Novel Group of AChE Reactivators—Synthesis, In Vitro Reactivation and Molecular Docking Study





Abstract
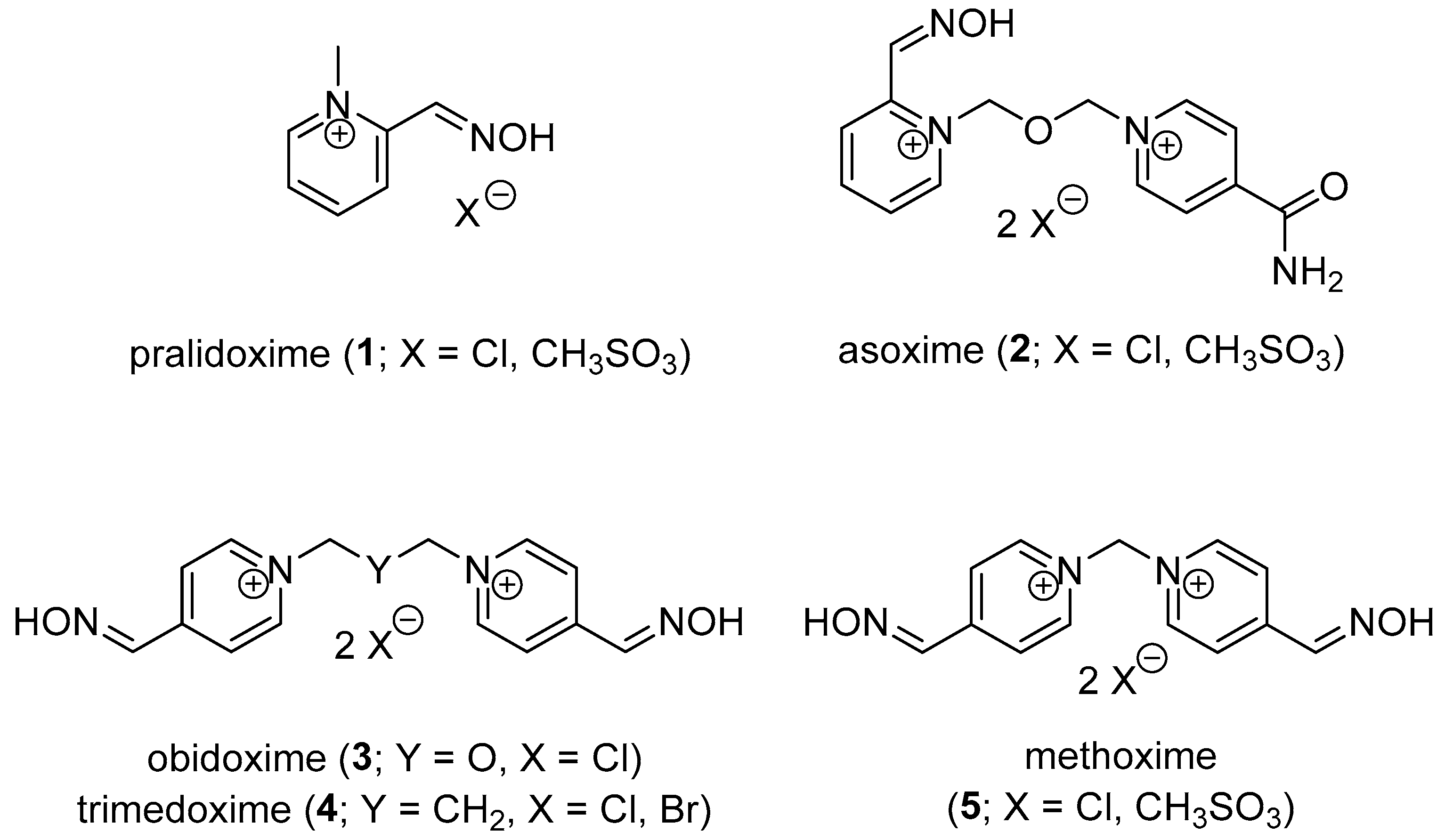
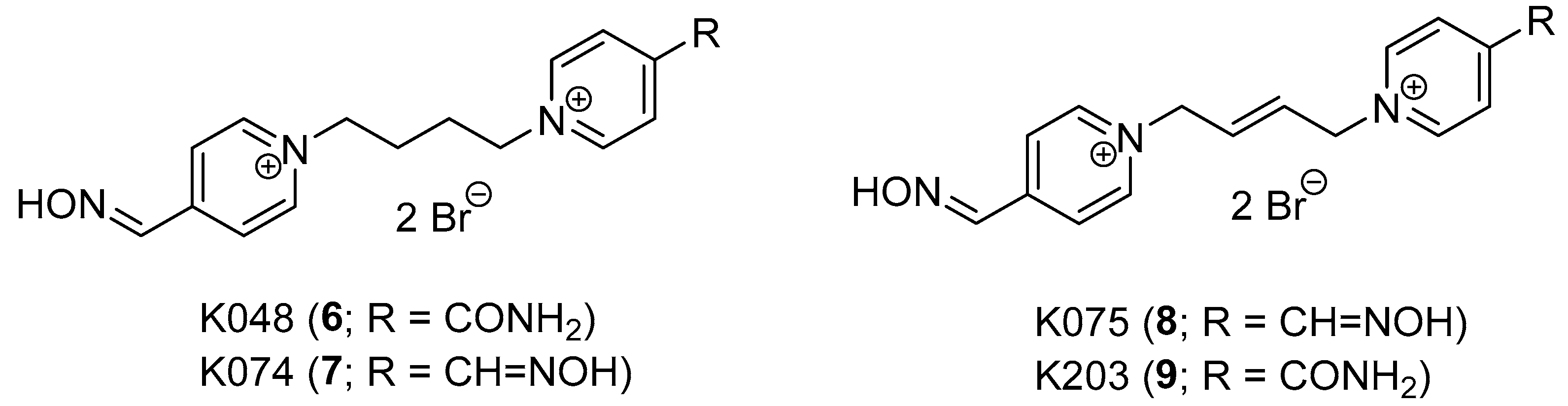
1. Introduction
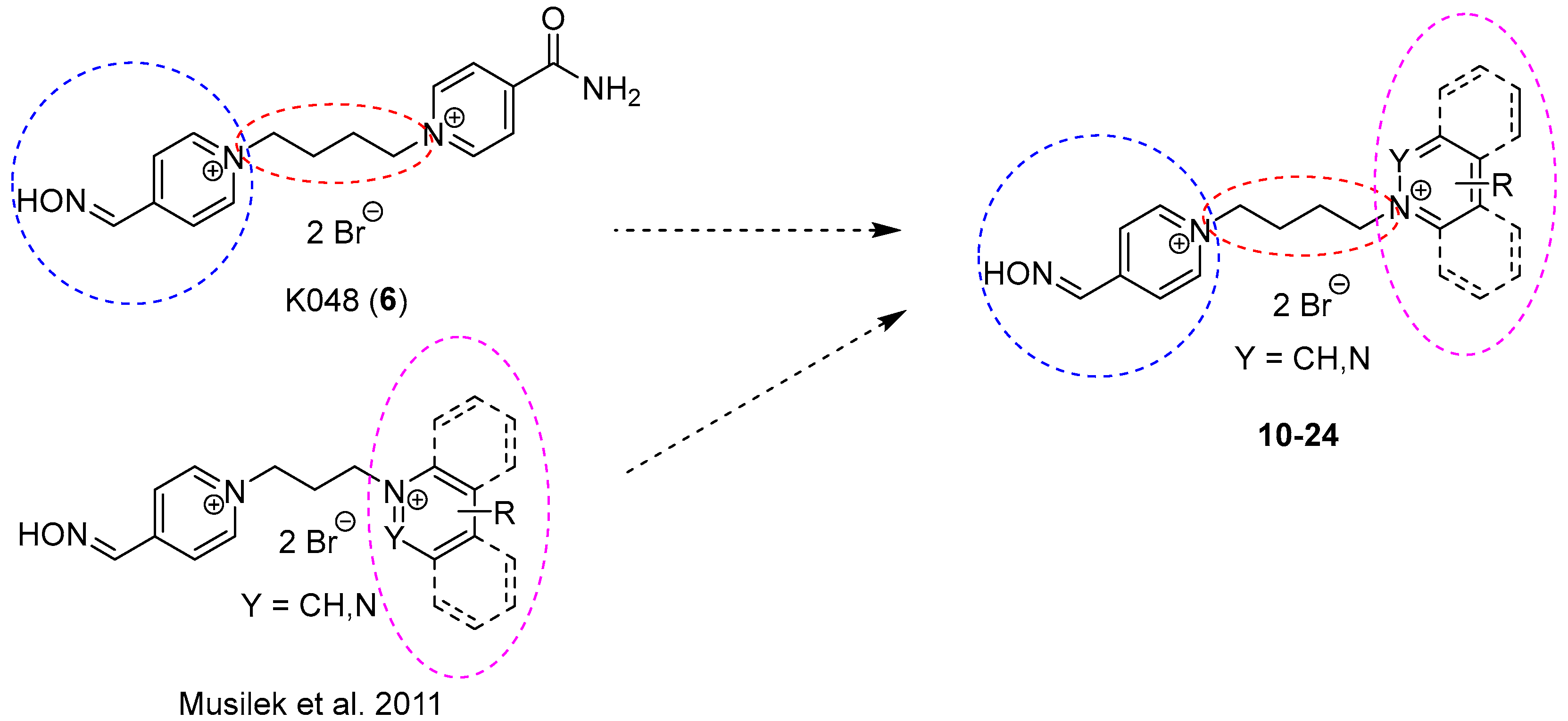
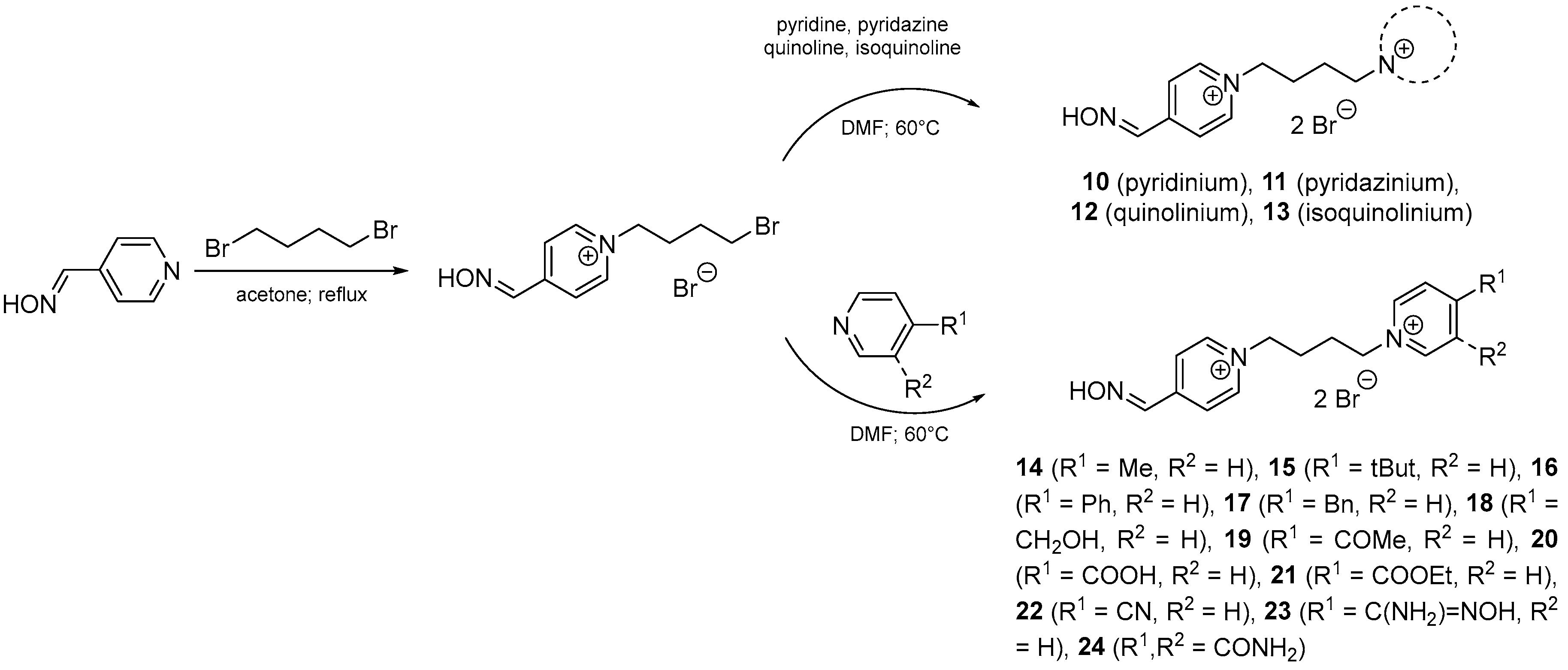
2. Design and Synthesis Mono-Oxime Bispyridinium Reactivators
3. Results and Discussion
3.1. HssAChE Reactivation Results
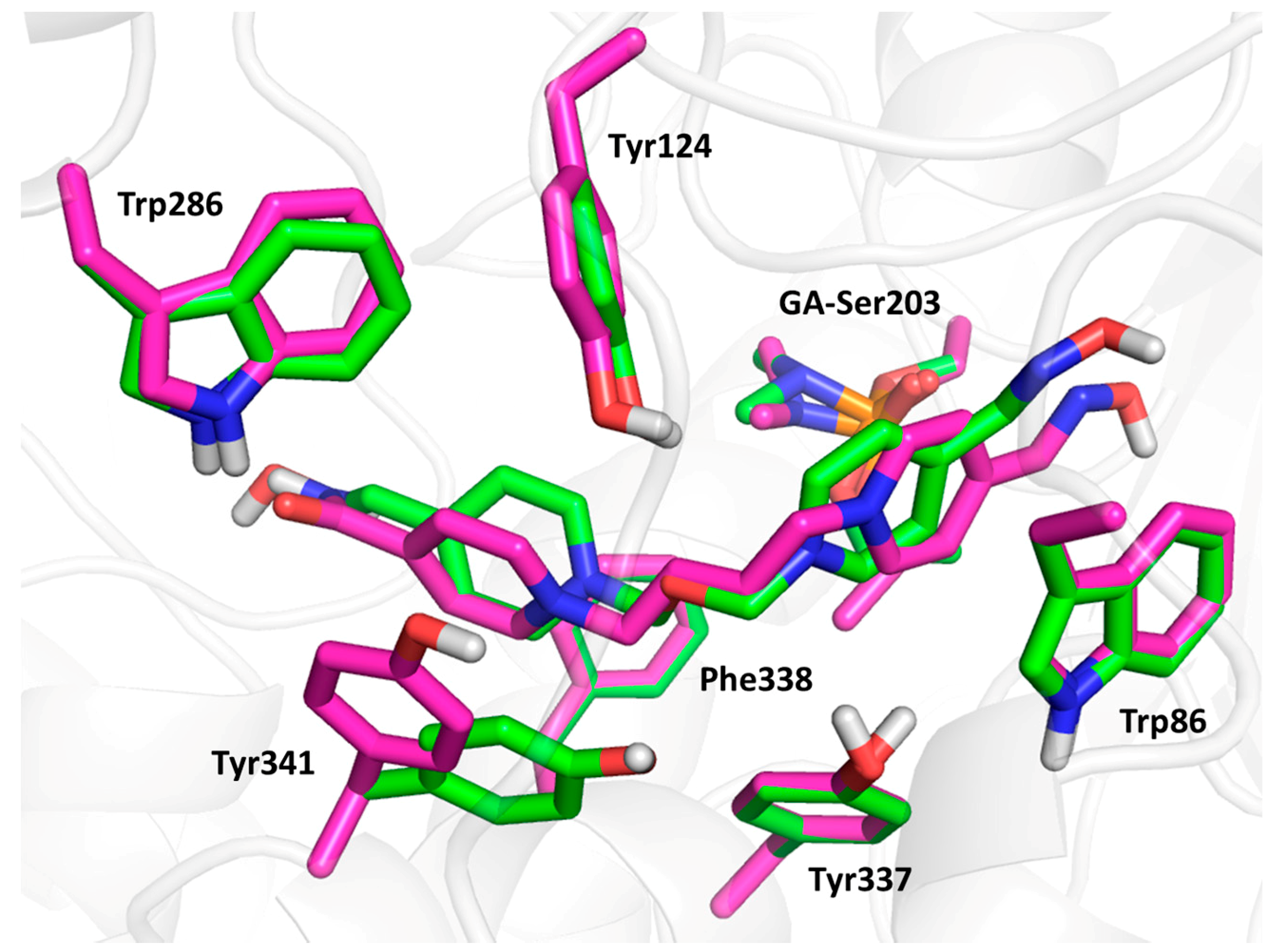
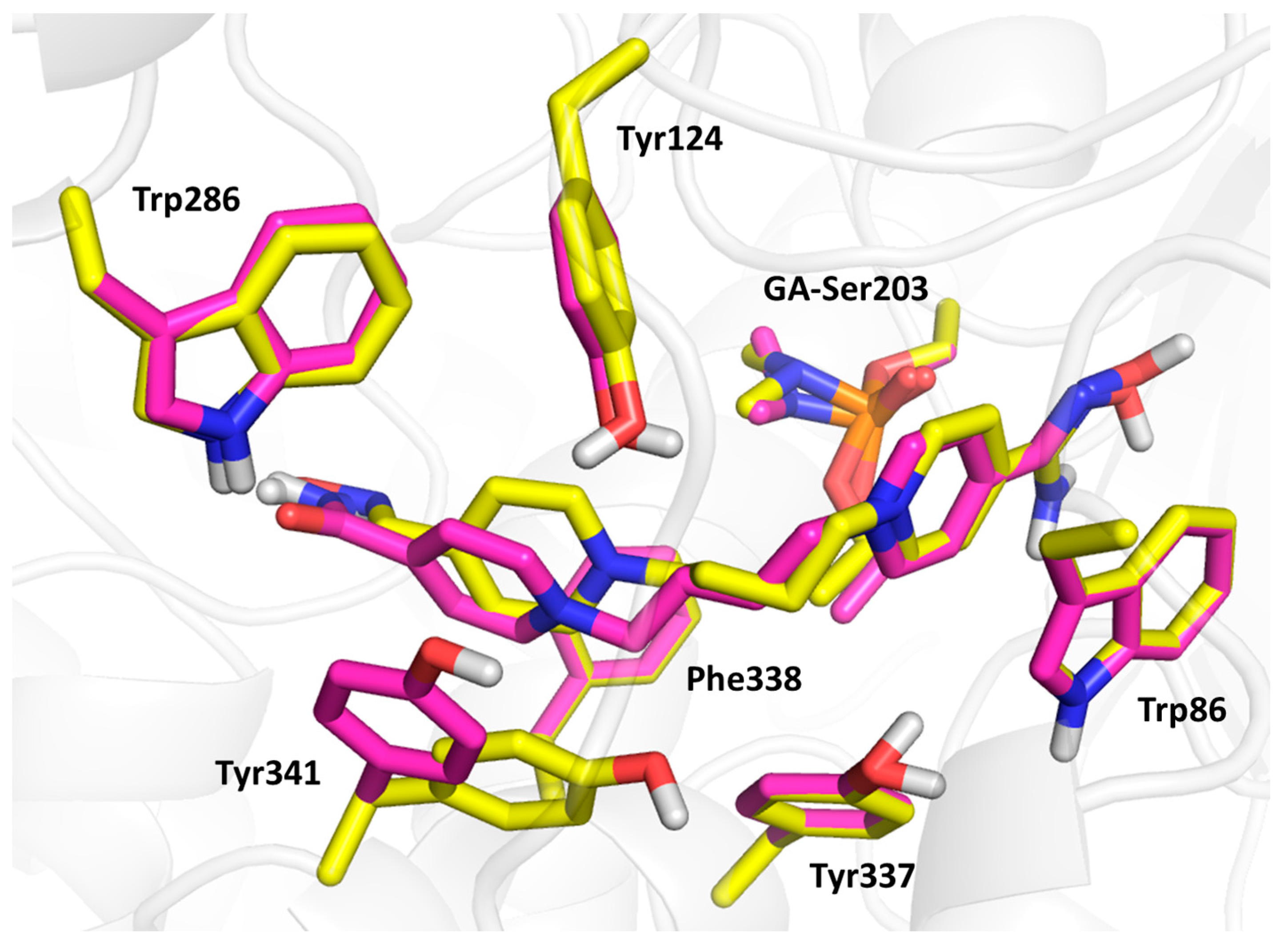
3.2. Molecular Modelling Results and SAR Discussion
4. Experimental Section
4.1. Chemical Preparation
4.2. Preparation of Bisquaternary Salts
4.3. Prepared Monoquaternary Salt
4.4. Prepared Bisquaternary Salts
4.5. In Vitro Reactivation Assay
4.6. In Vitro Inhibition Assay
4.7. Molecular Docking Study
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Marrs, T.C. Organophosphate poisoning. Pharmacol. Ther. 1993, 58, 51–66. [Google Scholar] [CrossRef]
- Bajgar, J. Organophosphates/nerve agent poisoning: Mechanism of action, diagnosis, prophylaxis, and teratment. Adv. Clin. Chem. 2004, 38, 151–193. [Google Scholar] [PubMed]
- Saxena, A.; Sun, W.; Luo, C.; Myers, T.M.; Koplovitz, I.; Lenz, D.E.; Doctor, B.P. Bioscavenger for protection from toxicity of organophosphorus compounds. J. Mol. Neurosci. 2006, 30, 145–147. [Google Scholar] [CrossRef]
- Newmark, J. Therapy for Nerve Agent Poisoning. Arch. Neurol. 2004, 61, 649–652. [Google Scholar] [CrossRef] [PubMed]
- Bajgar, J.; Fusek, J.; Kassa, J.; Kuca, K.; Jun, D. Chemical Aspects of Pharmacological Prophylaxis against Nerve Agent Poisoning. Curr. Med. Chem. 2009, 16, 2977–2986. [Google Scholar] [CrossRef] [PubMed]
