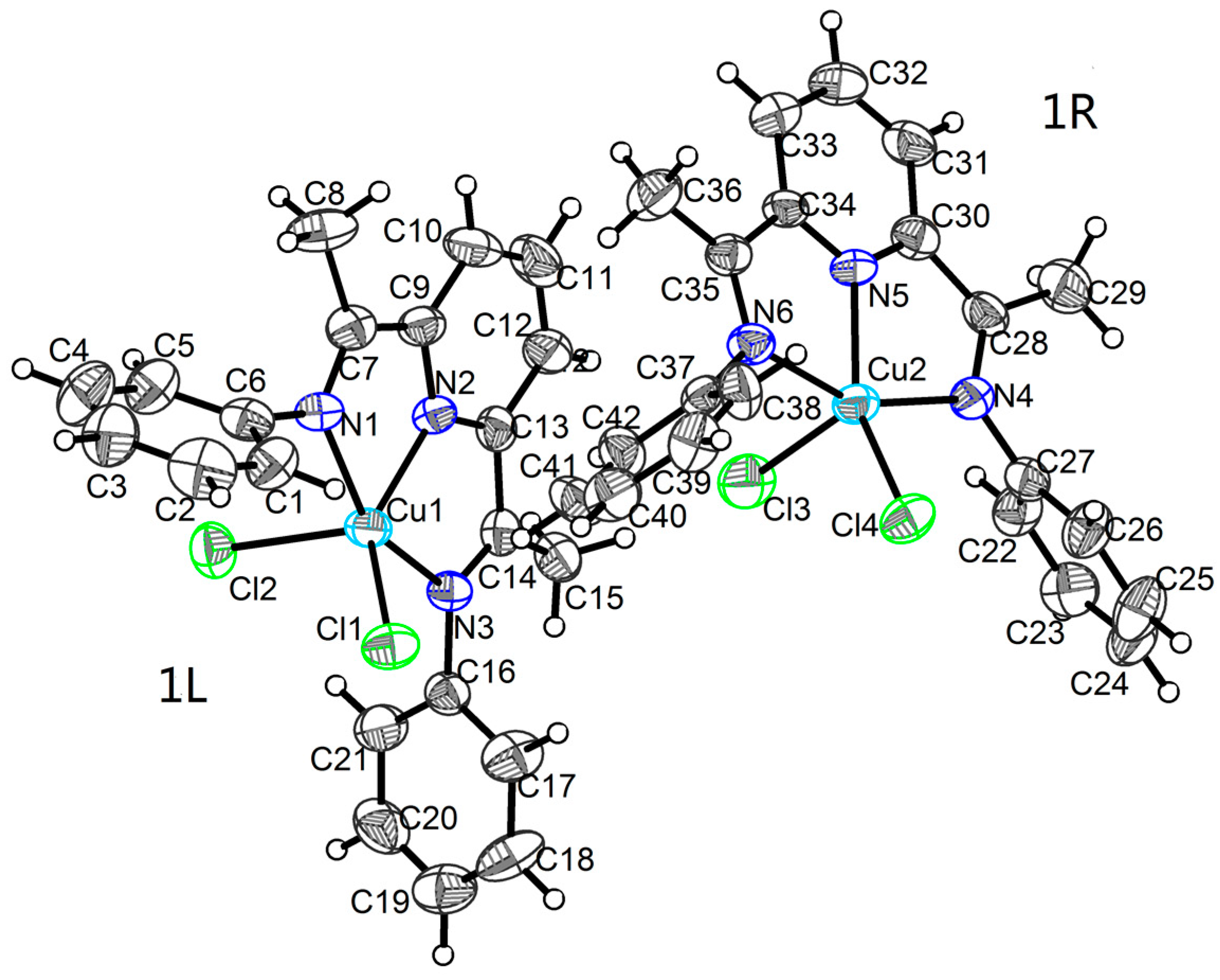
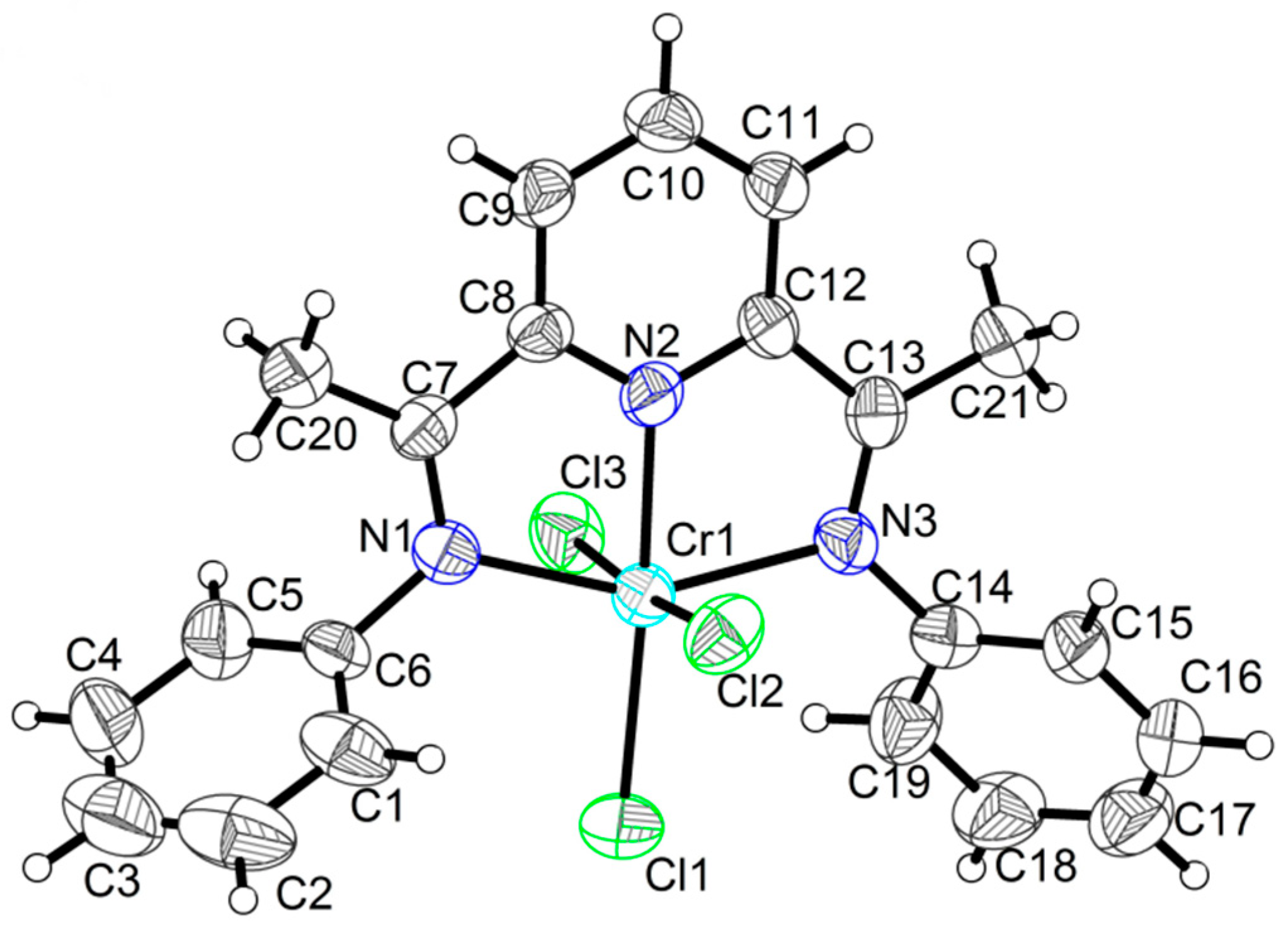
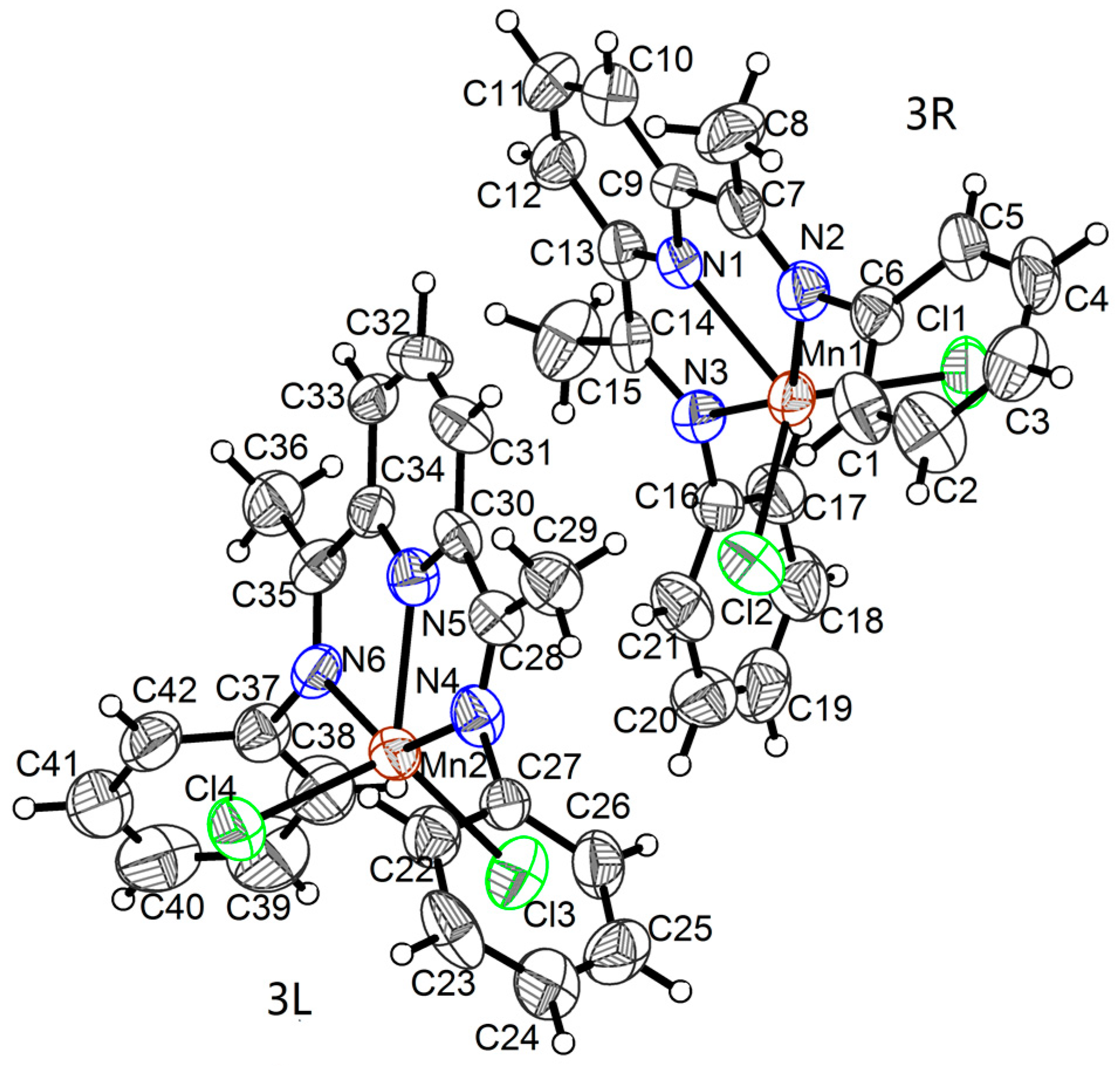
2.1. Crystal Structures
2.2. Cycloaddition of CO2 and Propylene Oxide
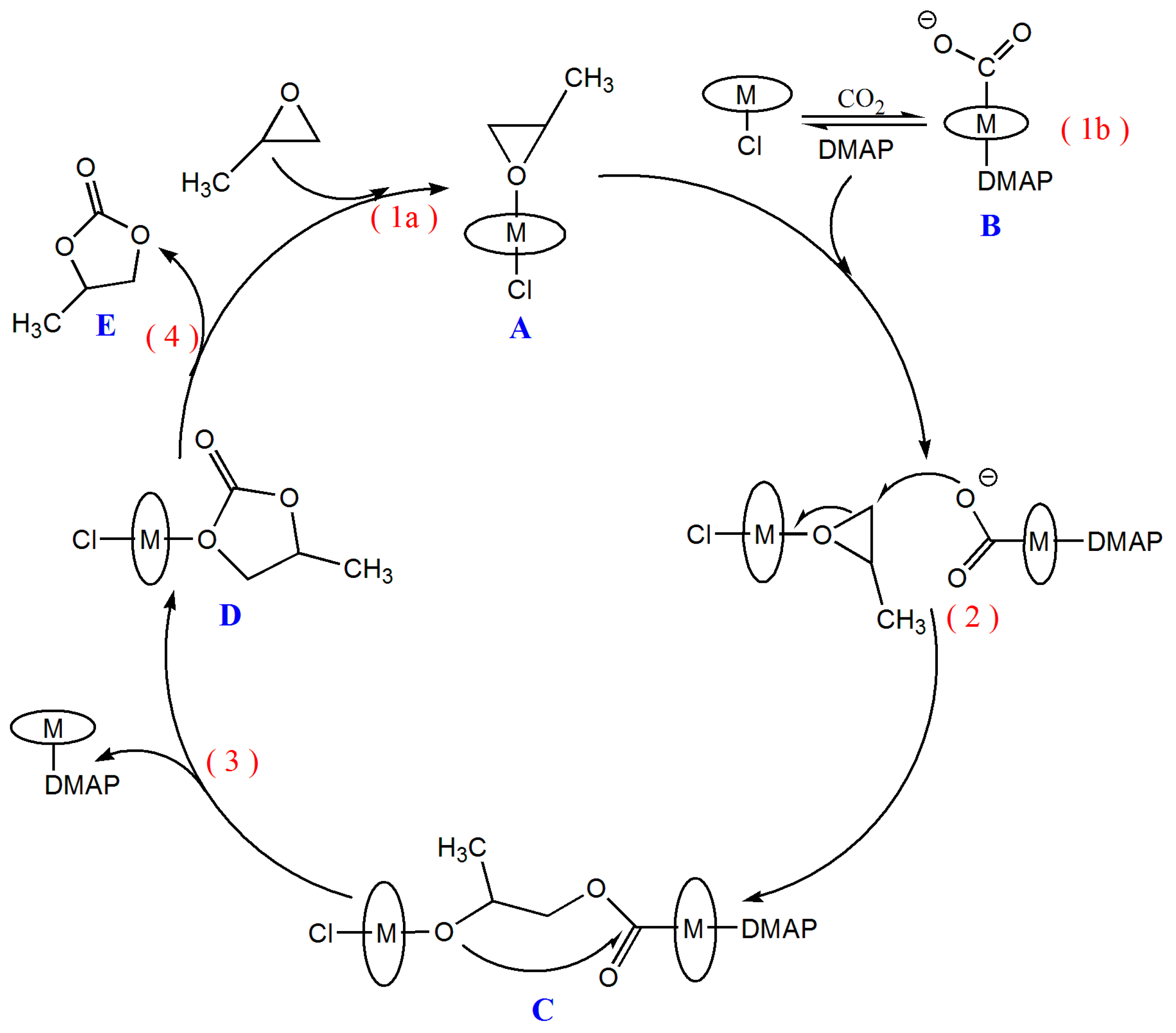
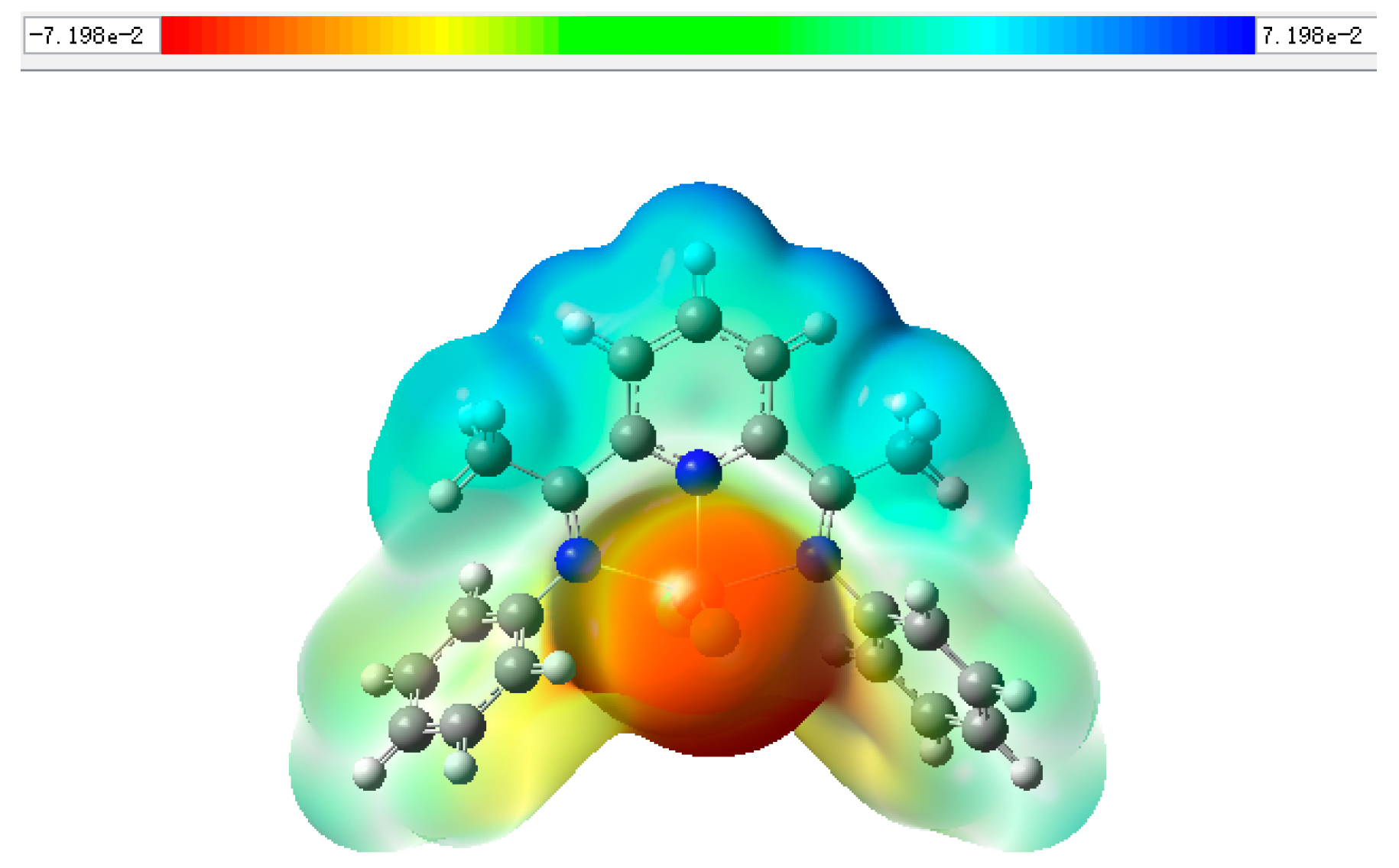
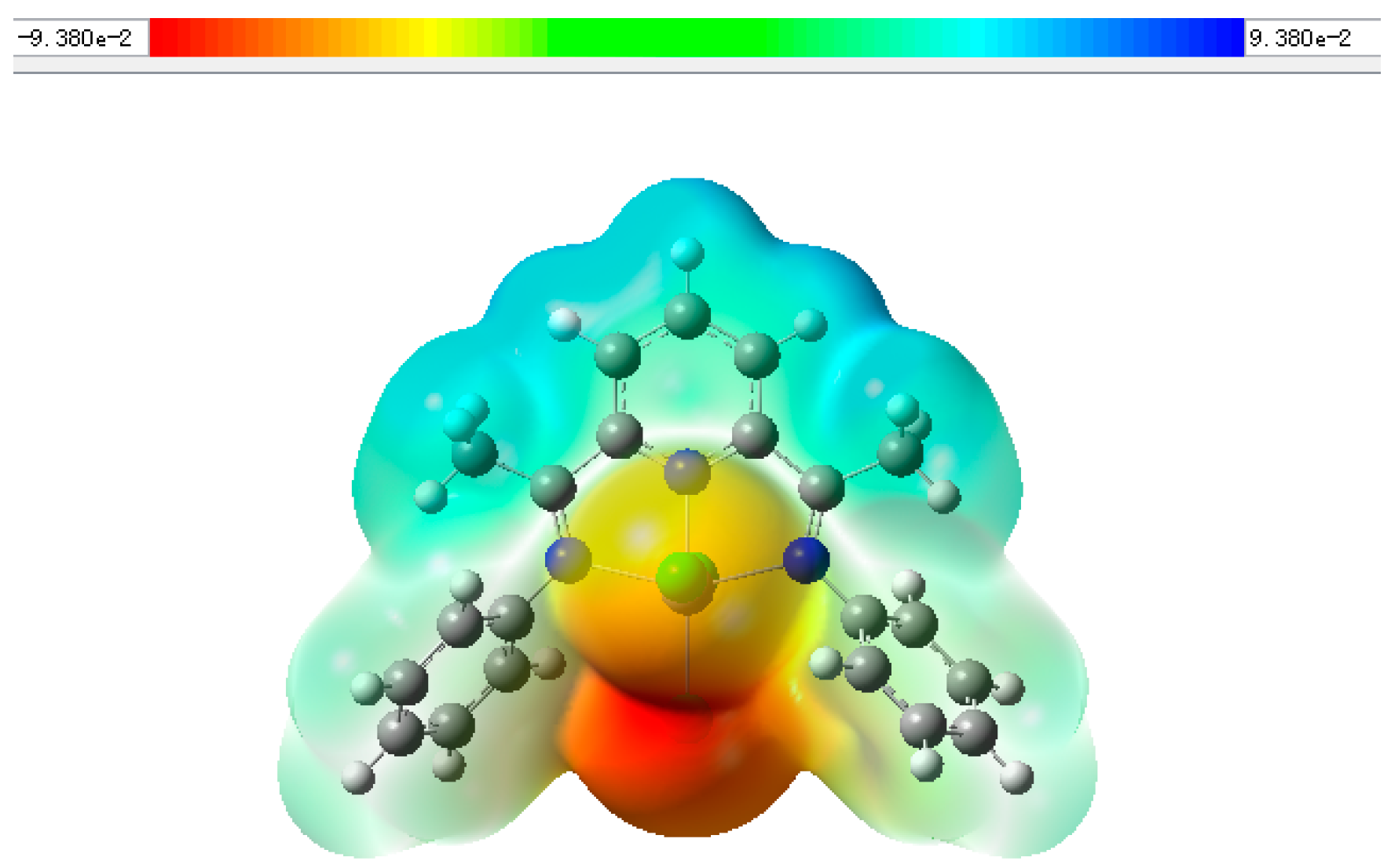
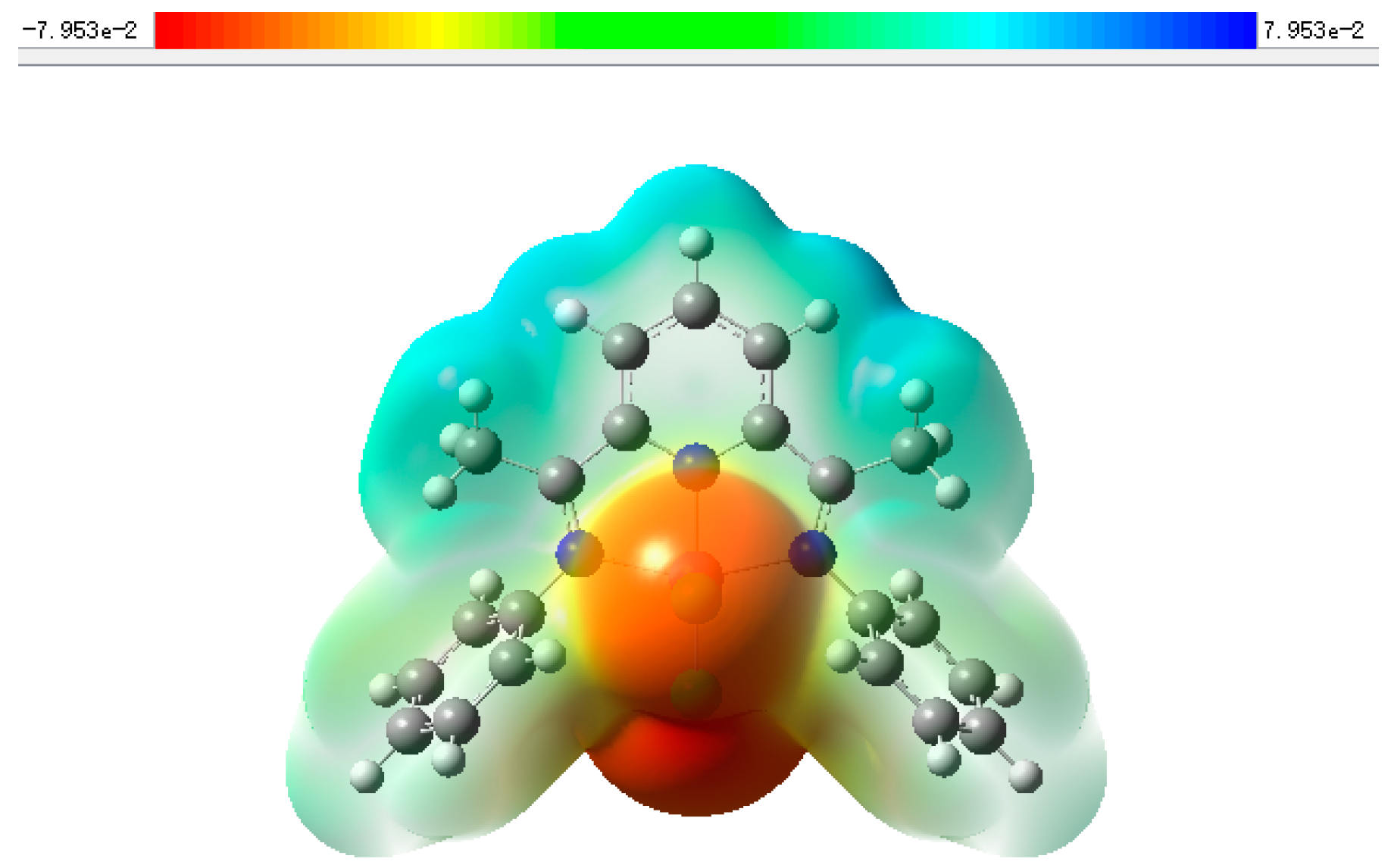
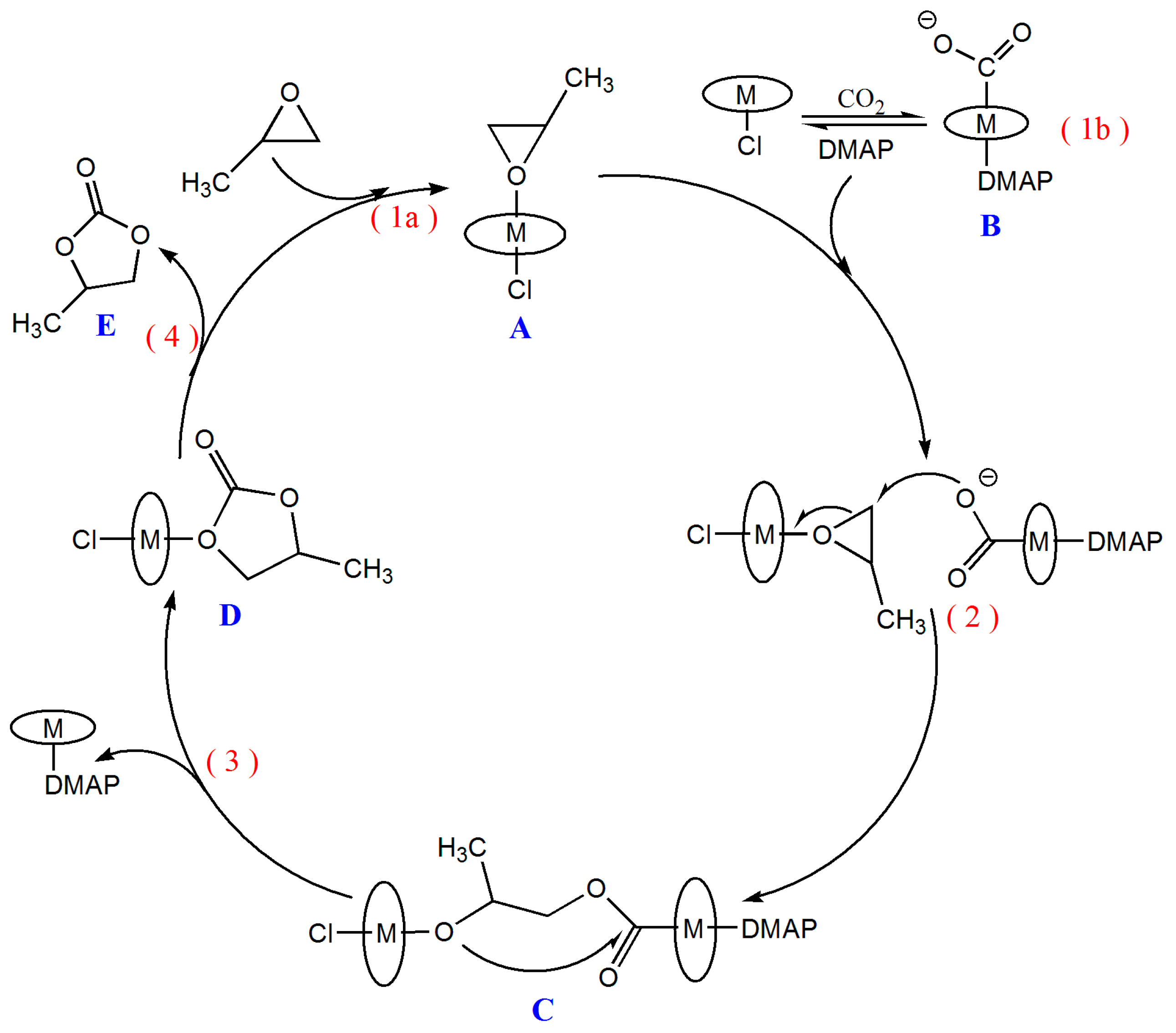
2.3. Mechanism for the Cycloaddition Reaction of Propylene Oxide and CO2
3. Materials and Methods
3.1. Chemical Materials
3.2. X-ray Crystallographic Studies
3.3. Catalyst Characterization
3.4. Catalyst Preparation
3.4.1. Synthesis of the Ligand (2,6-bis[1-(Phenylimino)ethyl] pyridine) (L)
3.4.2. Synthesis of Complexes
3.4.3. Catalytic Procedure
3.5. Gaussian Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Aresta, M.; Dibenedetto, A.; Angelini, A. Catalysis for the valorization of exhaust carbon: From CO2 to chemicals, materials, and fuels technological use of CO2. Chem. Rev. 2014, 114, 1709–1742. [Google Scholar] [CrossRef] [PubMed]
- Centi, G.; Quadrelli, E.A.; Perathoner, S. Catalysis for CO2 conversion: A key technology for rapid introduction of renewable energy in the value chain of chemical industries. Energy Environ. Sci. 2013, 6, 1711–1731. [Google Scholar] [CrossRef]
- Cokoja, M.; Bruckmeier, C.; Rieger, B.; Herrmann, W.A.; Kuhn, F.E. Transformation of carbon dioxide with homogeneous transition-metal catalysts: A molecular solution to a global challenge? Angew. Chem. Int. Ed. 2011, 50, 8510–8537. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.Y.; Ma, J.; Zhu, Q.G.; Qian, Q.L.; Han, H.L.; Mei, Q.Q.; Han, B.X. Zinc(II)-catalyzed reactions of carbon dioxide and propargylic alcohols to carbonates at room temperature. Green Chem. 2016, 18, 382–385. [Google Scholar] [CrossRef]
- Hu, J.Y.; Ma, J.; Zhu, Q.G.; Zhang, Z.F.; Wu, C.Y.; Han, B.X. Transformation of atmospheric CO2 catalyzed by protic ionic liquids: Efficient synthesis of 2-oxazolidinones. Angew. Chem. Int. Ed. 2015, 54, 5399–5403. [Google Scholar] [CrossRef] [PubMed]
- Von der Assen, N.; Jung, J.; Bardow, A. Life-cycle assessment of carbon dioxide capture and utilization: Avoiding the pitfalls. Energy Environ. Sci. 2013, 6, 2721–2734. [Google Scholar] [CrossRef]
