Structural Architectural Features of Cyclodextrin Oligoesters Revealed by Fragmentation Mass Spectrometry Analysis




Abstract

1. Introduction
2. Results and Discussion
2.1. Nuclear Magnetic Resonance (NMR) and Gel Permeation Chromatography (GPC) Characterization
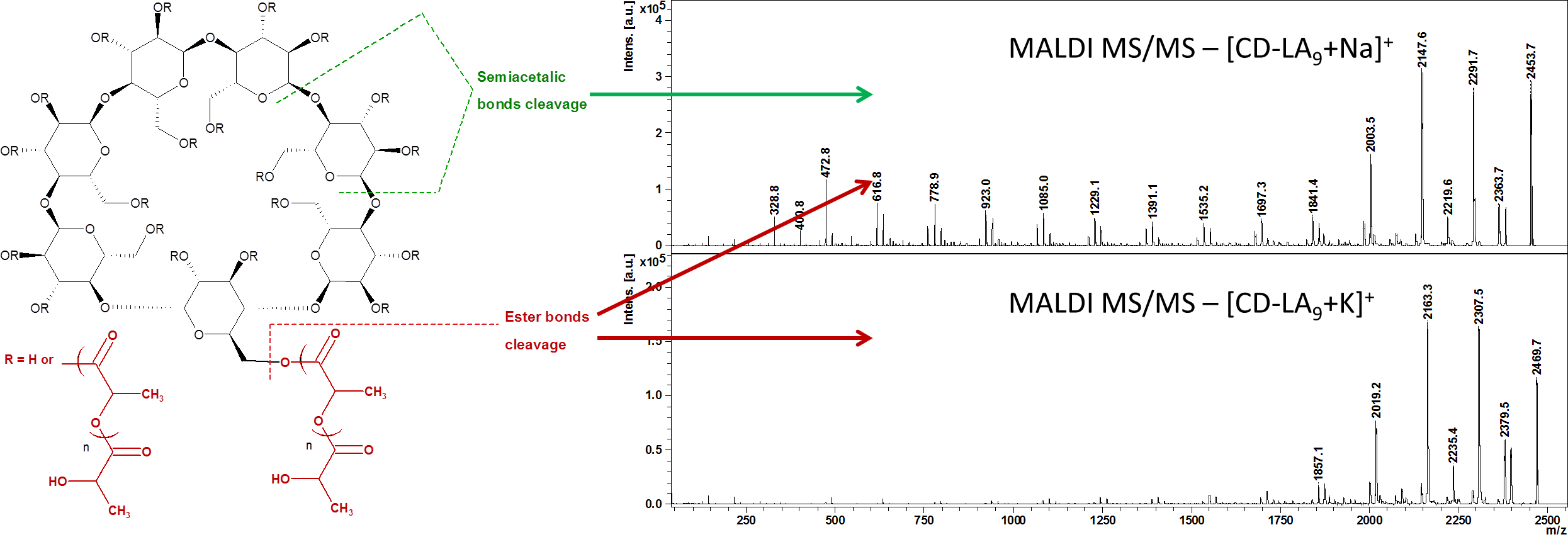
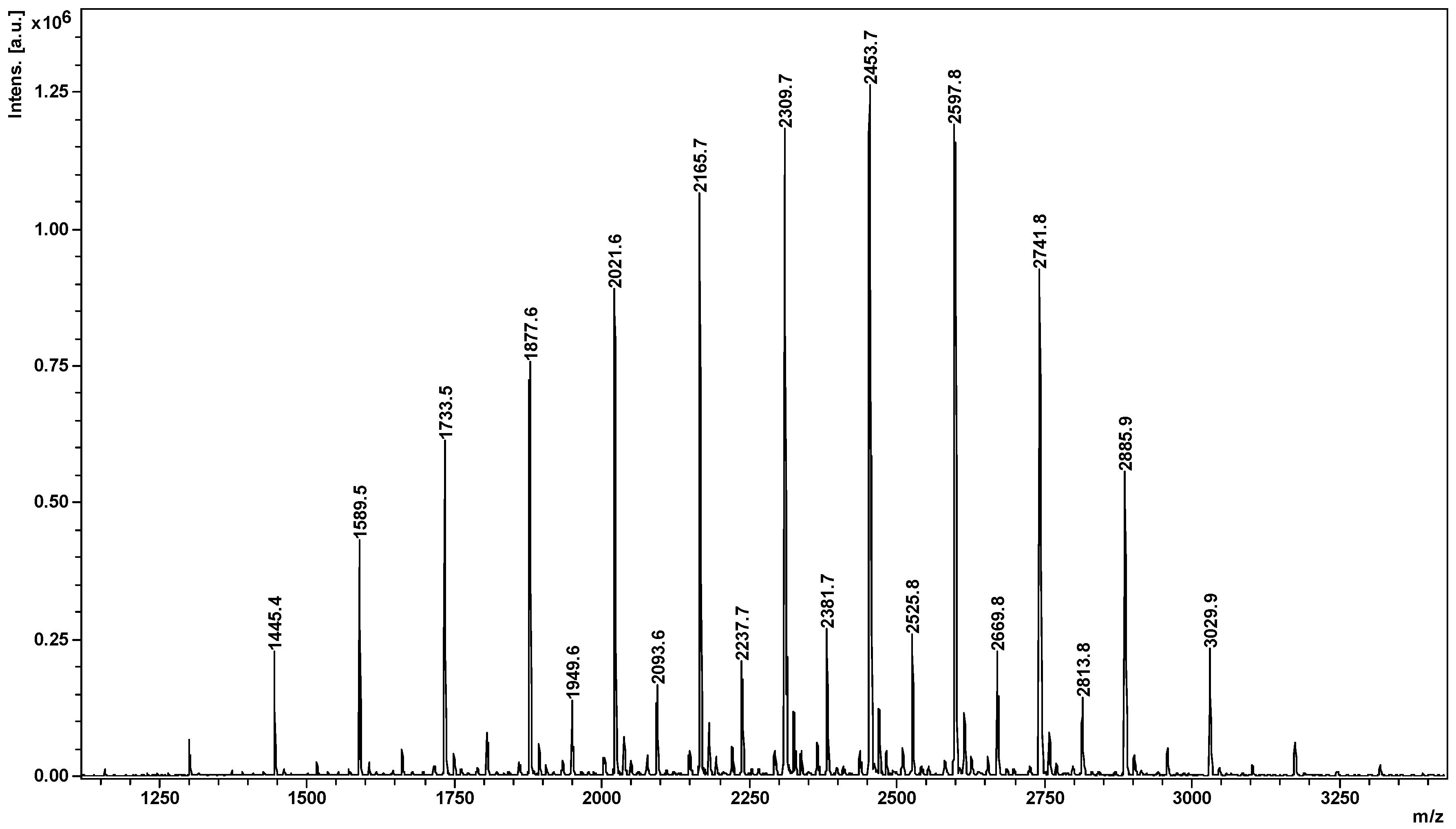
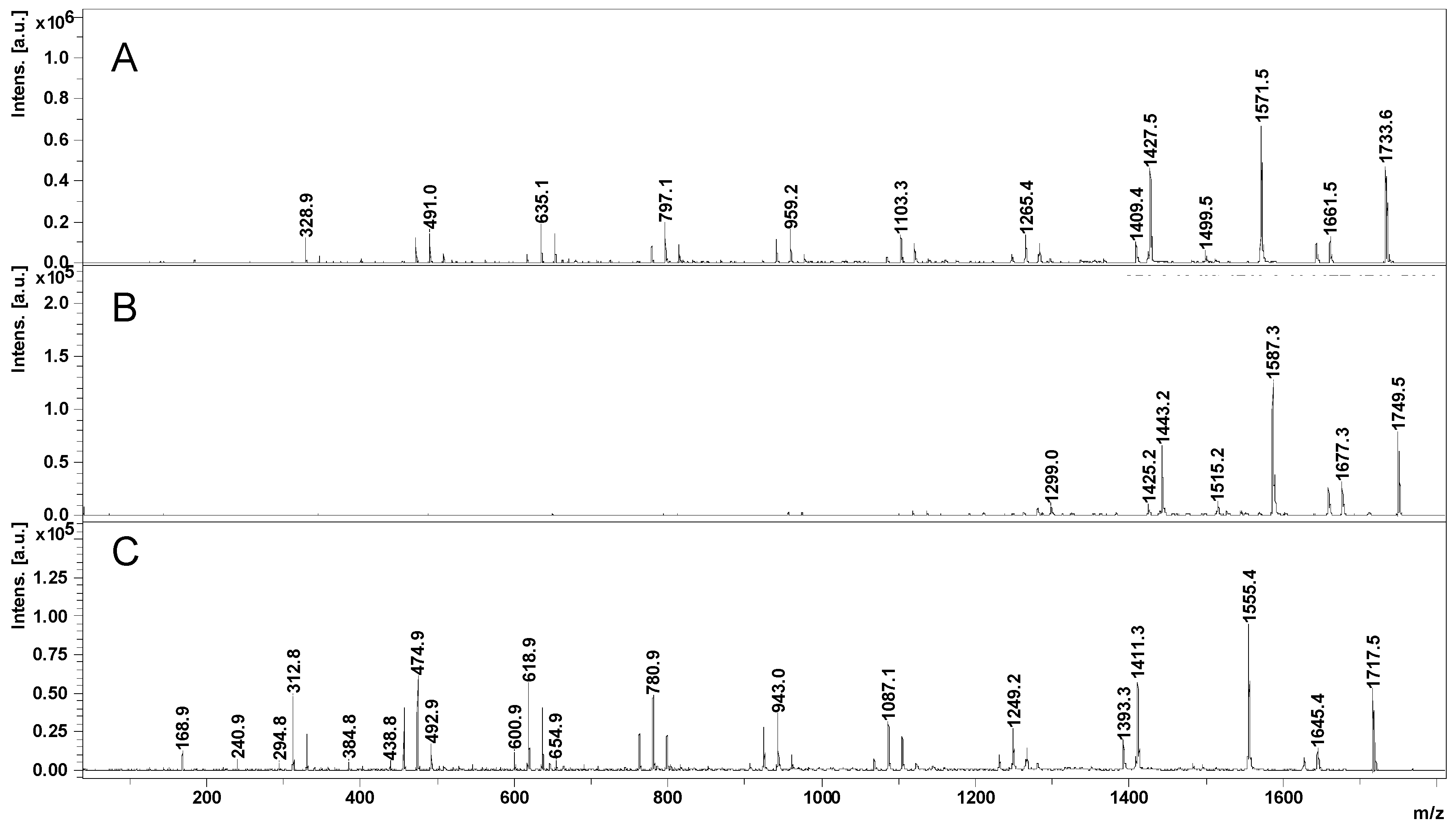
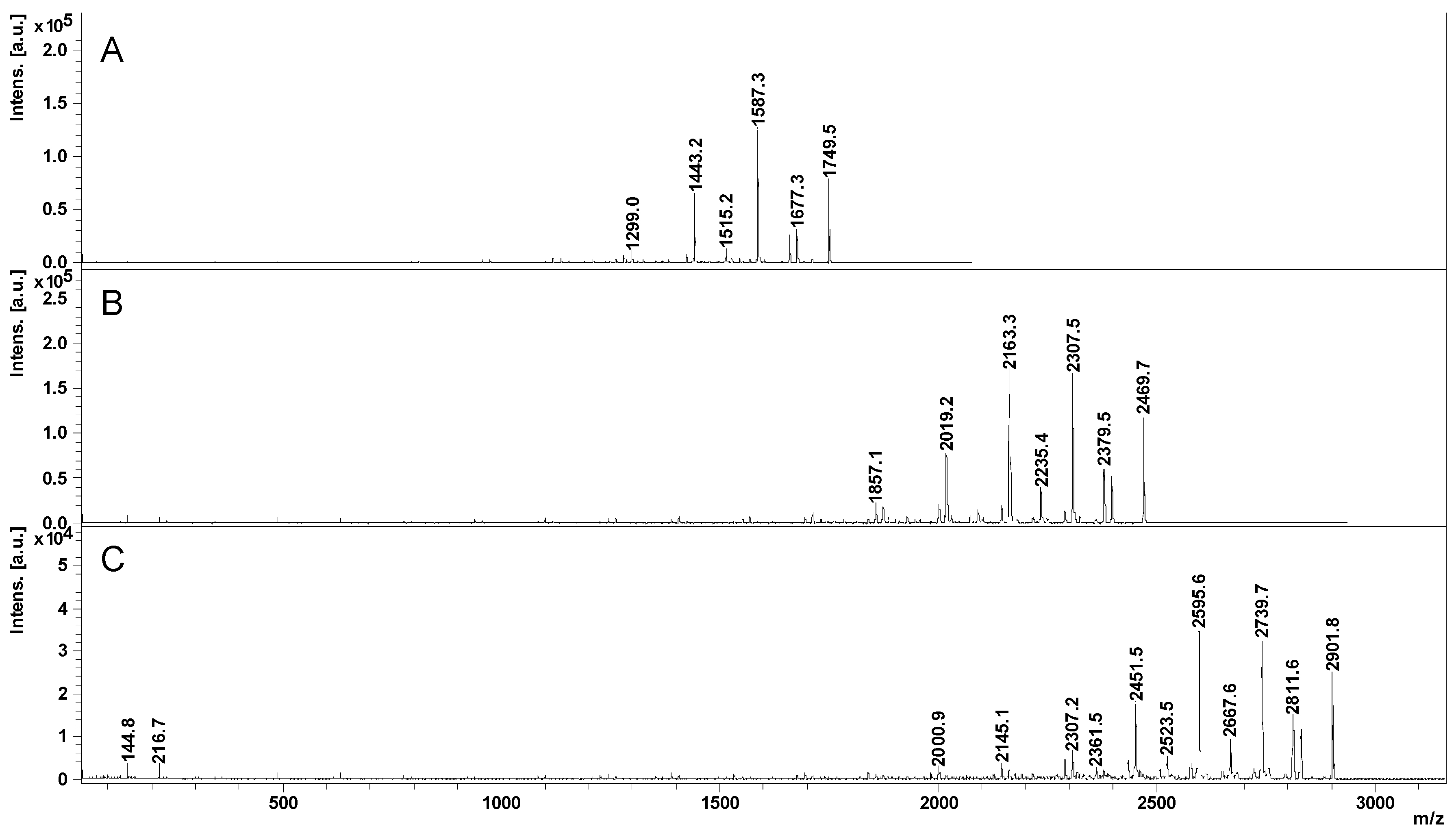
2.2. Mass Spectrometry (MS) Characterization
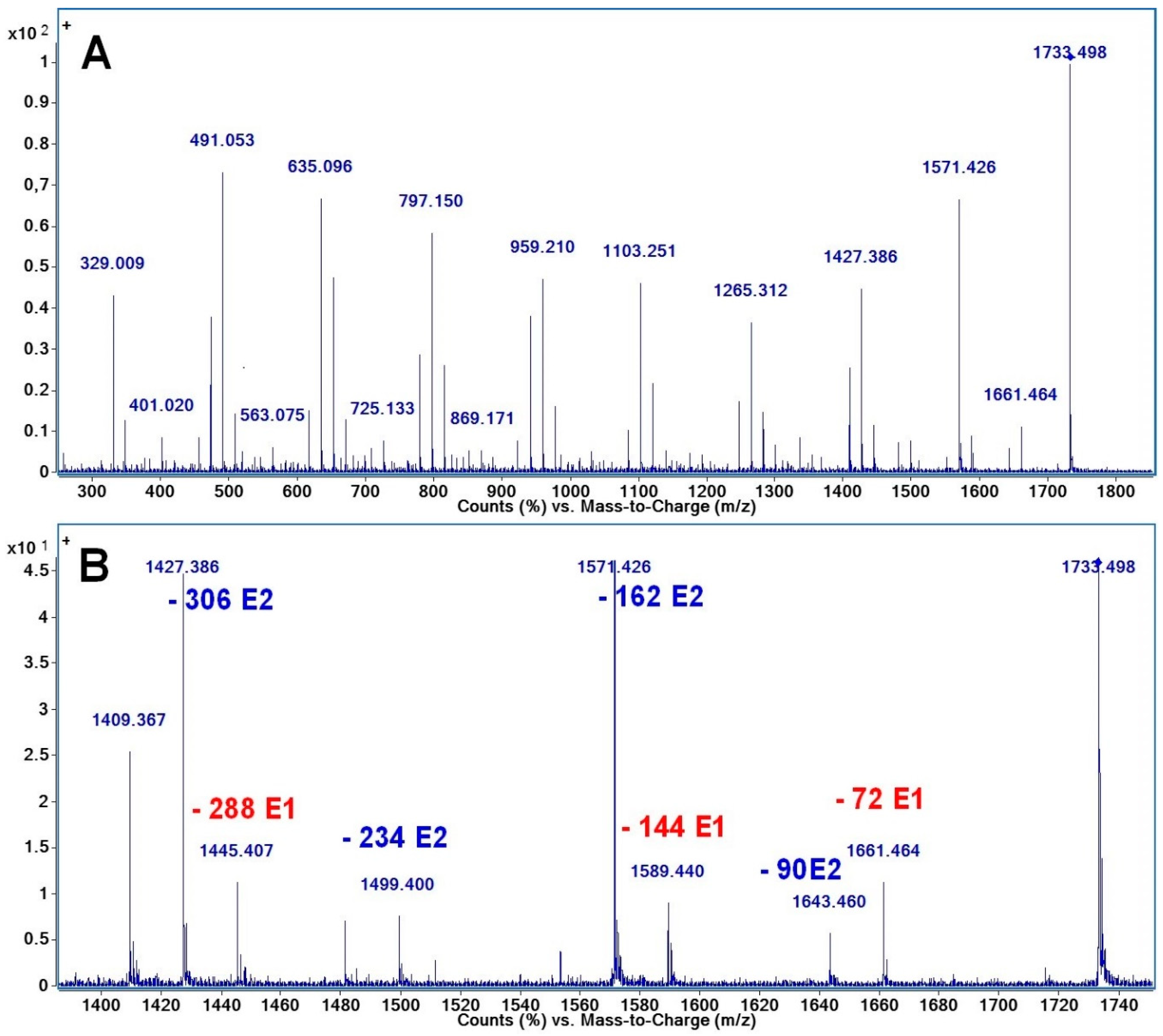
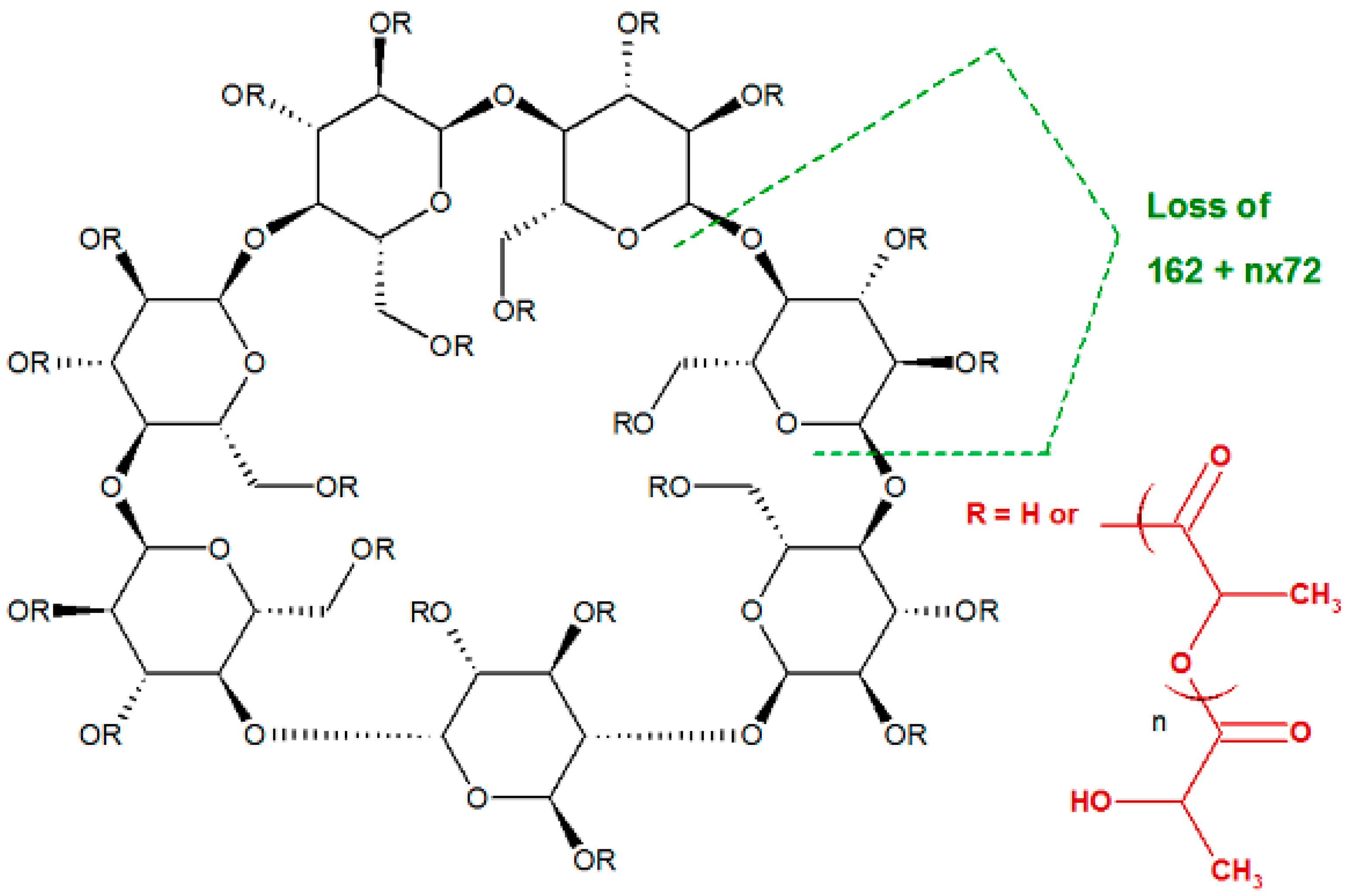
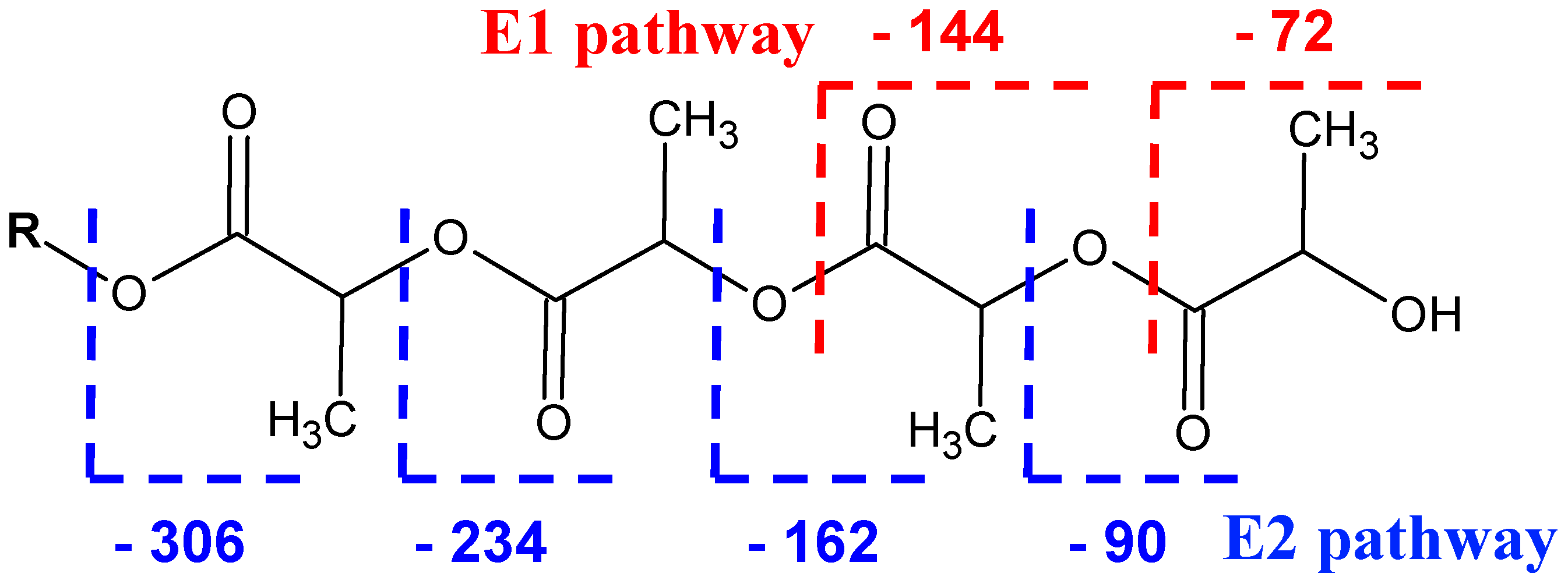
2.3. Fragmentation Studies by Collision-Induced Dissociation (CID) MS/MS on Electrospray (ESI) Q-TOF Instrument
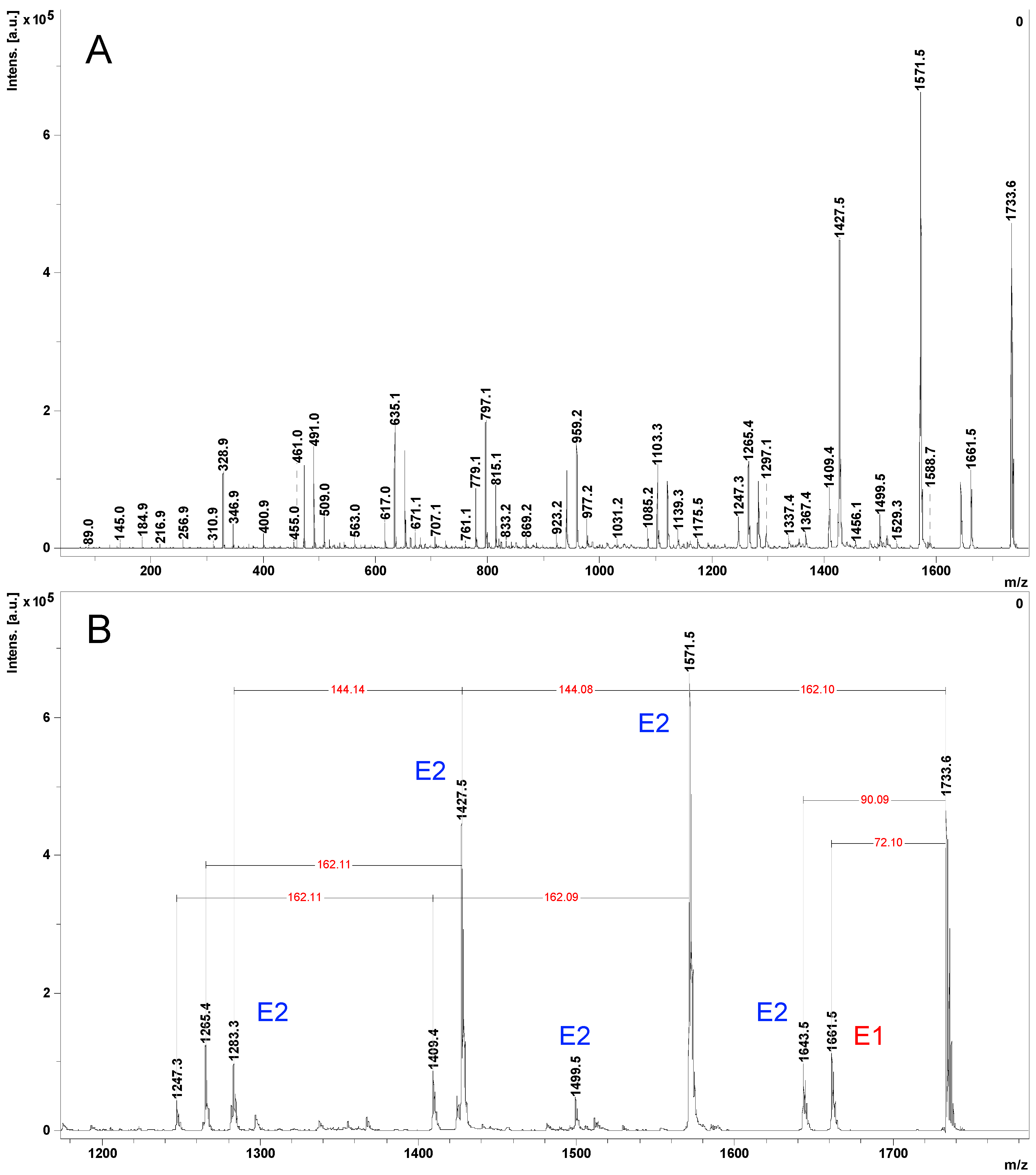
2.4. Fragmentation by MALDI Laser-Induced Dissociation (LID)
3. Materials and Methods
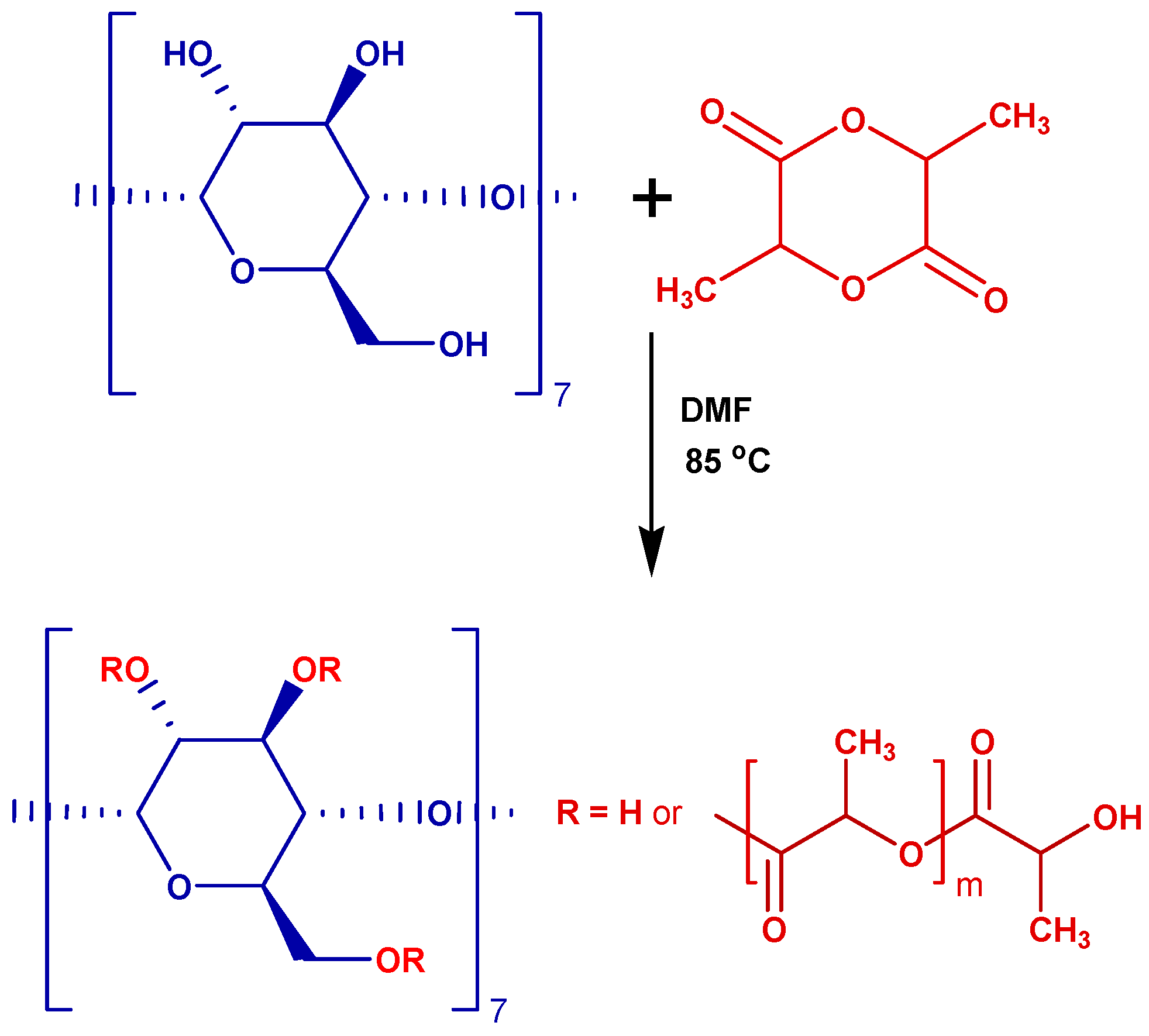
3.1. Chemicals
3.2. Solution Polymerization of L-LA in DMF
3.3. Matrix-Assisted Laser Desorption Ionization (MALDI MS)
3.4. Electrospray Ionization Mass Spectrometry (ESI MS)
3.5. Nuclear Magnetic Resonance (NMR)
3.6. Gel Permeation Chromatography (GPC)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Loftsson, T.; Duchêne, D. Cyclodextrins and their pharmaceutical applications. Int. J. Pharm. 2007, 329, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Szejtli, J. Introduction and General Overview of Cyclodextrin Chemistry. Chem. Rev. 1998, 98, 1743–1753. [Google Scholar] [CrossRef] [PubMed]