- Doctor, B.P.; Raveh, L.; Wolfe, A.D.; Maxwell, D.M.; Ashani, Y. Enzymes as pretreatment drugs for organophosphate toxicity. Neurosci. Biobehav. Rev. 1991, 15, 123–128. [Google Scholar] [CrossRef]
- Jokanovic, M.; Stojiljkovic, M.P. Current understanding of the application of pyridinium oximes as cholinesterase reactivators in treatment of organophosphate poisoning. Eur. J. Pharmacol. 2006, 553, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Bajgar, J.; Fusek, J.; Kuca, K.; Bartosova, L.; Jun, D. Treatment of Organophosphate Intoxication Using Cholinesterase Reactivators: Facts and Fiction. Mini-Rev. Med. Chem. 2007, 7, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Carletti, E.; Li, H.; Li, B.; Ekstrom, F.; Nicolet, Y.; Loiodice, M.; Gillon, E.; Froment, M.T.; Lockridge, O.; Schopfer, L.M.; et al. Aging of Cholinesterases Phosphylated by Tabun Proceeds through O-Dealkylation. J. Am. Chem. Soc. 2008, 130, 16011–16020. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Schopfer, L.M.; Nachon, F.; Froment, M.T.; Masson, P.; Lockridge, O. Aging Pathways for Organophosphate-Inhibited Human Butyrylcholinesterase, Including Novel Pathways for Isomalathion, Resolved by Mass Spectrometry. Toxicol. Sci. 2007, 100, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Kuca, K.; Musilek, K.; Jun, D.; Pohanka, M.; Zdarova-Karasova, J.; Novotny, L.; Musilova, L. Could oxime HI-6 really be considered as broad-spectrum antidote? J. Appl. Biomed. 2009, 7, 143–149. [Google Scholar]
- De Koning, M.C.; Joosen, M.J.A.; Noort, D.; van Zuylen, A.; Tromp, M.C. Peripheral site ligand-oxime conjugates: A novel concept towards reactivation of nerve agent-inhibited human acetylcholinesterase. Bioorg. Med. Chem. 2011, 19, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Mercey, G.; Verdelet, T.; Saint-Andre, G.; Gillon, E.; Wagner, A.; Baati, R.; Jean, L.; Nachon, F.; Renard, P.Y. First efficient uncharged reactivators for the dephosphylation of poisoned human acetylcholinesterase. Chem. Commun. 2011, 47, 5295–5297. [Google Scholar] [CrossRef] [PubMed]