- Von der Assen, N.; Sternberg, A.; Katelhon, A.; Bardow, A. Environmental potential of carbon dioxide utilization in the polyurethane supply chain. Faraday Discuss. 2015, 183, 291–307. [Google Scholar] [CrossRef] [PubMed]
- Kleij, A.W.; North, M.; Urakawa, A. CO2 catalysis. ChemSusChem 2017, 10, 1036–1038. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.W.; Chen, W.Q.; Ma, R.; Yu, A.; Li, Q.Y.; Chang, Y.; He, L.N. Bifunctional silver(I) complex-catalyzed CO2 conversion at ambient conditions: Synthesis of α-methylene cyclic carbonates and derivatives. ChemSusChem 2015, 8, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Shyeni, P.; Zhu, Y.; Charles, R. Ring-opening copolymerization (ROCOP): Synthesis and properties of polyesters and polycarbonates. Chem. Commun. 2015, 51, 6459–6479. [Google Scholar] [CrossRef]
- Qing, H.; Jeremy, W.O.; Kayla, A.K.; Lindsay, E.T.; Gregory, C.T.C.; Francesca, M.K. Synthesis of cyclic carbonates from CO2 and epoxides using ionic liquids and related catalysts including choline chloride-metal halide mixtures. Catal. Sci. Technol. 2014, 4, 1513–1528. [Google Scholar] [CrossRef]
- North, M.; Pasquale, R.; Young, C. Synthesis of cyclic carbonates from epoxides and CO2. Green Chem. 2010, 12, 1514–1539. [Google Scholar] [CrossRef]
- Shi, Y.L.; Zhang, P.; Liu, D.H. Homogenous dual-ligand zinc complex catalysts for chemical fixation of CO2 to propylene carbonate. Catal. Lett. 2015, 145, 1673–1682. [Google Scholar] [CrossRef]
- Zhao, Y.; Qi, X.H.; He, L.N. Green process for synthesis of propylene carbonate. Chem. World 2008, 11, 696–699. [Google Scholar] [CrossRef]
- Paolo, P.P.; Masoumeh, T. Challenges in the catalytic synthesis of cyclic and polymeric carbonates from epoxides and CO2. Catal. Sci. Technol. 2012, 2, 2169–2187. [Google Scholar] [CrossRef]
- Zhao, D.; Wang, W.Z.; Jia, X.G.; Li, H.Y. Progress in synthesis of organic carbonates and polycarbonates from carbon dioxide. Mod. Chem. Ind. 2015, 37, 32–38. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Yeung, A.D. A concise review of computational studies of the carbon dioxide-epoxide copolymerization reactions. Polym. Chem. 2014, 5, 3949–3962. [Google Scholar] [CrossRef]
