
The Targeted Pesticides as Acetylcholinesterase Inhibitors: Comprehensive Cross-Organism Molecular Modelling Studies Performed to Anticipate the Pharmacology of Harmfulness to Humans In Vitro

 ,
, 
 , ,
, , 
Abstract

1. Introduction
2. Results and Discussion
2.1. The Conformational Analysis of Pesticides in Aqueous Solution
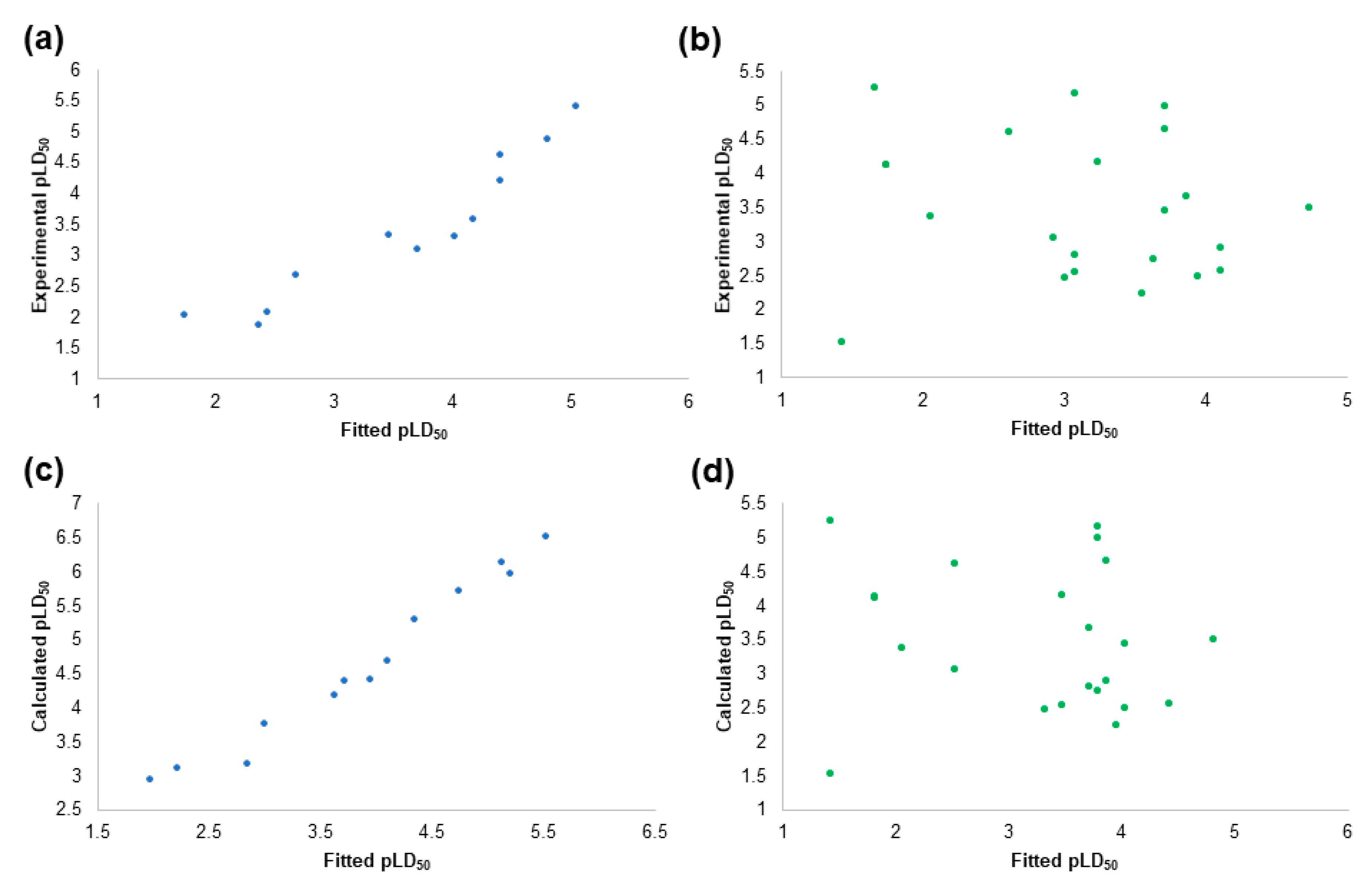
2.2. The Prediction of Pesticides Structures in Aqueous Solution
2.3. The Prediction of Externally Evaluated Pesticides Structures in Aqueous Solution
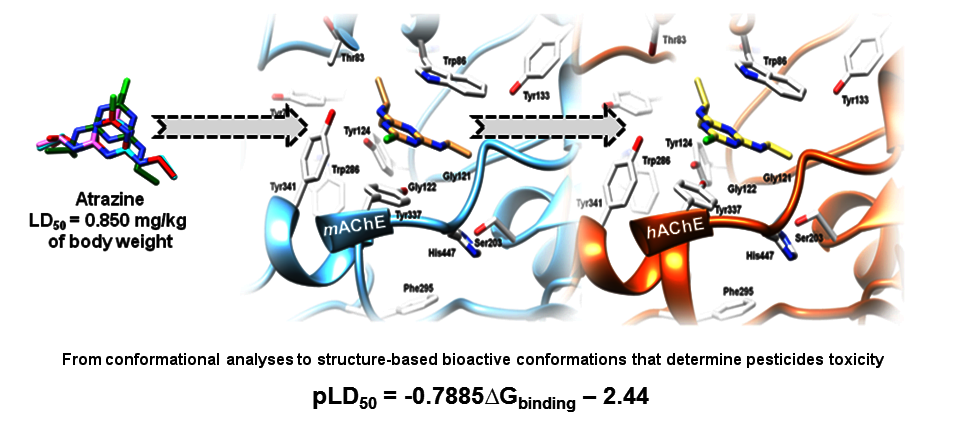