- Musilek, K.; Holas, O.; Jun, D.; Dohnal, V.; Gunn-Moore, F.; Opletalova, V.; Dolezal, M.; Kuca, K. Monooxime reactivators of acetylcholinesterase with (E)-but-2-ene linker—Preparation and reactivation of tabun- and paraoxon-inhibited acetylcholinesterase. Bioorg. Med. Chem. 2007, 15, 6733–6741. [Google Scholar] [CrossRef] [PubMed]
- Musilek, K.; Komloova, M.; Holas, O.; Horova, A.; Pohanka, M.; Gunn-Moore, F.; Dohnal, V.; Dolezal, M.; Kuca, K. Mono-oxime bisquaternary acetylcholinesterase reactivators with prop-1,3-diyl linkage—Preparation, in vitro screening and molecular docking. Bioorg. Med. Chem. 2011, 19, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Kuca, K.; Kassa, J. A Comparison of the Ability of a New Bispyridinium Oxime—1-(4-hydroxyiminomethylpyridinium)-4-(4-carbamoylpyridinium)butane Dibromide and Currently used Oximes to Reactivate Nerve Agent-inhibited Rat Brain Acetylcholinesterase by In Vitro Methods. J. Enzym. Inhib. Med. Chem. 2003, 18, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Musilek, K.; Jun, D.; Cabal, J.; Kassa, J.; Gunn-Moore, F.; Kuca, K. Design of a Potent Reactivator of Tabun-Inhibited Acetylcholinesterase Synthesis and Evaluation of (E)-1-(4-Carbamoylpyridinium)-4-(4-hydroxyiminomethylpyridinium)-but-2-ene Dibromide (K203). J. Med. Chem. 2007, 50, 5514–5518. [Google Scholar] [CrossRef] [PubMed]