- Li, H.; Zhong, S. Dimethyl carbonate synthesis from carbon dioxide and methanol. Prog. Chem. 2002, 14, 368–373. [Google Scholar]
- Kruper, W.J.; Dellar, D.V. Catalytic formation of cyclic carbonates from epoxides and CO2 with chromium metalloporphyrinates. J. Org. Chem. 1995, 60, 725–727. [Google Scholar] [CrossRef]
- Darensbourg, D.J. Making Plastics from Carbon Dioxide: Salen Metal Complexes as Catalysts for the Production of Polycarbonates from Epoxides and CO2. Chem. Rev. 2007, 107, 2388–2410. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Kim, J.J.; Lee, B.G.; Jung, O.S.; Jang, H.G.; Kang, S.O. Isolation of a pyridinium alkoxy ion bridged dimeric zinic complex for the coupling reaction of CO2 and epoxides. Angew. Chem. Int. Ed. 2000, 39, 4096–4098. [Google Scholar] [CrossRef]
- Kim, H.S.; Kim, J.J.; Lee, S.D.; Lah, M.S.; Jang, D.M.H.G. New mechanistic insight into the coupling reactions of CO2 and epoxides in the presence of zinc complexes. Chem. Eur. J. 2003, 9, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Bae, J.Y.; Lee, J.S.; Kwon, O.-S.; Palgunadi, J.; Lee, S.D.; Lee, S.-H. Phosphine-bound zinc halide complexes for the coupling reaction of ethylene oxide and carbon dioxide. J. Catal. 2005, 232, 80–84. [Google Scholar] [CrossRef]
- Huang, J.W.; Shi, M. Chemical fixation of carbon dioxide by NaI/PPh3/PhOH. J. Org. Chem. 2003, 68, 6705–6709. [Google Scholar] [CrossRef] [PubMed]
- Meléndez, D.O.; Lara-Sánchez, A.; Martínez, J.; Wu, X.; Otero, A.; Castro-Osma, J.A.; North, M.; Rojas, R.S. Amidinate aluminium complexes as catalysts for carbon dioxide fixation into cyclic carbonates. ChemCatChem 2018, 10, 2271–2277. [Google Scholar] [CrossRef]
- Xu, B.H.; Wang, J.Q.; Sun, J.; Huang, Y.; Zhang, J.P.; Zhang, X.P.; Zhang, S.J. Fixation of CO2 into cyclic carbonates catalyzed by ionic liquids: A multi-scale approach. Green Chem. 2015, 17, 108–122. [Google Scholar] [CrossRef]
- D’Elia, V.; Pelletier, J.D.A.; Basset, J.M. Cycloadditions to epoxides catalyzed by group III–V transition-metal complexes. ChemCatChem 2015, 7, 1906–1917. [Google Scholar] [CrossRef]
- Rintjema, J.; Kleij, A.W. Aluminum-mediated formation of cyclic carbonates: Benchmarking catalytic performance metrics. ChemSusChem 2017, 10, 1274–1282. [Google Scholar] [CrossRef] [PubMed]