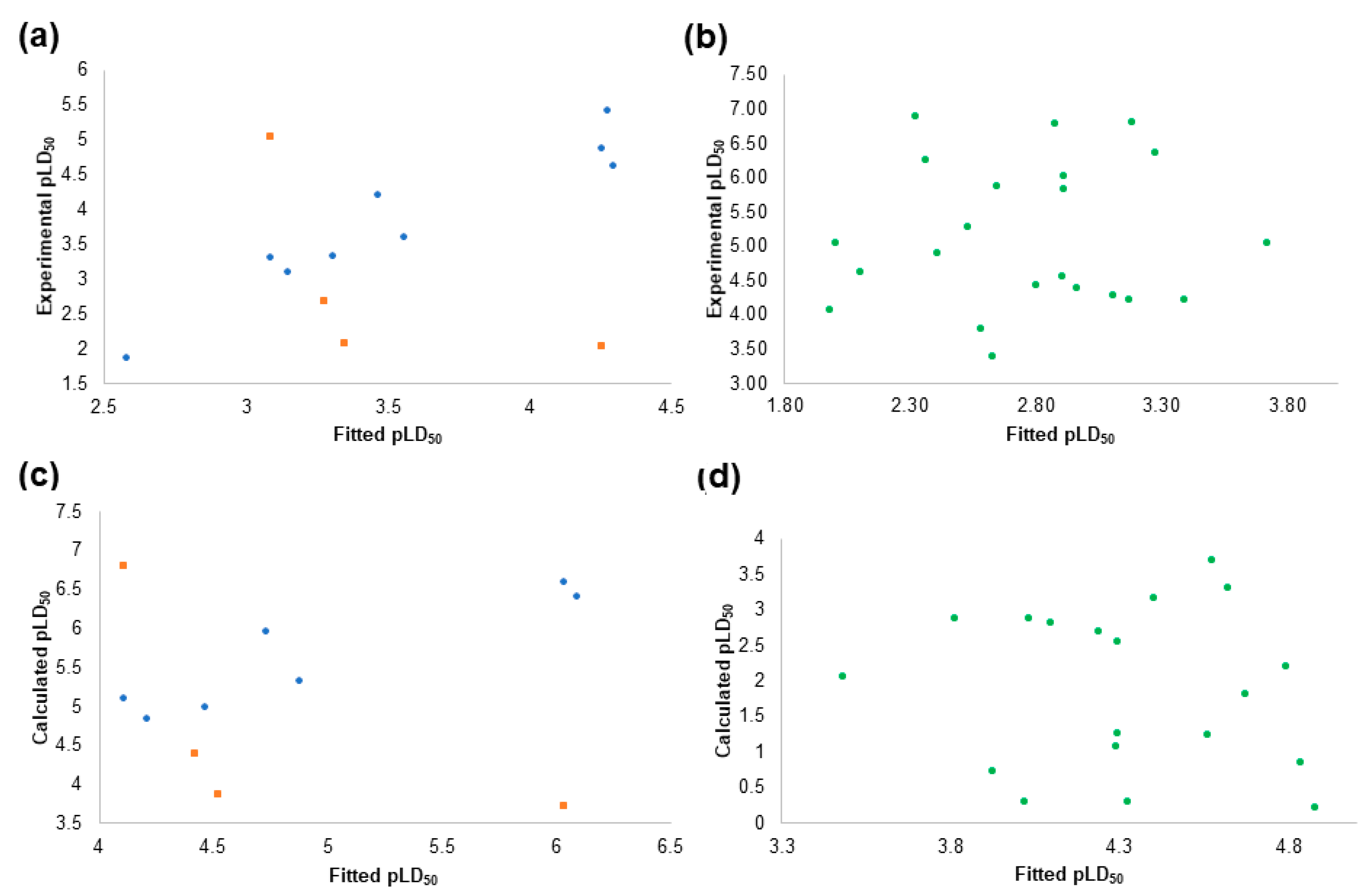
2.4. The Pesticides Acute Toxicity Anticipation through the Pre-Bound Conformations
2.5. The Structure-Based Studies
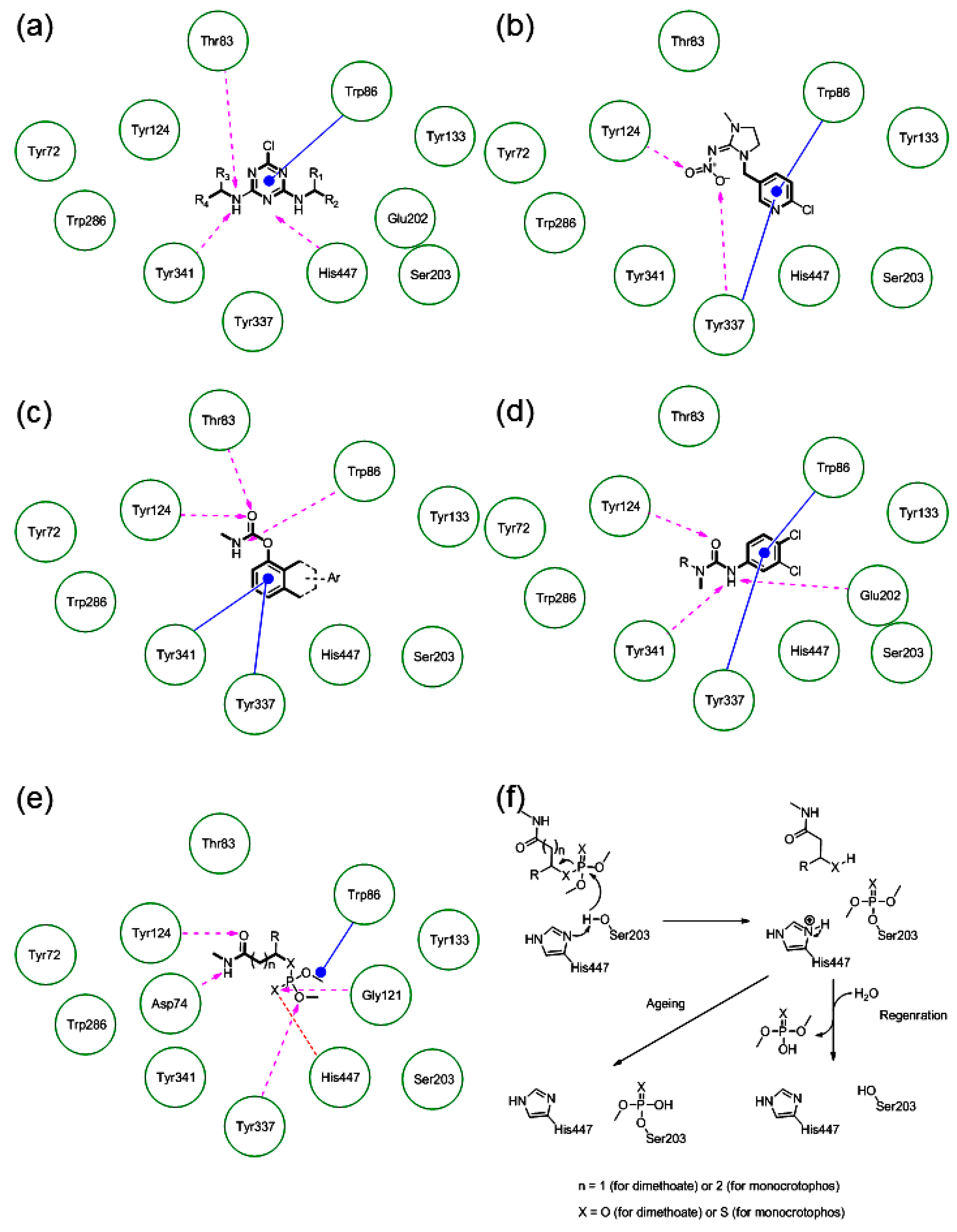
2.5.1. The Alignment Rules Assessment
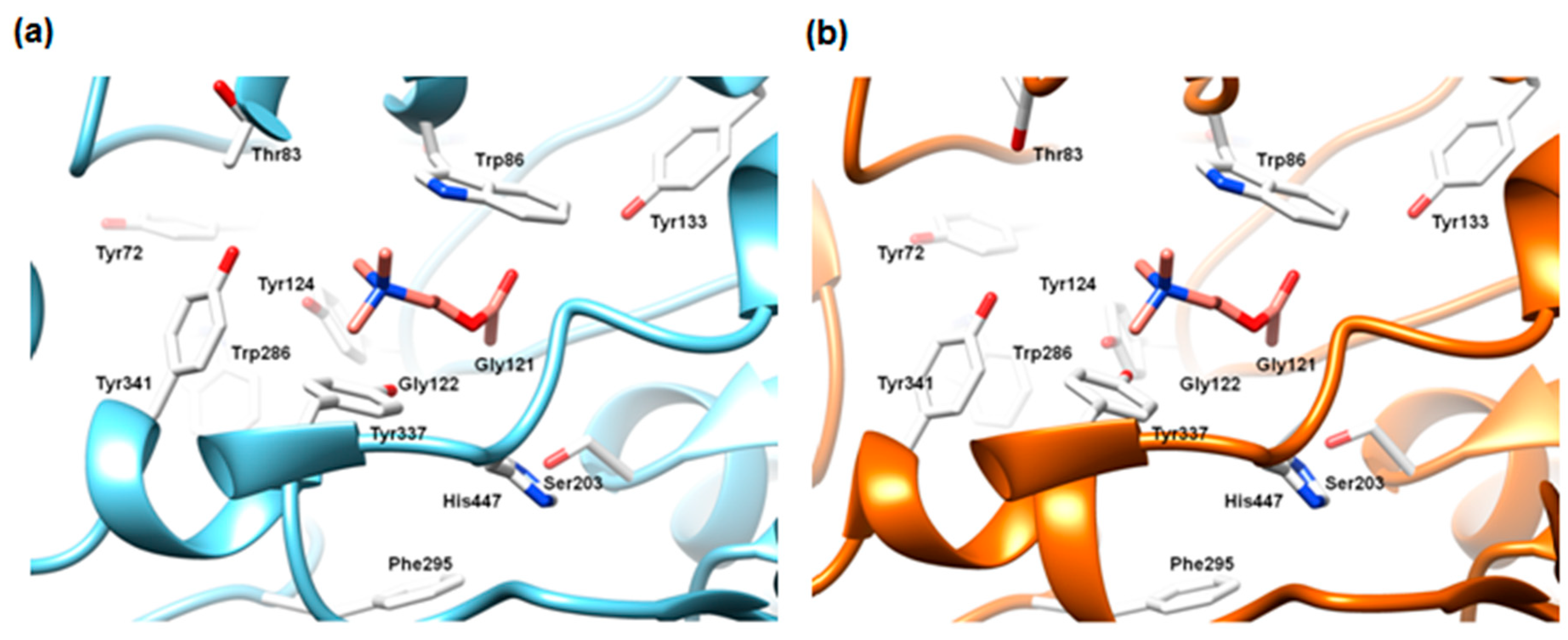
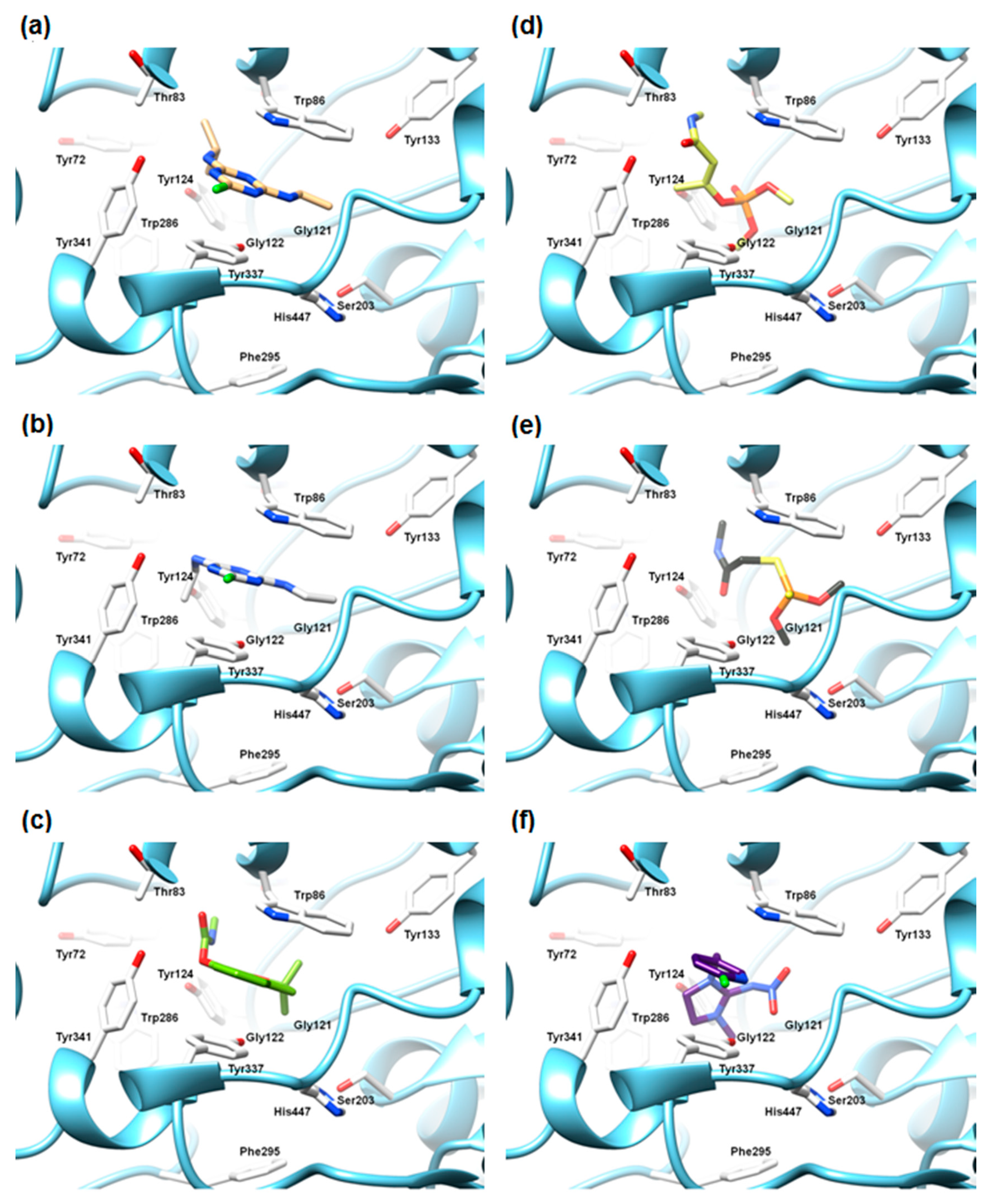
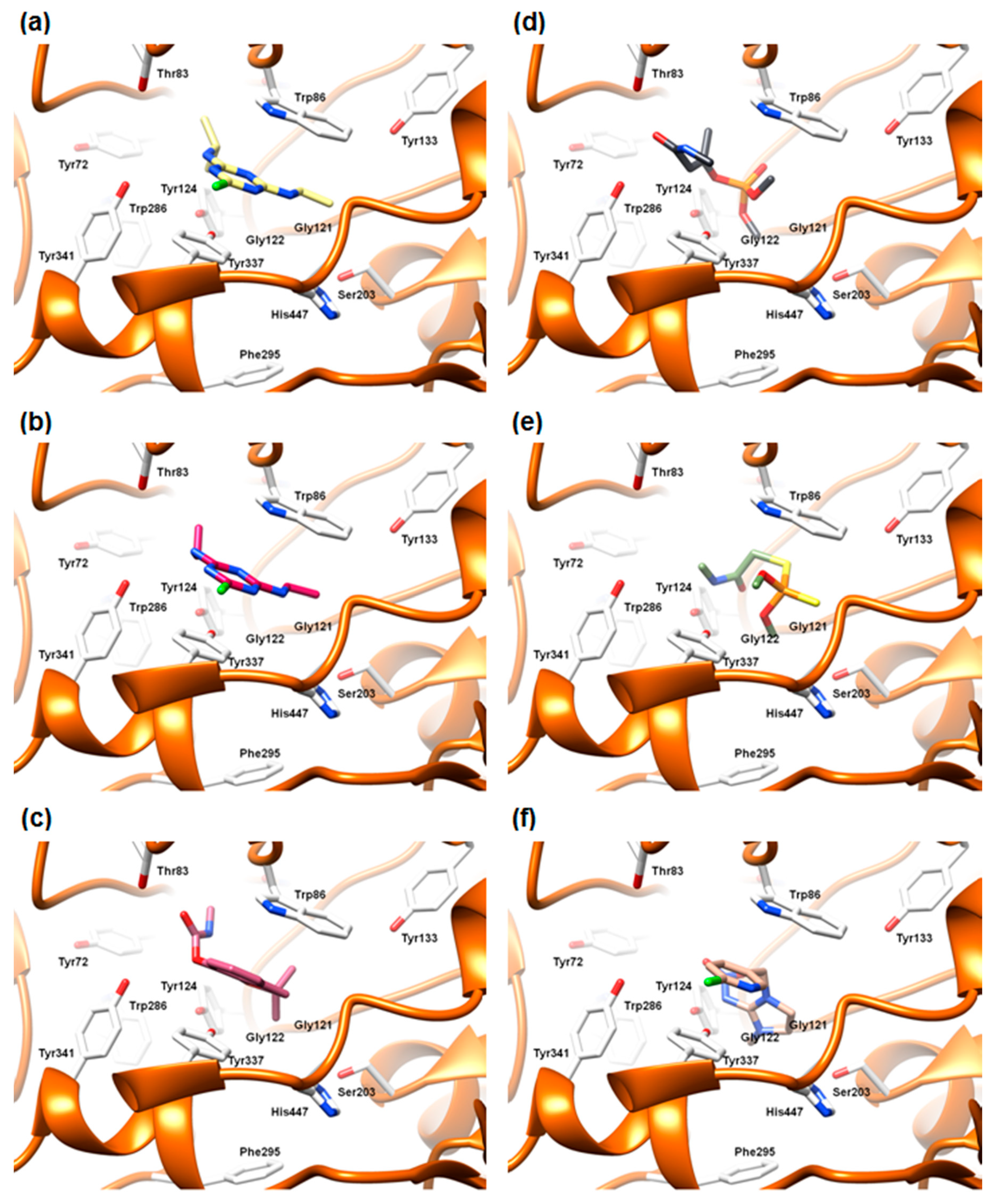
2.5.2. The Cross-Docking and Molecular Dynamics Studies of ACh
2.5.3. The Cross-Docking and Molecular Dynamics Studies of Pesticides 2-chloro-1,3,5-triazine Group