- Kuca, K.; Jun, D.; Musilek, K. Structural Requirements of Acetylcholinesterase Reactivators. Mini Rev. Med. Chem. 2006, 6, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Kuca, K.; Cabal, J.; Musilek, K.; Jun, D.; Bajgar, J. Effective bisquaternary reactivators of tabun-inhibited AChE. J. Appl. Toxicol. 2005, 25, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Lorke, D.E.; Nurulain, S.M.; Hasan, M.Y.; Kuca, K.; Musilek, K.; Petroianu, G.A. Eight new bispyridinium oximes in comparison with the conventional oximes pralidoxime and obidoxime: In vivo efficacy to protect from diisopropylfluorophosphate toxicity. J. Appl. Toxicol. 2008, 28, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Sinko, G.; Brglez, J.; Kovarik, Z. Interactions of pyridinium oximes with acetylcholinesterase. Chem. Biol. Interact. 2010, 187, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Pohanka, M.; Jun, D.; Kuca, K. Improvement of acetylcholinesterase-based assay for organophosphates in way of identification by reactivators. Talanta 2008, 77, 451–454. [Google Scholar] [CrossRef] [PubMed]
- Calic, M.; Vrdoljak, A.L.; Radic, M.; Jelic, D.; Jun, D.; Kuca, K.; Kovarik, Z. In vitro and in vivo evaluation of pyridinium oximes: Mode of interaction with acetylcholinesterase, effect on tabun- and soman-poisoned mice and their cytotoxicity. Toxicology 2006, 219, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Kovarik, Z.; Vrdoljak, A.L.; Berend, S.; Katalinic, M.; Kuca, K.; Musilek, K.; Radic, B. Evaluation of oxime K203 as antidote in tabun poisoning. Arh. Hig. Rada Toksikol. 2009, 60, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Tattersall, J.E.H. Ion channel blockade by oximes and recovery of diaphragm muscle from soman poisoning in vitro. Brit. J. Pharmacol. 1993, 108, 1006–1015. [Google Scholar] [CrossRef]