- Catalina, R.P.; Joaquín, S. Ferromagnetism in Malonato-bridged copper(II) complexes. synthesis, crystal structures, and magnetic properties of {[Cu(H2O)3][Cu(mal)2(H2O)]}n and {[Cu(H2O)4]2[Cu(mal)2 (H2O)][Cu(mal)2(H2O)2]}, {[Cu(H2O)4][Cu(mal)2(H2O)2]} (H2mal) malonic Acid). Inorg. Chem. 2000, 39, 1363–1370. [Google Scholar] [CrossRef]
- Addison, A.W.; Rao, T.N. Synthesis, structure, and spectroscopic properties of copper (II) compounds containing nitrogen-sulphur donor ligands; the crystal and molecular structure of aqua[l,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane] copper (II) perchlorate. Dalton Trans. 1984, 1349–1356. [Google Scholar] [CrossRef]
- Paddock, R.L.; Nguyen, S.T. Chemical CO2 fixation: Cr (III) Salen complexes as highly efficient catalysts for the coupling of CO2 and epoxides. J. Am. Chem. Soc. 2001, 123, 11498–11499. [Google Scholar] [CrossRef] [PubMed]
- Darensbourg, D.J.; Holtcamp, M.W. Catalysts for the reactions of epoxides and carbon dioxide. Coord. Chem. Rev. 1996, 153, 155. [Google Scholar] [CrossRef]
- Wing, N.S.; Siu, M.N.; Kar, Y.K.; Chak, P.L. Coupling reactions of CO2 with neat epoxides catalyzed by PPN salts to yield cyclic carbonates. J. Org. Chem. 2005, 70, 8583. [Google Scholar] [CrossRef]
- Tian, D.; Liu, B.; Zhang, L.; Wang, X.; Zhang, W.; Han, L.; Park, D.-W. Coupling reaction of carbon dioxide and epoxides efficiently catalyzed by one-component aluminum–salen complex under solvent-free conditions. J. Ind. Eng. Chem. 2012, 18, 1332–1338. [Google Scholar] [CrossRef]
- Ramin, M.; Jutz, F.; Grunwaldt, J.D.; Baiker, A. Solventless synthesis of propylene carbonate catalysed by chromium–salen complexes: Bridging homogeneous and heterogeneous catalysis. J. Mol. Catal. A Chem. 2005, 242, 32–39. [Google Scholar] [CrossRef]
- Hansen, K.B.; Leighton, J.L.; Jacobsen, E.N. On the Mechanism of asymmetric nucleophilic ring-opening of epoxides catalyzed by (Salen)CrIII Complexes. J. Am. Chem. Soc. 1996, 118, 10924–10925. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Ebitani, K.; Yoshida, T.; Yoshida, T.; Yoshida, H.; Kaneda, K. Mg−Al mixed oxides as highly active acid−base catalysts for cycloaddition of carbon dioxide to epoxides. J. Am. Chem. Soc. 1999, 121, 4526–4527. [Google Scholar] [CrossRef]