2.5.4. The Cross-Docking and Molecular Dynamics Studies of Pesticides Amide Group
2.5.5. The Cross-Docking and Molecular Dynamics Studies of Pesticides (6-Chloropyridin-3-Yl)Methanamine and 1-(4-Chlorophenyl)-3-methylurea Groups
2.5.6. The Cross-Docking of Externally Evaluated Pesticides
2.6. The Pesticides Acute Toxicity Anticipation through the Bioactive Conformations
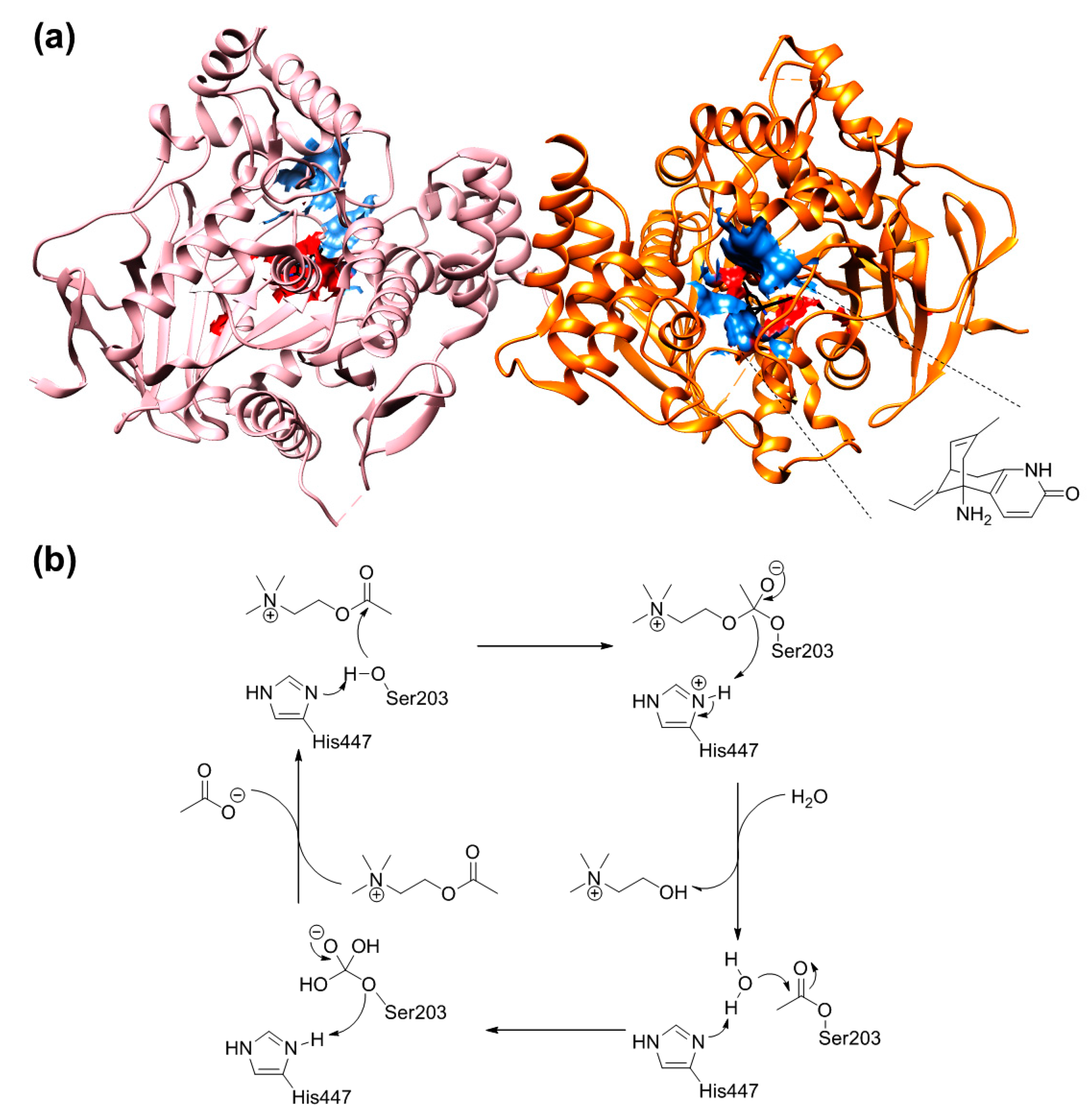
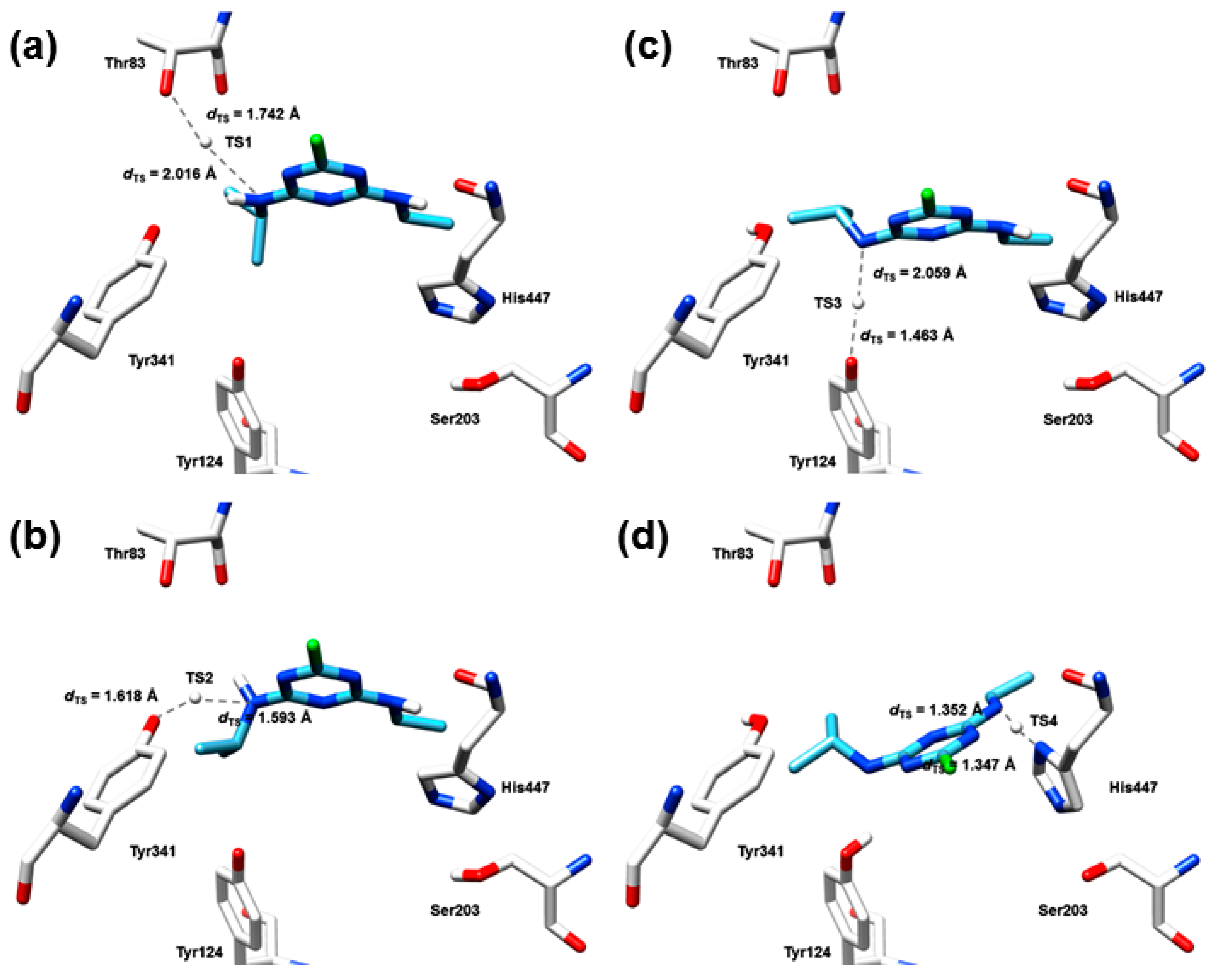
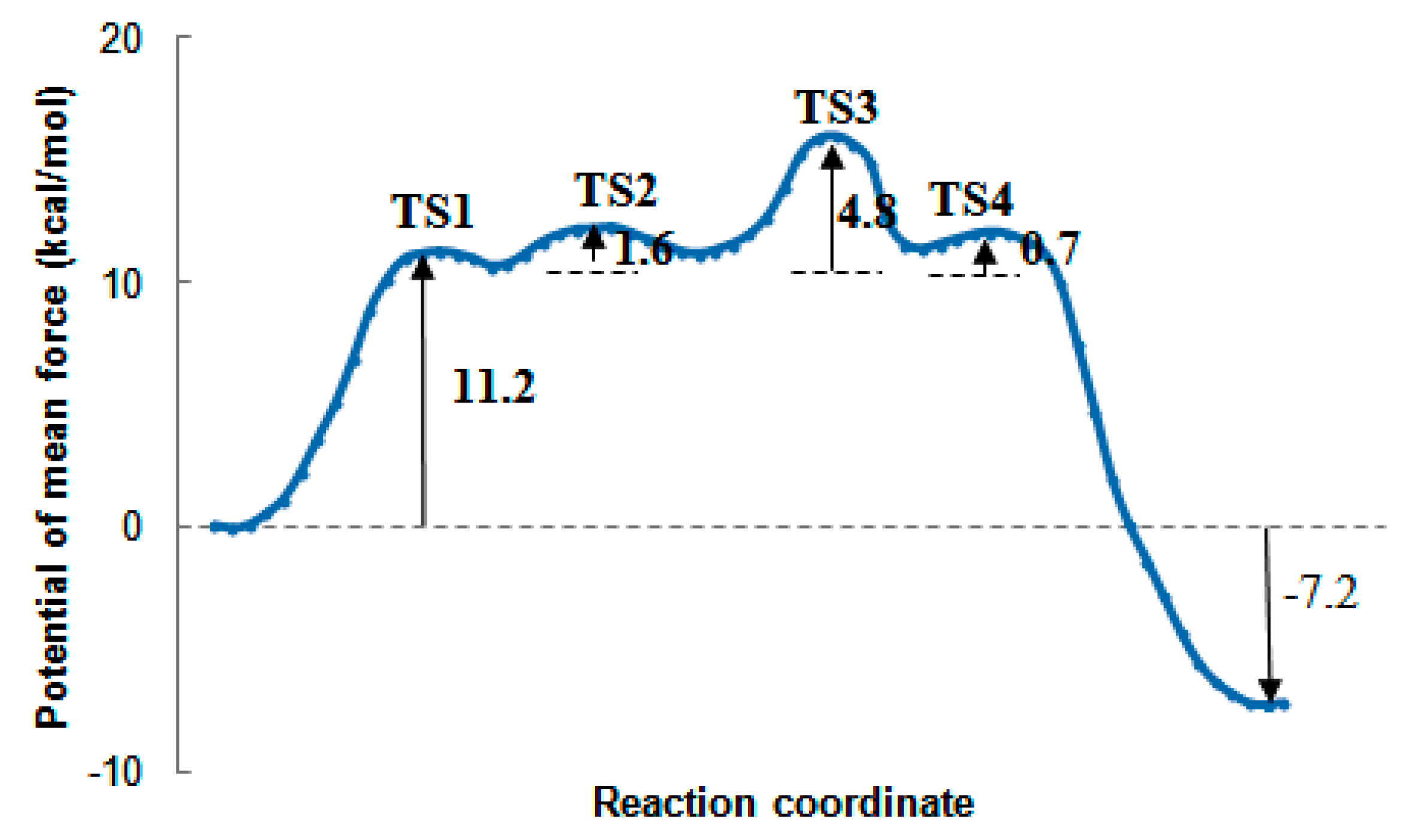
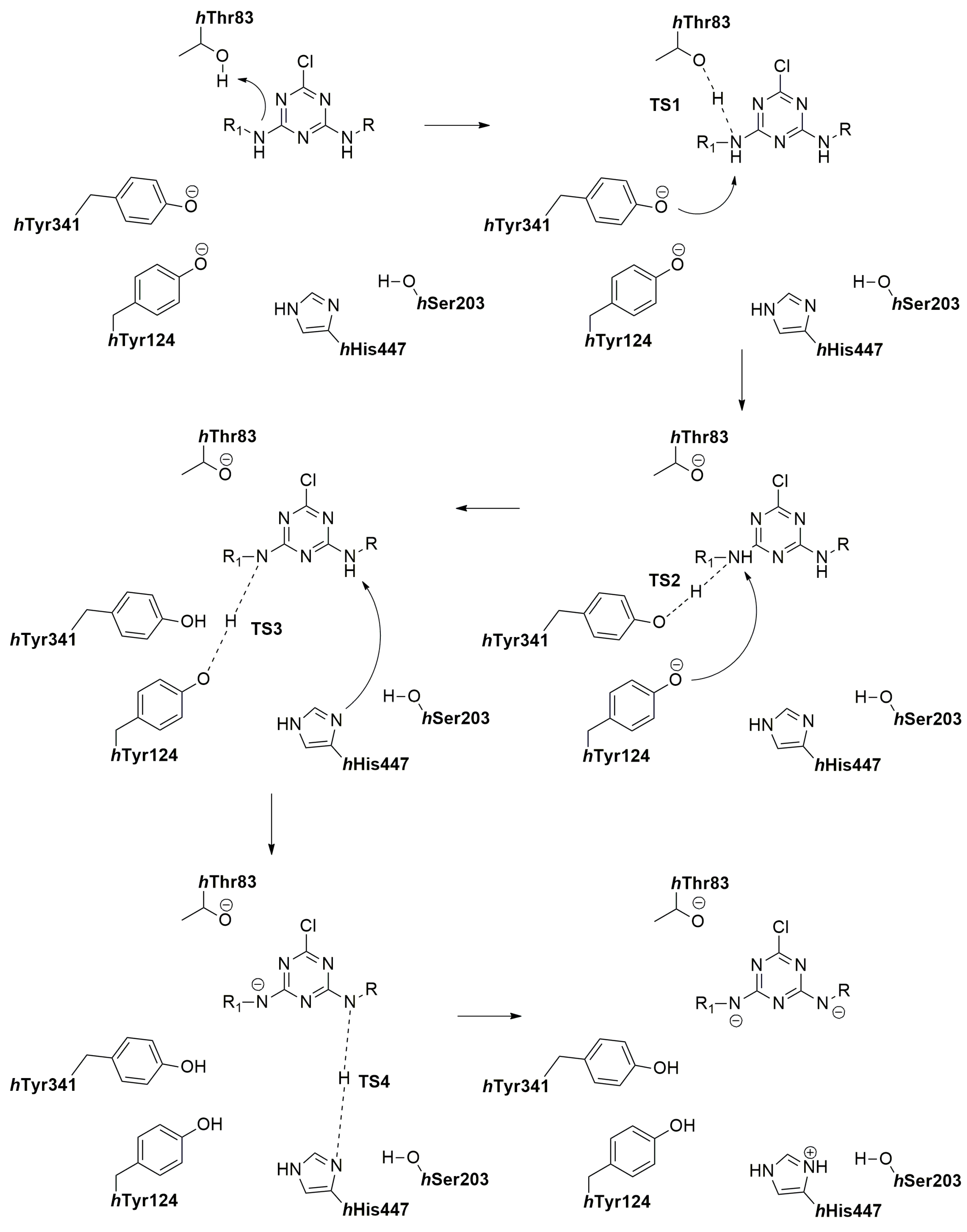