- Worek, F.; Aurbek, N.; Wille, T.; Eyer, P.; Thiermann, H. Kinetic analysis of interactions of paraoxon and oximes with human, Rhesus monkey, swine, rabbit, rat and guinea pig acetylcholinesterase. Toxicol. Lett. 2011, 200, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Lorke, D.E.; Hasan, M.Y.; Arafat, K.; Kuca, K.; Musilek, K.; Schmitt, A.; Petroianu, G.A. In vitro oxime protection of human red blood cell acetylcholinesterase inhibited by diisopropyl-fluorophosphate. J. Appl. Toxicol. 2008, 28, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Odzak, R.; Calic, M.; Hrenar, T.; Primozic, I.; Kovarik, Z. Evaluation of monoquaternary pyridinium oximes potency to reactivate tabun-inhibited human acetylcholinesterase. Toxicology 2007, 233, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Musilek, K.; Holas, O.; Misik, J.; Pohanka, M.; Novotny, L.; Dohnal, V.; Opletalova, V.; Kuca, K. Monooxime-monocarbamoyl Bispyridinium Xylene-Linked Reactivators of Acetylcholinesterase—Synthesis, In vitro and Toxicity Evaluation, and Docking Studies. ChemMedChem 2010, 5, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Worek, F.; Aurbek, N.; Thiermann, H. Reactivation of organophosphate-inhibited human AChE by combinations of obidoxime and HI 6 in vitro. J. Appl. Toxicol. 2007, 27, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Kryger, G.; Harel, M.; Giles, K.; Toker, L.; Velan, B.; Lazar, A.; Kronman, C.; Barak, D.; Ariel, N.; Shafferman, A.; et al. Structures of recombinant native and E202Q mutant human acetylcholinesterase complexed with the snake-venom toxin fasciculin-II. Acta Crystallogr. Sect. D 2000, 56, 1385–1394. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Mercey, G.; Verdelet, T.; Renou, J.; Kliachyna, M.; Baati, R.; Nachon, F.; Jean, L.; Renard, P.-Y. Reactivators of acetylcholinesterase inhibited by organophosphorus nerve agents. Acc. Chem. Res. 2012, 45, 756–766. [Google Scholar] [CrossRef] [PubMed]