- Wenz, G. Cyclodextrins as Building Blocks for Supramolecular Structures and Functional Units. Angew. Chem. Int. Ed. Engl. 1994, 33, 803–822. [Google Scholar] [CrossRef]
- Yhaya, F.; Gregory, A.M.; Stenzel, M.H. Polymers with Sugar Buckets—The Attachment of Cyclodextrins onto Polymer Chains. Aust. J. Chem. 2010, 63, 195–210. [Google Scholar] [CrossRef]
- Van de Manakker, F.; Vermonden, T.; van Nostrum, C.F.; Hennink, W.E. Cyclodextrin-Based Polymeric Materials: Synthesis, Properties, and Pharmaceutical/Biomedical Applications. Biomacromolecules 2009, 10, 3157–3175. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Zinck, P. Ring-opening polymerization of cyclic esters initiated by cyclodextrins. Polym. Chem. 2012, 3, 1119–1122. [Google Scholar] [CrossRef]
- Huin, C.; Eskandani, Z.; Badi, N.; Farcas, A.; Bennevault-Celton, V.; Guégan, P. Anionic ring-opening polymerization of ethylene oxide in DMF with cyclodextrin derivatives as new initiators. Carbohydr. Polym. 2013, 94, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Adeli, M.; Kalantari, M.; Zarnega, Z.; Kabiri, R. Cyclodextrin-based dendritic supramolecules; new multivalent nanocarriers. RSC Adv. 2012, 2, 2756–2758. [Google Scholar] [CrossRef]