2.7. The Quantum Mechanical Studies of Acetylcholinesterase Inhibition by 2-Chloro-1,3,5-triazine-based Pesticides
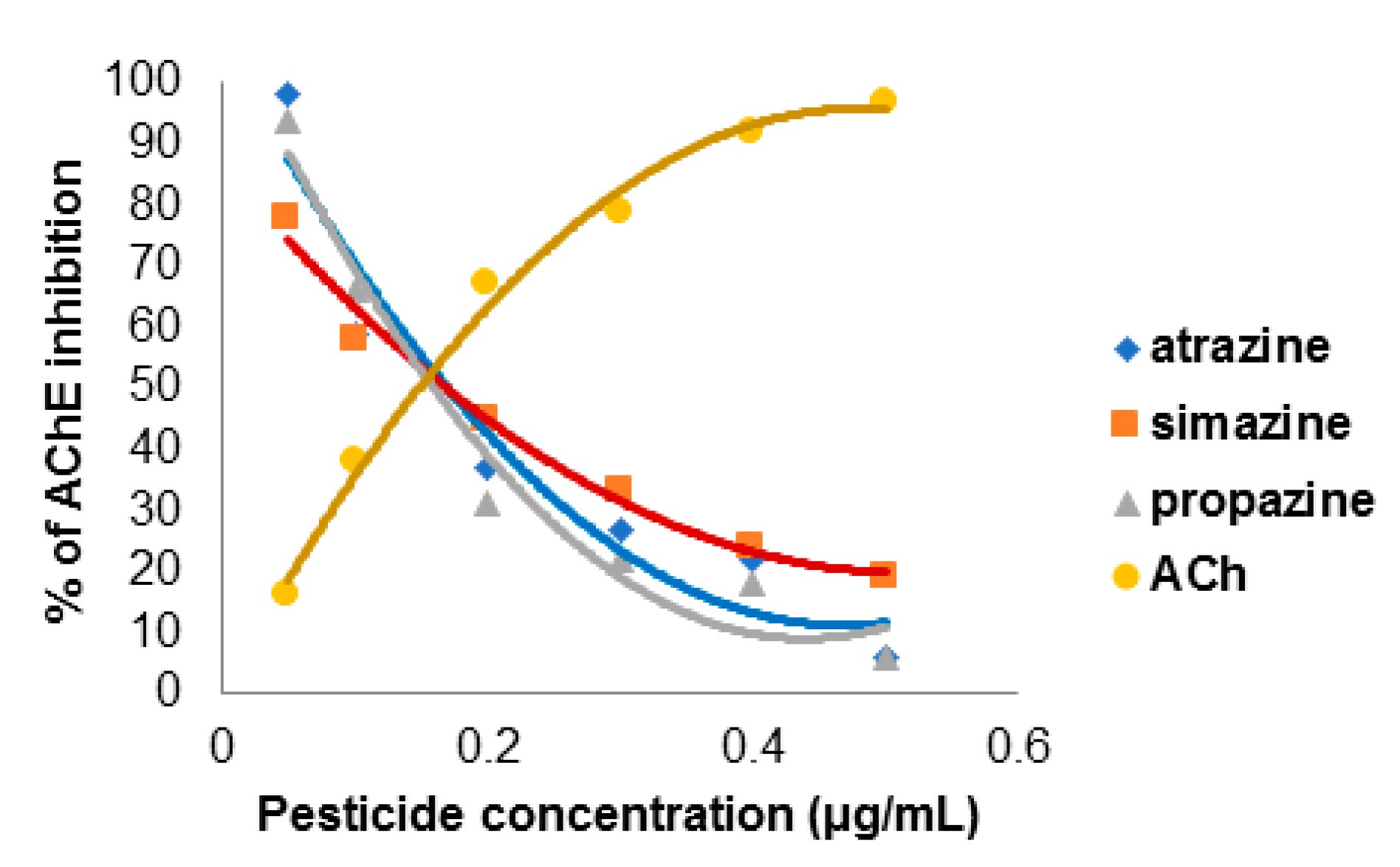
2.8. The Concentration-Dependent Kinetics of Human Acetylcholinesterase Inhibition by 2-Chloro-1,3,5-triazine-based Pesticides
3. Materials and Methods
3.1. Materials
3.2. The Generation of Pesticides Structures
3.3. The Conformational Analysis
3.4. The Mus musculus and Homo sapiens AChE Sequences Alignment
3.5. The Mus musculus and Homo sapiens AChE Complexes Preparation
3.6. The Structure-Based Alignment Assessment
3.6.1. The AutoDock Settings