- Yano, T.; Matsui, H.; Koike, T.; Ishiguro, H.; Fujihara, H.; Yoshihara, M.; Maeshima, T. Magnesium oxide-catalysed reaction of carbon dioxide with an epoxide with retention of stereochemistry. Chem. Commun. 1997, 1129–1130. [Google Scholar] [CrossRef]
- Srivastava, R.; Bennur, T.H.; Srinivas, D. Factors affecting activation and utilization of carbon dioxide in cyclic carbonates synthesis over Cu and Mn peraza macrocyclic complexes. J. Mol. Catal. A Chem. 2005, 226, 199. [Google Scholar] [CrossRef]
- Wang, L.; Li, J.F.; Zhang, J.L. Insight into the role of weak interaction played in the fixation of CO2 catalyzed by the amino-functionalized imidazolium-based ionic liquids. J. CO2 Utilization 2017, 18, 156–163. [Google Scholar] [CrossRef]
- Alan, E.R.; Robert, B.W.; Frank, W. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Cao, B.; Du, J.; Liu, S.; Zhu, X.; Sun, X.; Sun, H.; Fu, H. Carbon dioxide capture by amino-functionalized ionic liquids: DFT based theoretical analysis substantiated by FT-IR investigation. RSC Adv. 2016, 6, 10462–10470. [Google Scholar] [CrossRef]
- Jiang, Q.; Zhao, G.Z. Study on the catalyst for the cycloaddition of carbon dioxide and propylene oxide. Nat. Gas Chem. Ind. 1996, 6, 22–26. [Google Scholar]
- Sheldrick, G.M. SHELXL97: Program for the refinement of crystal structures. Sci. Res. 1997. Available online: http://www.scirp.org/(S(oyulxb452alnt1aej1nfow45))/reference/ReferencesPapers.aspx?ReferenceID=1586897 (accessed on 31 August 2018).
- Wang, W.Z.; Wu, Y. An efficient catalyst system at mild reaction conditions containing rare earth metal complexes. J. Chin. Chem. Soc. 2013, 60, 1463. [Google Scholar] [CrossRef]
- Alan, E.R.; Larry, A.C.; Frank, W. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
Sample Availability: All samples of the compounds are available from the authors. |
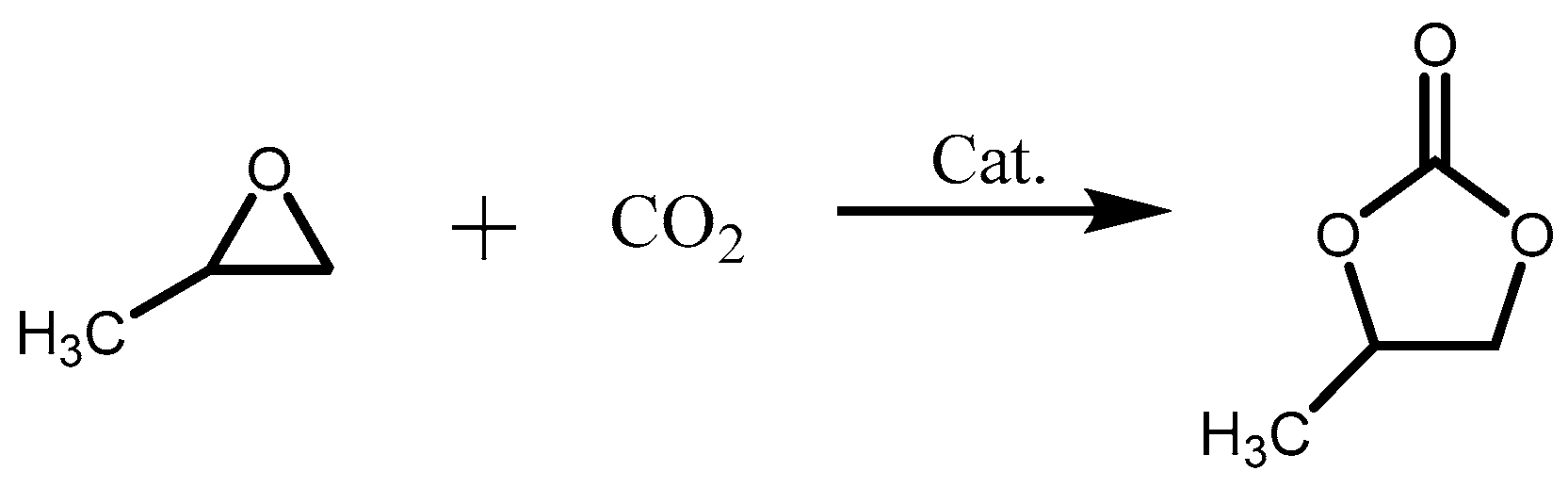


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
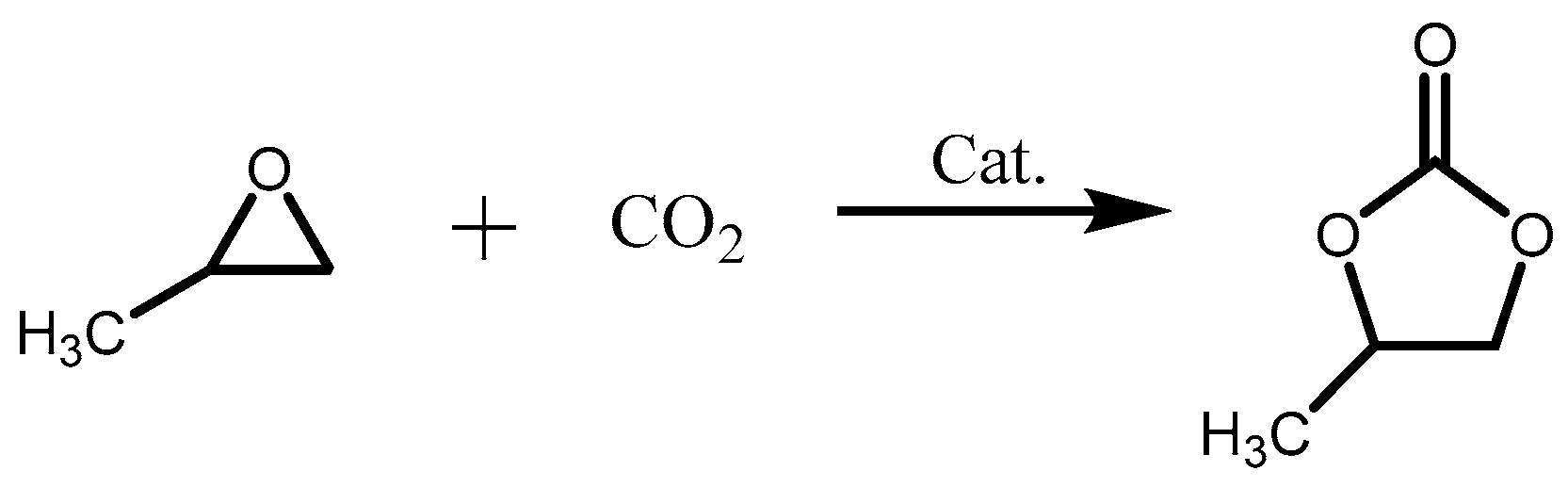{kind=link}
{kind=link}
| Compound | 1 | 2 | 3 |
|---|---|---|---|
| Empirical formula | C21H19Cl2CuN3 | C21H19Cl3CrN3 | C22H20.5Cl2MnN3.5 |