- Gou, P.-F.; Zhu, W.-P.; Xu, N.; Shen, Z.-Q. Synthesis and characterization of well-defined cyclodextrin-centered seven-arm star poly(ε-caprolactone)s and amphiphilic star poly(ε-caprolactone-b-ethylene glycol). J. Polym. Sci. Part A Polym. Chem. 2008, 46, 6455–6465. [Google Scholar] [CrossRef]
- Adeli, M.; Zarnegar, Z.; Kabiri, R. Amphiphilic star copolymer containing cyclodextrin core and their application as nanocarrier. Eur. Polym. J. 2008, 44, 1921–1930. [Google Scholar] [CrossRef]
- Mooguee, M.; Omidi, Y.; Davaran, S. Synthesis and in vitro release of adriamycin from star-shaped poly(lactide-co-glycolide) nano- and microparticles. J. Pharm. Sci. 2010, 99, 3389–3397. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Rousseau, C.; Mortreux, A.; Martin, P.; Zinck, P. Access to new carbohydrate-functionalized polylactides via organocatalyzed ring-opening polymerization. Polymer 2011, 52, 5018–5026. [Google Scholar] [CrossRef]
- Normand, M.; Kirillov, E.; Carpentier, J.-F.; Guillaume, S.M. Cyclodextrin-Centered Polyesters: Controlled Ring-Opening Polymerization of Cyclic Esters from β-Cyclodextrin-Diol. Macromolecules 2012, 45, 1122–1130. [Google Scholar] [CrossRef]