3.6.2. The Autodock Vina Settings
3.6.3. The DOCK6 Settings
3.7. The Ligand-Based and Structure-Based Alignment Accuracy
3.8. The Structure-Based Alignment of Pesticides
3.9. The Pesticide-AChE Complexes Molecular Dynamics Simulations
3.10. The MM-GBSA Calculations and Free Energy Decomposition
3.11. The Quantum Mechanical Mechanistic Studies
3.12. The Assay of the AChE Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gilden, R.C.; Huffling, K.; Sattler, B. Pesticides and health risks. J. Obstet. Gynecol. Neonatal Nurs. 2010. [Google Scholar] [CrossRef] [PubMed]
- Arduini, F.; Ricci, F.; Tuta, C.S.; Moscone, D.; Amine, A.; Palleschi, G. Detection of carbamic and organophosphorous pesticides in water samples using a cholinesterase biosensor based on Prussian Blue-modified screen-printed electrode. Anal. Chim. Acta 2006. [Google Scholar] [CrossRef] [PubMed]
- U.S. Environmental Protection Agency, Washington, DC (1994) Pesticide Industry Sales and Usage, 1992 and 1993 Market Estimates. Available online: https://nepis.epa.gov/Exe/ZyPURL.cgi?Dockey=200001EL.TXT (accessed on 15 June 2018).