| Formula weight | 447.83 | 471.74 | 459.76 |
| Crystal system | Orthorhombic | Tetragonal | Triclinic |
| Space group | Pbca | I41/a | P |
| a (Å) | 15.632(8) | 15.775(4) | 8.988(3) |
| b (Å) | 20.298(11) | 15.775(4) | 12.730(5) |
| c (Å) | 25.070(13) | 33.247(8) | 20.143(7) |
| α (°) | 90.00 | 90.00 | 80.318(8) |
| β (°) | 90.00 | 90.00 | 79.252(7) |
| γ (°) | 90.00 | 90.00 | 86.277(8) |
| V (Å3) | 7955(7) | 8274(3) | 2230.7(14) |
| Z | 16 | 16 | 4 |
| ρcalcd (Mg·m−3) | 1.496 | 1.515 | 1.369 |
| μ (mm−1) | 1.377 | 0.953 | 0.845 |
| F(000) | 3664 | 3856 | 944 |
| Crystal size (mm) | 0.35 × 0.28 × 0.21 | 0.33 × 0.26 × 0.14 | 0.31 × 0.24 × 0.13 |
| θ range for data collection (°) | 2.31–18.37 | 1.43–25.10 | 2.07–25.10 |
| Reflections collected | 38,290 | 20,559 | 11,205 |
| Independent reflections | 7082 | 3688 | 7858 |
| Rint | 0.1513 | 0.1302 | 0.0723 |
| Final R indices [(I > 2σ(I)] | R1 = 0.0571 wR1 = 0.1052 | R1 = 0.0613 wR1 = 0.1305 | R1 = 0.0807 wR1 = 0.1638 |
| R indices (all data) | R2 = 0.1291 wR2 = 0.1316 | R2 = 0.1296 wR2 = 0.1651 | R2 = 0.1985 wR2 = 0.2245 |
| GOF | 0.958 | 1.011 | 0.951 |
| 1 | |||||
| Cu1―N1 | 2.121(4) | Cu1―Cl2 | 2.3857(19) | N2―C13 | 1.335(6) |
| Cu1―N2 | 1.950(5) | N1―C6 | 1.429(7) | N3―C14 | 1.277(6) |
| Cu1―N3 | 2.078(4) | N1―C7 | 1.296(7) | N3―C16 | 1.434(6) |
| Cu1―Cl1 | 2.2340(18) | N2―C9 | 1.337(6) | ||
| Cu2―N4 | 2.107(4) | Cu2―Cl3 | 2.388(2) | N5―C34 | 1.324(6) |
| Cu2―N5 | 1.936(4) | N4―C27 | 1.429(7) | N6―C35 | 1.277(6) |
| Cu2―N6 | 2.086(4) | N4―C28 | 1.285(6) | N6―C37 | 1.435(6) |
| Cu2―Cl4 | 2.2076(18) | N5―C30 | 1.344(7) | ||
| N1―Cu1―N3 | 155.34(18) | N2―Cu1―N3 | 77.64(18) | N3―Cu1―Cl2 | 95.23(13) |
| N1―Cu1―Cl1 | 100.62(13) | N2―Cu1―Cl1 | 141.29(13) | Cl1―Cu1―Cl2 | 109.23(7) |
| N1―Cu1―Cl2 | 94.55(13) | N2―Cu1―Cl2 | 109.45(13) | ||
| N2―Cu1―N1 | 77.76(18) | N3―Cu1―Cl1 | 97.41(13) | ||
| N4―Cu2―N6 | 154.87(17) | N5―Cu2―N6 | 77.50(18) | N6―Cu2―Cl3 | 98.07(13) |
| N4―Cu2―Cl4 | 101.51(13) | N5―Cu2―Cl4 | 149.59(14) | Cl4―Cu2―Cl3 | 106.88(7) |
| N4―Cu2―Cl3 | 93.57(13) | N5―Cu2―Cl3 | 103.48(14) | ||
| N5―Cu2―N4 | 78.17(19) | N6―Cu2―Cl4 | 96.31(13) | ||
| 2 | |||||
| Cl1―Cr1 | 2.3002(19) | Cr1―N2 | 2.001(5) | N2―C8 | 1.333(7) |