- Xu, Z.; Liu, S.; Liu, H.; Yang, C.; Kang, Y.; Wang, M. Unimolecular micelles of amphiphilic cyclodextrin-core star-like block copolymers for anticancer drug delivery. Chem. Commun. 2015, 51, 15768–15771. [Google Scholar] [CrossRef] [PubMed]
- Peptu, C.; Nicolescu, A.; Peptu, C.A.; Harabagiu, V.; Simionescu, B.C.; Kowalczuk, M. Mass spectrometry characterization of 3-OH butyrated β-cyclodextrin. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 5581–5592. [Google Scholar] [CrossRef]
- Takashima, Y.; Osaki, M.; Harada, A. Cyclodextrin-Initiated Polymerization of Cyclic Esters in Bulk: Formation of Polyester-Tethered Cyclodextrins. J. Am. Chem. Soc. 2004, 126, 13588–13589. [Google Scholar] [CrossRef] [PubMed]
- Osaki, M.; Takashima, Y.; Yamaguchi, H.; Harada, A. Polymerization of Lactones and Lactides Initiated by Cyclodextrins. Kobunshi Ronbunshu 2007, 64, 607–616. [Google Scholar] [CrossRef]
- Peptu, C.; Balan-Porcarasu, M.; Šišková, A.; Škultéty, L.; Mosnáček, J. Cyclodextrins tethered with oligolactides—Green synthesis and structural assessment. Beilstein J. Org. Chem. 2017, 13, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Hao, A.; Du, G.; Zhang, H.; Sun, H. A convenient preparation of 6-oligo(lactic acid)cyclomaltoheptaose as kinetically degradable derivative for controlled release of amoxicillin. Carbohydr. Res. 2008, 343, 2517–2522. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.A.; Simonsick, W.J. Application of mass spectrometry to the characterization of polymers. Curr. Opin. Solid State Mater. Sci. 1997, 2, 661–667. [Google Scholar] [CrossRef]
- Jacquet, R.; Favetta, P.; Elfakir, C.; Lafosse, M. Characterization of a new methylated beta-cyclodextrin with a low degree of substitution by matrix-assisted laser desorption/ionization mass spectrometry and liquid chromatography using evaporative light scattering detection. J. Chromatogr. A 2005, 1083, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Kieken, F.; West, C.; Keddadouche, K.; Elfakir, C.; Choisnard, L.; Gèze, A.; Wouessidjewe, D. Characterisation of complex amphiphilic cyclodextrin mixtures by high-performance liquid chromatography and mass spectrometry. J. Chromatogr. A 2008, 1189, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, R.; Elfakir, C.; Lafosse, M. Characterization of a new methylated Beta-cyclodextrin with a low degree of substitution by electrospray ionization mass spectrometry and liquid chromatography/mass spectrometry. Rapid Commun. Mass Spectrom. 2005, 19, 3097–3102. [Google Scholar] [CrossRef] [PubMed]
- Peptu, C.; Kwiecien, I.; Harabagiu, V. Modification of β-cyclodextrin through solution ring-opening oligomerization of β-butyrolactone. Cell. Chem. Technol. 2014, 48, 1–10. [Google Scholar]
- Peptu, C.; Harabagiu, V. Tandem mass spectrometry characterization of esterified cyclodextrins. Dig. J. Nanomater. Biostruct. 2013, 8, 1551–1561. [Google Scholar]
- Koster, S.; Duursma, M.C.; Boon, J.J.; Nielen, M.W.; de Koster, C.G.; Heeren, R.M. Structural analysis of synthetic homo- and copolyesters by electrospray ionization on a fourier transform ion cyclotron resonance mass spectrometer. J. Mass Spectrom. 2000, 35, 739–748. [Google Scholar] [CrossRef]
- Wesdemiotis, C.; Solak, N.; Polce, M.J.; Dabney, D.E.; Chaicharoen, K.; Katzenmeyer, B.C. Fragmentation pathways of polymer ions. Mass Spectrom. Rev. 2011, 30, 523–559. [Google Scholar] [CrossRef] [PubMed]
- De Winter, J.; Lemaur, V.; Marsal, P.; Coulembier, O.; Cornil, J.; Dubois, P.; Gerbaux, P. Mechanistic study of the collision-induced dissociation of sodium-cationized polylactide oligomers: A joint experimental and theoretical investigation. J. Am. Soc. Mass Spectrom. 2010, 21, 1159–1168. [Google Scholar] [CrossRef] [PubMed]
- Peptu, C.; van den Brink, O.F.; Harabagiu, V.; Simionescu, B.C.; Kowalczuk, M.; Silberring, J. Molecular level differentiation between end-capped and intramolecular azofunctional oligo(ε-caprolactone) positional isomers through liquid chromatography multistage mass spectrometry. J. Polym. Sci. A Polym. Chem. 2012, 50, 2421–2431. [Google Scholar] [CrossRef]