- Bolton, T.B.; Lim, S.P. Action of acetylcholine on smooth muscle. Z. Kardiol. 1991, 80 (Suppl. 7), 73–77. [Google Scholar] [PubMed]
- Jones, B.E. From waking to sleeping: Neuronal and chemical substrates. Trends Pharmacol. Sci. 2005, 26, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Chapalamadugu, S.; Chaudhry, G.R. Microbiological and biotechnological aspects of metabolism of carbamates and organophosphates. Crit. Rev. Biotechnol. 1992. [Google Scholar] [CrossRef] [PubMed]
- Đorđević, J.; Vladisavljević, G.T.; Trtić-Petrović, T. Liquid-phase membrane extraction of targeted pesticides from manufacturing wastewaters in a hollow fibre contactor with feed-stream recycle. Environ. Technol. 2017, 38, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Đorđević, J.S.; Vladisavljević, G.T.; Trtić-Petrović, T.M. Removal of the selected pesticides from a water solution by applying hollow fiber liquid–liquid membrane extraction. Ind. Eng. Chem. Res. 2014, 53, 4861–4870. [Google Scholar] [CrossRef]
- Brown, J.E.; Baughman, R.G. N-Ethyl-6-ethylamino-4-oxo-1,3,5-triazin-2-aminium chloride (Oxysimazine·HCl). Acta Crystallogr. Sect. E 2010. [Google Scholar] [CrossRef] [PubMed]
- Cheon, S.; Kim, T.H.; Park, K.-M.; Kim, J. Monocrotophos: Dimethyl (E)-1-methyl-2-(methylcarbamoyl)ethenyl phosphate. Acta Crystallogr. Sect. E 2011, 67, o584. [Google Scholar] [CrossRef] [PubMed]
- Lapp, R.L.; Jacobson, R.A. Crystal and molecular structures of organophosphorus insecticides. 13. S-isopropyl O-methyl O-(3,5,6-trichloro-2-pyridyl) phosphoramidothioate and dimethoate. J. Agric. Food Chem. 1980, 28, 755–759. [Google Scholar] [CrossRef]
- Chopra, D.; Mohan, T.P.; Rao, K.S.; Guru Row, T.N. IUCr (E)-N1-[(6-Chloropyridin-3-yl)methyl]-N2-cyano-N1-methylacetamidine. Acta Crystallogr. Sect. E 2004, 60, o2374–o2375. [Google Scholar] [CrossRef]
- Boudina, A.; Emmelin, C.; Baaliouamer, A.; Grenier-Loustalot, M.F.T.; Chovelon, J.M. Photochemical behaviour of carbendazim in aqueous solution. Chemosphere 2003. [Google Scholar] [CrossRef]
- World Health Organization. The WHO Recommended Classification of Pesticides by Hazard and Guidelines to Classification 2009; World Health Organization: Geneva, Switzerland, 2010; pp. 1–60. [Google Scholar]
- Bravo, R.; Caltabiano, L.M.; Weerasekera, G.; Whitehead, R.D.; Fernandez, C.; Needham, L.L.; Bradman, A.; Barr, D.B. Measurement of dialkyl phosphate metabolites of organophosphorus pesticides in human urine using lyophilization with gas chromatography-tandem mass spectrometry and isotope dilution quantification. J. Expo. Anal. Environ. Epidemiol. 2004. [Google Scholar] [CrossRef] [PubMed]
- Catalá-Icardo, M.; López-Paz, J.L.; Choves-Barón, C.; Peña-Bádena, A. Native vs. photoinduced chemiluminescence in dimethoate determination. Anal. Chim. Acta 2012. [Google Scholar] [CrossRef] [PubMed]
- Park, C. A Dictionary of Environment and Conservation, 1st ed.; Oxford University Press: Oxford, UK, 2007. [Google Scholar]
- Elbert, A.; Haas, M.; Springer, B.; Thielert, W.; Nauen, R. Applied aspects of neonicotinoid uses in crop protection. Pest Manag. Sci. 2008, 64, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- USEPA. Reregistration Eligibility Decision (RED), Linuron. 1995. Available online: https://archive.epa.gov/pesticides/reregistration/web/pdf/0047.pdf (accessed on 15 June 2018).
- Gallo, M.A.; Lawryk, N.J. Organic phosphorus pesticides. In Handbook of Pesticide Toxicology, Hayes, W.J., Laws, E.R.J., Eds.; Academic Press: San Diego, CA, USA, 1991. [Google Scholar]
- U.S. Environmental Protection Agency. Propazine: Health Advisory. Office of Drinking Water, US EPA, Washington, DC, USA. 1988. Available online: https://www.epa.gov (accessed on 16 March 2016).
- Extension Toxicology Network. Pesticide Information Profiles, Oregon State University, Diuron. 1996. Available online: http:// www.extoxnet.orst.edu (accessed on 19 March 2016).
- Worthing, C.R.; Walker, S.B. The Pesticide Manual: A World Compendium, 8th ed.; The British Crop. Protection Council: Thornton Heath, UK, 1987. [Google Scholar]
- U.S. Environmental Protection Agency. Simazine: Health advisory. Office of Drinking Water, US EPA, Washington, DC, USA. August 1987. Available online: https://www.epa.gov (accessed on 16 March 2016).
- Hayes, W.J.J.; Laws, E.R.J. Handbook of Pesticide Toxicology; Academic Press: New York, NY, USA, 1991. [Google Scholar]
- Rotterdam Convention—Operation of the Prior Informed Consent (PIC) Procedure for Banned or Severely Restricted Chemicals in International Trade, Decision Guidance Document, Monocrotophos. Available online: http://www.pic.int (accessed on 16 March 2016).
- Occupational Health Services, Inc. MSDS for Dimethoate. OHS Inc., Secaucus, NJ, USA. 1991. Available online: https://hr.umich.edu (accessed on 17 March 2016).
- U.S. Environmental Protection Agency, Office of Drinking Water (1987) Carbaryl Health Advisory. Draft Report. Available online: https://www.epa.gov (accessed on 16 March 2016).
- Morrison, R.D.; Hamilton, J.D. RH-75992 Technical: Acute Oral Toxicity Study in Male and Female Mice. Unpublished Report No. 91R-003. Rohm Haas Co., Toxicology Department: Philadelphia, PA, USA. Submitted to WHO by Rohm & Haas Co., Philadelphia, PA, USA. Available online: www.inchem.org/documents/jmpr/jmpmono/v96pr11.htm (accessed on 14 June 2018).
- Kidd, H.; James, D. Agrochemicals Handbook, 3rd ed.; Royal Society of Chemistry: Cambridge, UK, 1991. [Google Scholar]
- O’Neil, M.J. The Merck Index—An Encyclopedia of Chemicals, Drugs, and Biologicals, 13th ed.; Merck Research Laboratories: Whitehouse Station, NJ, USA, 2001; Available online: https://www.rsc.org/merck-index (accessed on 19 March 2016).