| Cl2―Cr1 | 2.3106(19) | Cr1―N3 | 2.111(5) | N2―C12 | 1.336(7) |
| Cl3―Cr1 | 2.3239(19) | N1―C6 | 1.441(7) | N3―C13 | 1.295(7) |
| Cr1―N1 | 2.109(5) | N1―C7 | 1.281(7) | N3―C14 | 1.448(7) |
| N1―Cr1―N3 | 153.68(19) | N2―Cr1―N3 | 77.1(2) | N3―Cr1―Cl2 | 88.06(14) |
| N1―Cr1―Cl1 | 100.10(14) | N2―Cr1―Cl1 | 176.62(16) | N3―Cr1―Cl3 | 89.96(14) |
| N1―Cr1―Cl2 | 90.44(14) | N2―Cr1―Cl2 | 87.15(14) | Cl1―Cr1―Cl2 | 92.43(7) |
| N1―Cr1―Cl3 | 89.14(14) | N2―Cr1―Cl3 | 87.58(14) | Cl1―Cr1―Cl3 | 92.88(7) |
| N2―Cr1―N1 | 76.55(19) | N3―Cr1―Cl1 | 106.21(14) | Cl2―Cr1―Cl3 | 174.67(8) |
| 3 | |||||
| Mn1―N1 | 2.186(6) | Mn1―Cl2 | 2.331(3) | N2―C7 | 1.278(10) |
| Mn1―N2 | 2.297(7) | N1―C9 | 1.350(10) | N3―C14 | 1.290(9) |
| Mn1―N3 | 2.303(7) | N1―C13 | 1.347(10) | N3―C16 | 1.417(8) |
| Mn1―Cl1 | 2.337(3) | N2―C6 | 1.449(10) | ||
| Mn2―N5 | 2.200(7) | Mn2―Cl3 | 2.341(3) | N4―C28 | 1.280(10) |
| Mn2―N4 | 2.258(7) | N5―C30 | 1.352(10) | N6―C35 | 1.293(10) |
| Mn2―N6 | 2.271(7) | N5―C34 | 1.324(10) | N6―C37 | 1.451(10) |
| Mn2―Cl4 | 2.328(3) | N4―C27 | 1.445(10) | ||
| N1―Mn1―N3 | 71.0(3) | N2―Mn1―N3 | 142.3(2) | N3―Mn1―Cl2 | 100.37(18) |
| N1―Mn1―Cl1 | 116.29(10) | N2―Mn1―Cl1 | 99.24(19) | Cl1―Mn1―Cl2 | 118.28(10) |
| N1―Mn1―Cl2 | 125.43(19) | N2―Mn1―Cl2 | 99.6(2) | ||
| N2―Mn1―N1 | 71.3(3) | N3―Mn1―Cl1 | 98.97(18) | ||
| N5―Mn2―N6 | 71.0(3) | N4―Mn2―N6 | 141.6(6) | N6―Mn2―Cl3 | 102.94(18) |
| N5―Mn2―Cl4 | 116.26(18) | N4―Mn2―Cl4 | 97.37(4) | Cl4―Mn2―Cl3 | 116.91(11) |
| N5―Mn2―Cl3 | 126.84(19) | N4―Mn2―Cl3 | 101.9(3) | ||
| N4―Mn2―N5 | 70.3(3) | N6―Mn2―Cl4 | 96.89(4) | ||
| Entry | Catalyst | Co-Catalyst | Conversion a (%) | TOF b (h−1) |
|---|---|---|---|---|
| 1 | none | DMAP | 0 | 0 |
| 2 | 1 | DMAP | 43.1 | 216 |
| 3 | 2 | DMAP | 74.6 | 373 |
| 4 | 3 | DMAP | 62.3 | 312 |
| 5 | 2 | none | 0 | 0 |
| 6 | none | TBAB | 28.6 | 143 |
| 7 | 1 | TBAB | 40.8 | 204 |
| 8 | 2 | TBAB | 69.5 | 348 |
| 9 | 3 | TBAB | 61.4 | 307 |
| Entry | [PO]/[2]/[DMAP] | P (MPa) | Time (h) | Temp (°C) | Conversion a (%) | TOF b (h−1) |
|---|---|---|---|---|---|---|
| 1 | 1000:1:1 | 2.0 | 2 | 120 | 67.3 | 337 |
| 2 | 1000:1:1 | 2.5 | 2 | 120 | 74.6 | 373 |
| 3 | 1000:1:1 | 3.0 | 2 | 120 | 82.8 | 429 |
| 4 | 1000:1:1 | 3.5 | 2 | 120 | 84.2 | 421 |