- Sforza, S.; Galaverna, G.; Corradini, R.; Dossena, A.; Marchelli, R. ESI-Mass Spectrometry Analysis of Unsubstituted and Disubstituted-Cyclodextrins: Fragmentation Mode and Identification of the AB, AC, AD Regioisomers. J. Am. Soc. Mass Spectrom. 2003, 14, 124–135. [Google Scholar] [CrossRef]
- Macht, M.; Asperger, A.; Deininger, S. Comparison of laser-induced dissociation and high-energy collision-induced dissociation using matrix-assisted laser desorption/ionization tandem time-of-flight (MALDI-TOF/TOF) for peptide and protein identification. Rapid Commun. Mass Spectrom. 2004, 18, 2093–2105. [Google Scholar] [CrossRef] [PubMed]
- Frański, R.; Gierczyk, B.; Schroeder, G.; Beck, S.; Springer, A.; Linscheid, M. Mass spectrometric decompositions of cationized β-cyclodextrin. Carbohydr. Res. 2005, 340, 1567–1572. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| G */la ** | G1 | G2 | G3 | G4 | G5 | G6 | G7 |
|---|---|---|---|---|---|---|---|
| la0 | 185 | 347 | 509 | 671 | 833 | 995 | |
| la1 | 257 | 419 | 581 | 743 | 905 | 1067 | |
| la2 | 329 | 491 | 653 | 815 | 977 | 1139 | |
| la3 | 401 | 563 | 725 | 887 | 1049 | 1211 | |