- Acharya, J.; Dubey, D.K.; Srivastava, A.K.; Raza, S.K. In vitro reactivation of sarin-inhibited human acetylcholinesterase (AChE) by bis-pyridinium oximes connected by xylene linkers. Toxicol. In Vitro 2011, 25, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Musilek, K.; Dolezal, M.; Gunn-Moore, F.; Kuca, K. Design, evaluation and structure—Activity relationship studies of the AChE reactivators against organophosphorus pesticides. Med. Res. Rev. 2011, 31, 548–575. [Google Scholar] [CrossRef] [PubMed]
- Musilek, K.; Roder, J.; Komloova, M.; Holas, O.; Hrabinova, M.; Pohanka, M.; Dohnal, V.; Opletalova, V.; Kuca, K.; Jung, Y.S. Preparation, in vitro screening and molecular modelling of symmetrical 4-tert-butylpyridinium cholinesterase inhibitors—Analogues of SAD-128. Bioorg. Med. Chem. Lett. 2011, 21, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Musilek, K.; Holas, O.; Kuca, K.; Jun, D.; Dohnal, V.; Dolezal, M. Synthesis of asymmetrical bispyridinium compounds bearing cyano-moiety and evaluation of their reactivation activity against tabun and paraoxon-inhibited acetylcholinesterase. Bioorg. Med. Chem. Lett. 2006, 16, 5673–5676. [Google Scholar] [CrossRef] [PubMed]
- Kuca, K.; Jun, D.; Junova, L.; Musilek, K.; Hrabinova, M.; da Silva, J.A.V.; Ramalho, T.C.; Valko, M.; Wu, Q.; Nepovimova, E.; et al. Synthesis, Biological Evaluation, and Docking Studies of Novel Bisquaternary Aldoxime Reactivators on Acetylcholinesterase and Butyrylcholinesterase Inhibited by Paraoxon. Molecules 2018, 23, 1103. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The PyMOL Molecular Graphics System. 2002. Available online: http://www.pymol.org (accessed on 6 September 2018).