| 5 | 2000:1:1 | 2.5 | 2 | 100 | 69.5 | 695 |
| 6 | 2000:1:1 | 2.5 | 3 | 100 | 83.1 | 554 |
| 7 | 2000:1:1 | 2.5 | 4 | 100 | 86.7 | 434 |
| 8 | 2000:1:1 | 2.5 | 2 | 120 | 62.2 | 622 |
| 9 | 2000:1:1 | 2.5 | 1 | 200 | 51.3 | 1026 |
| 10 | 2000:1:1 | 2.5 | 1 | 220 | 41.6 | 832 |
| 11 | 3000:1:1 | 2.5 | 2 | 120 | 25.4 | 381 |
| 12 | 4000:1:1 | 2.5 | 2 | 120 | 14.8 | 296 |
| 13 | 2000:1:2 | 2.5 | 2 | 120 | 72.3 | 723 |
| 14 | 2000:1:4 | 2.5 | 2 | 120 | 15.3 | 153 |
| Complex | Metal Atom Charge Distribution | LUMO/eV | HOMO/eV | ΔE/eV |
|---|---|---|---|---|
| 1 | 0.460 | −0.03090 | −0.24552 | 0.21463 |
| 2 | 0.454 | −0.15833 | −0.16290 | 0.00457 |
| 3 | 0.697 | −0.10583 | −0.22280 | 0.11697 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, L.; Wang, W.-Z.; Liu, S.; Jia, X.-G.; Zhang, Y.-H.; Li, L.-L.; Wu, Y.; Su, B.-Y.; Geng, S.-B.; Fan, W. New Coordination Complexes Based on the 2,6-bis[1-(Phenylimino)ethyl] Pyridine Ligand: Effective Catalysts for the Synthesis of Propylene Carbonates from Carbon Dioxide and Epoxides. Molecules 2018, 23, 2304. https://doi.org/10.3390/molecules23092304
Xia L, Wang W-Z, Liu S, Jia X-G, Zhang Y-H, Li L-L, Wu Y, Su B-Y, Geng S-B, Fan W. New Coordination Complexes Based on the 2,6-bis[1-(Phenylimino)ethyl] Pyridine Ligand: Effective Catalysts for the Synthesis of Propylene Carbonates from Carbon Dioxide and Epoxides. Molecules. 2018; 23(9):2304. https://doi.org/10.3390/molecules23092304
Chicago/Turabian StyleXia, Li, Wen-Zhen Wang, Shuang Liu, Xin-Gang Jia, Ying-Hui Zhang, Lei-Lei Li, Ya Wu, Bi-Yun Su, Shu-Bo Geng, and Wei Fan. 2018. "New Coordination Complexes Based on the 2,6-bis[1-(Phenylimino)ethyl] Pyridine Ligand: Effective Catalysts for the Synthesis of Propylene Carbonates from Carbon Dioxide and Epoxides" Molecules 23, no. 9: 2304. https://doi.org/10.3390/molecules23092304
APA StyleXia, L., Wang, W.-Z., Liu, S., Jia, X.-G., Zhang, Y.-H., Li, L.-L., Wu, Y., Su, B.-Y., Geng, S.-B., & Fan, W. (2018). New Coordination Complexes Based on the 2,6-bis[1-(Phenylimino)ethyl] Pyridine Ligand: Effective Catalysts for the Synthesis of Propylene Carbonates from Carbon Dioxide and Epoxides. Molecules, 23(9), 2304. https://doi.org/10.3390/molecules23092304