| la4 | 473 | 635 | 797 | 959 | 1121 | 1283 | |
| la5 | 545 | 707 | 869 | 1031 | 1193 | 1355 | |
| la6 | 617 | 779 | 941 | 1103 | 1265 | 1427 | |
| la7 | 689 | 851 | 1013 | 1175 | 1337 | 1499 | |
| la8 | 761 | 923 | 1085 | 1247 | 1409 | 1571 | 1733 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peptu, C.; Danchenko, M.; Škultéty, Ľ.; Mosnáček, J. Structural Architectural Features of Cyclodextrin Oligoesters Revealed by Fragmentation Mass Spectrometry Analysis. Molecules 2018, 23, 2259. https://doi.org/10.3390/molecules23092259
Peptu C, Danchenko M, Škultéty Ľ, Mosnáček J. Structural Architectural Features of Cyclodextrin Oligoesters Revealed by Fragmentation Mass Spectrometry Analysis. Molecules. 2018; 23(9):2259. https://doi.org/10.3390/molecules23092259
Chicago/Turabian StylePeptu, Cristian, Maksym Danchenko, Ľudovít Škultéty, and Jaroslav Mosnáček. 2018. "Structural Architectural Features of Cyclodextrin Oligoesters Revealed by Fragmentation Mass Spectrometry Analysis" Molecules 23, no. 9: 2259. https://doi.org/10.3390/molecules23092259
APA StylePeptu, C., Danchenko, M., Škultéty, Ľ., & Mosnáček, J. (2018). Structural Architectural Features of Cyclodextrin Oligoesters Revealed by Fragmentation Mass Spectrometry Analysis. Molecules, 23(9), 2259. https://doi.org/10.3390/molecules23092259