Sample Availability: Samples of the compounds are available from the authors. |








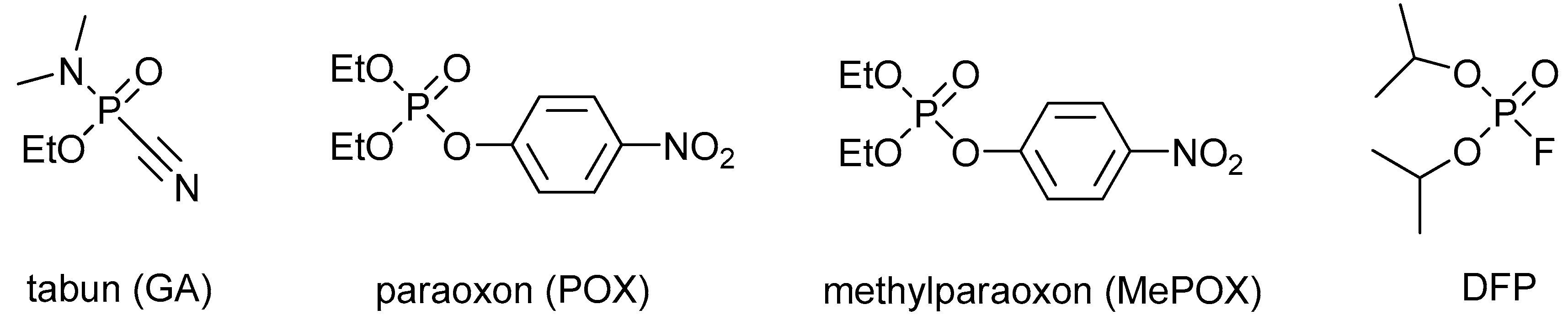{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Non-Reactivating Moiety | GA-HssAChE Reactivation (% ± SD) | POX-HssAChE Reactivation (% ± SD) | MePOX-HssAChE Reactivation (% ± SD) | DFP-HssAChE Reactivation (% ± SD) | HssAChE IC50 ± SD | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | 100 μM | 10 μM | 100 μM | 10 μM | 100 μM | 10 μM | 100 μM | 10 μM | µM | |
| 1 (2-PAM) | - | 3.3 ± 0.5 | 2.4 ± 0.2 | 10.7 ± 0.3 | 2.1 ± 0.1 | 30.2 ± 0.3 | 22.4 ± 0.7 | 1.3 ± 0.6 | 0.1 ± 0.4 | 878 ± 171 |
| 2 (HI-6) | CONH2 | 0.9 ± 0.6 | 0.8 ± 0.3 | 6.2 ± 0.6 | 1.7 ± 0.1 | 13.6 ± 0.2 | 17.9 ± 0.4 | 0.7 ± 0.1 | 1.4 ± 0.1 | 203 ± 39 |
| 3 (obidoxime) | CH=NOH | 15.1 ± 0.9 | 7.9 ± 0.5 | 59.7 ± 1.0 | 22.4 ± 0.4 | 61.7 ± 0.3 | 45.3 ± 0.9 | 7.6 ± 0.7 | 3.3 ± 0.4 | 577 ± 113 |
| 4 (trimedoxime) | CH=NOH | 31.5 ± 1.2 | 16.9 ± 0.2 | 44.3 ± 0.6 | 22.5 ± 1.3 | 51.4 ± 0.9 | 59.5 ± 0.7 | 10.0 ± 0.5 | 2.7 ± 0.3 | 167 ± 33 |
| 5 (methoxime) | CH=NOH | 2.9 ± 0.01 | 2.1 ± 0.5 | 16.1 ± 0.5 | 1.8 ± 0.3 | 14.2 ± 0.1 | 14.3 ± 0.2 | 2.4 ± 0.1 | 0.6 ± 0.3 | 2010 ± 391 |
| 6 (K048) | CONH2 | 27.4 ± 1.1 | 10.0 ± 0.6 | 25.7 ± 0.7 | 12.5 ± 0.2 | 54.4 ± 0.9 | 29.1 ± 0.4 | 3.8 ± 0.1 | 1.4 ± 0.1 | 349 ± 68 |
| 7 (K074) | CH=NOH | 32.1 ± 0.6 | 24.6 ± 0.2 | 31.1 ± 0.4 | 13.9 ± 0.8 | 17.9 ± 0.1 | 21.2 ± 0.4 | 1.4 ± 0.5 | 1.4 ± 0.2 | 29 ± 6 |
| 8 (K075) | CH=NOH | 19.5 ± 1.0 | 18.1 ± 0.2 | 28.5 ± 1.1 | 14.1 ± 0.3 | 19.2 ± 0.9 | 22.8 ± 0.2 | 1.1 ± 0.3 | 1.5 ±0.2 | 80 ± 16 |
| 9 (K203) | CONH2 | 48.1 ± 1.5 | 21.2 ± 0.3 | 39.3 ± 0.4 | 13.1 ± 0.4 | 55.9 ± 0.5 | 41.1 ± 0.1 | 4.4 ± 0.1 | 2.1 ± 0.4 | 566 ± 110 |
| 10 | pyridinium | 3.9 ± 0.1 | 4.5 ± 0.3 | 16.6 ± 1.0 | 3.5 ± 0.3 | 14.3 ± 0.8 | 13.2 ± 0.1 | 2.4 ± 0.5 | 0.4 ± 0.3 | - |
| 11 | pyridazinium | 1.5 ± 0.5 | 4.1 ± 0.2 | 6.3 ± 0.8 | 3.0 ± 0.3 | 7.1 ± 0.7 | 11.8 ± 0.4 | 1.2 ± 0.7 | 1.0 ± 0.4 | - |
| 12 | quinolinium | 0.6 ± 0.4 | 3.1 ± 0.3 | 0.6 ± 0.5 | 2.0 ± 0.1 | 2.2 ± 0.5 | 8.6 ± 0.4 | 0.2 ± 1.0 | 0.8 ± 0.2 | - |
| 13 | isoquinolinium | 3.5 ± 0.7 | 6.0 ± 0.2 | 9.1 ± 0.3 | 8.7 ± 0.3 | 8.2 ± 0.8 | 16.9 ± 0.4 | 0.7 ± 0.2 | 1.1 ± 0.2 | - |
| 14 | Me | 3.4 ± 0.4 | 6.3 ± 0.3 | 24.5 ± 0.3 | 7.6 ± 0.6 | 9.5 ± 0.5 | 9.4 ± 0.2 | 1.1 ± 0.4 | 0.8 ± 0.3 | - |
| 15 | tBut | 1.7 ± 0.4 | 3.9 ± 0.3 | 18.7 ± 0.2 | 58.7 ± 4.0 | 4.0 ± 0.5 | 7.6 ± 0.5 | 1.7 ± 0.5 | 0.8 ± 0.3 | 24 ± 6 |
| 16 | Ph | 0.7 ± 0.4 | 3.5 ± 0.3 | 6.9 ± 0.4 | 8.9 ± 0.5 | 1.7 ± 0.3 | 6.2 ± 0.4 | 0.7 ± 0.2 | 1.0 ± 0.3 | 6 ± 1 |
| 17 | Bn | 0.3 ± 0.2 | 1.9 ± 0.6 | 1.2 ± 0.1 | 4.7 ± 0.3 | 0.0 ± 0.1 | 3.8 ± 0.4 | 0.0 ± 0.5 | 0.8 ± 0.2 | - |
| 18 | CH2OH | 2.6 ± 0.3 | 4.0 ± 0.3 | 20.6 ± 0.1 | 8.0 ± 0.1 | 6.7 ± 0.3 | 8.1 ± 0.3 | 3.8 ± 1.0 | 1.4 ± 0.4 | - |
| 19 | COMe | 1.7 ± 0.3 | 3.1 ± 0.1 | 11.9 ± 1.0 | 3.9 ± 0.3 | 5.2 ± 0.4 | 7.5 ± 0.1 | 1.5 ± 0.3 | 1.0 ± 0.2 | - |
| 20 | COOH | 3.3 ± 0.5 | 6.2 ± 0.3 | 14.5 ± 0.6 | 4.2 ± 0.4 | 9.5 ± 0.3 | 8.8 ± 0.4 | 1.1 ± 0.5 | 0.4 ± 0.1 | - |
| 21 | COOEt | 1.7 ± 0.5 | 4.1 ± 0.2 | 7.1 ± 0.7 | 4.0 ± 0.6 | 3.8 ± 0.4 | 7.2 ± 0.2 | 0.8 ± 0.3 | 1.1 ± 0.2 | - |
| 22 | CN | 3.6 ± 0.4 | 5.2 ± 0.2 | 10.1 ± 0.8 | 42.6 ± 0.1 | 11.1 ± 0.1 | 13.8 ± 0.3 | 1.7 ± 0.5 | 0.4 ± 0.2 | 45 ± 9 |
| 23 | C(NH2)=NOH | 12.8 ± 0.9 | 13.1 ± 0.4 | 19.1 ± 0.8 | 7.4 ± 0.3 | 20.2 ± 1.0 | 20.7 ± 0.4 | 3.1 ± 0.8 | 2.0 ± 0.2 | 90 ± 17 |
| 24 | 3,4-CONH2 | 1.8 ± 0.6 | 4.5 ± 0.2 | 2.0 ± 0.4 | 2.2 ± 0.8 | 4.2 ± 0.5 | 10.1 ± 0.2 | 0.7 ± 0.4 | 0.7 ± 1.0 | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malinak, D.; Nepovimova, E.; Jun, D.; Musilek, K.; Kuca, K. Novel Group of AChE Reactivators—Synthesis, In Vitro Reactivation and Molecular Docking Study. Molecules 2018, 23, 2291. https://doi.org/10.3390/molecules23092291
Malinak D, Nepovimova E, Jun D, Musilek K, Kuca K. Novel Group of AChE Reactivators—Synthesis, In Vitro Reactivation and Molecular Docking Study. Molecules. 2018; 23(9):2291. https://doi.org/10.3390/molecules23092291
Chicago/Turabian StyleMalinak, David, Eugenie Nepovimova, Daniel Jun, Kamil Musilek, and Kamil Kuca. 2018. "Novel Group of AChE Reactivators—Synthesis, In Vitro Reactivation and Molecular Docking Study" Molecules 23, no. 9: 2291. https://doi.org/10.3390/molecules23092291
APA StyleMalinak, D., Nepovimova, E., Jun, D., Musilek, K., & Kuca, K. (2018). Novel Group of AChE Reactivators—Synthesis, In Vitro Reactivation and Molecular Docking Study. Molecules, 23(9), 2291. https://doi.org/10.3390/molecules23092291