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.E.; Freed, V.H. An Agromedical Approach to Pesticide Management Some Health and Environmental Considerations; Consortium for International Crop. Protection: Berkeley, CA, USA, 1981. [Google Scholar]
- Carvalho, A.T.P.; Barrozo, A.; Doron, D.; Vardi Kilshtain, A.; Major, D.T.; Kamerlin, S.C.L. Challenges in computational studies of enzyme structure, function and dynamics. J. Mol. Graph. Model. 2014. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.S.; Jørgensen, F.S.; Klemmensen, P.D.; Hacksel, U.; Pettersson, I. Conformational analysis of the fungicide fenpropimorph by molecular mechanics calculations and NMR spectroscopy. Pestic. Sci. 1992, 36, 309–318. [Google Scholar] [CrossRef]
- Oakeshott, J.G.; Devonshire, A.L.; Claudianos, C.; Sutherland, T.D.; Horne, I.; Campbell, P.M.; Ollis, D.L.; Russell, R.J. Comparing the organophosphorus and carbamate insecticide resistance mutations in cholin-and carboxyl-esterases. Chem. Biol. Interact. 2005, 157, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Rathod, A.L.; Garg, R.K. Chlorpyrifos poisoning and its implications in human fatal cases: A forensic perspective with reference to Indian scenario. J. Forensic Leg. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Sabater, C.; Carrasco, J.M. Effects of the organophosphorous insecticide fenotrothion on growth in five freshwater species of phytoplankton. Environ. Toxicol. 2001, 16, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Buratti, F.M.; Volpe, M.T.; Meneguz, A.; Vittozzi, L.; Testai, E. CYP-specific bioactivation of four organophosphorothioate pesticides by human liver microsomes. Toxicol. Appl. Pharmacol. 2003, 186, 143–154. [Google Scholar] [CrossRef]
- Howard, J.J.; Oliver, J. Impact of naled (Dibrom 14) on the mosquito vectors of eastern equine encephalitis virus. J. Am. Mosq. Control Assoc. 1997, 13, 315–325. [Google Scholar] [PubMed]
- Davis, M.K.; Boone, J.S.; Moran, J.E.; Tyler, J.W.; Chambers, J.E. Assessing intermittent pesticide exposure from flea control collars containing the organophosphorus insecticide tetrachlorvinphos. J. Expo. Sci. Environ. Epidemiol. 2008, 18, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Garcia, S.; Abu-Qare, A.; Meeker-O’Connell, W.; Borton, A.; Abou-Donia, M. Methyl parathion: A review of health effects. J. Toxicol. Environ. Health Part B 2003, 6, 185–210. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, J.G.; Takhar, H.S.; Ruffalo, C.A.; Zwass, M. Health effects of diazinon on a family. J. Toxicol. Clin. Toxicol. 2004, 42, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Committee for Veterinary Medicinal Products Azamethiphos Summary Report (2). 1999. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Maximum_Residue_Limits_-_Report/2009/11/WC500010779.pdf (accessed on 14 June 2018).
- Bonner, M.R.; Williams, B.A.; Rusiecki, J.A.; Blair, A.; Beane Freeman, L.E.; Hoppin, J.A.; Dosemeci, M.; Lubin, J.; Sandler, D.P.; Alavanja, M.C.R. Occupational exposure to terbufos and the incidence of cancer in the Agricultural Health Study. Cancer Causes Control 2010, 21, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.P.; Antoniou, M.N.; Blumberg, B.; Carroll, L.; Colborn, T.; Everett, L.G.; Hansen, M.; Landrigan, P.J.; Lanphear, B.P.; Mesnage, R.; et al. Concerns over use of glyphosate-based herbicides and risks associated with exposures: A consensus statement. Environ. Health 2016, 15, 19. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.; Saavedra-Rodriguez, K.; Perera, R.; Paine, M.; Black, W.C.; Delgoda, R.; Delgoda, R. Insecticide resistance to permethrin and malathion and associated mechanisms in Aedes aegypti mosquitoes from St. Andrew Jamaica. PLoS ONE 2017, 12, e0179673. [Google Scholar] [CrossRef]
- Trachantong, W.; Saenphet, S.; Saenphet, K.; Chaiyapo, M. Lethal and sublethal effects of a methomyl-based insecticide in Hoplobatrachus rugulosus. J. Toxicol. Pathol. 2017, 30, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Hofmeister, M.V.; Bonefeld-Jørgensen, E.C. Effects of the pesticides prochloraz and methiocarb on human estrogen receptor α and β mRNA levels analyzed by on-line RT-PCR. Toxicol. In Vitro 2004, 18, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, G.L. Acute toxicity studies with oxamyl. Fundam. Appl. Toxicol. 1986, 6, 423–429. [Google Scholar] [CrossRef]
- Turusov, V.; Rakitsky, V.; Tomatis, L. Dichlorodiphenyltrichloroethane (DDT): Ubiquity, persistence, and risks. Environ. Health Perspect. 2002, 110, 125–128. [Google Scholar] [CrossRef] [PubMed]
- Legeay, S.; Clere, N.; Hilairet, G.; Do, Q.-T.; Bernard, P.; Quignard, J.-F.; Apaire-Marchais, V.; Lapied, B.; Faure, S. The insect repellent N,N-diethyl-m-toluamide (DEET) induces angiogenesis via allosteric modulation of the M3 muscarinic receptor in endothelial cells. Sci. Rep. 2016, 6, 28546. [Google Scholar] [CrossRef] [PubMed]
- Phosmet. Available online: http://pmep.cce.cornell.edu/profiles/extoxnet/metiram-propoxur/phosmet-ext.html (accessed on 7 April 2018).
- Sanderson, D.M. Treatment of poisoning by anticholinesterase insecticides in the rat. J. Pharm. Pharmacol. 1961, 13, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Reregistration Eligibility Decision (RED). Available online: https://www3.epa.gov/pesticides/chem_search/reg_actions/reregistration/red_PC-080301_1-Apr-98.pdf (accessed on 12 June 2018).
- Chlorpyrifos; No Health Council of The Netherlands: Committee on Updating of Occupational Exposure Limits. Chlorpyrifos; Health-Based Reassessment of Administrative Occupational Exposure Limits. The Hague: Health Council of The Netherlands, 2003; 2000/15OSH/067. 2000, 067; Available online: https://www.gezondheidsraad.nl/sites/default/files/0015067osh.pdf (accessed on 15 June 2018).
- Extoxnet Pip Terbufos. Available online: http://extoxnet.orst.edu/pips/terbufos.htm (accessed on 4 July 2018).
- Dichlorvos NIOSH Publications and Products. Available online: https://www.cdc.gov/niosh/idlh/62737.html (accessed on 2 July 2018).
- Methiocarb Chemical Profile 6/86. Available online: http://pmep.cce.cornell.edu/profiles/insect-mite/fenitrothion-methylpara/methiocarb/methio_prf_0686.html (accessed on 4 July 2018).
- Diazinon. Available online: http://www.herbiguide.com.au/Descriptions/hg_DIAZINON.htm (accessed on 2 July 2018).
- Ellenhorn, M.J.; Schonwald, S.; Ordog, G.J.; Wasserberger, J. Ellenhorn’s Medical Toxicology: Diagnosis and Treatment of Human Poisoning; Williams Wilkins: Baltimore, MD, USA, 1997; ISBN 068323093X. [Google Scholar]
- Pearce, E.M. Kirk-OTHMER encyclopedia of Chemical Technology, 3rd ed.; Wiley-Interscience: New York, NY, USA, 1978. [Google Scholar]
- Extoxnet Pip Oxamyl. Available online: http://extoxnet.orst.edu/pips/oxamyl.htm (accessed on 4 July 2018).
- “Glyphosate Technical Fact Sheet (Revised June 2015)”. National Pesticide Information Center. 2010. Available online: http://npic.orst.edu/factsheets/archive/glyphotech.html (accessed on 10 May 2018).
- Material Safety Data Sheet Malathion sc-211768 Section 1—Chemical Product and Company Identification NFPA Supplier. Available online: https://www.caymanchem.com/msdss/22998m.pdf (accessed on 11 April 2018).
- US EPA 2,4-D Reregistration Eligibility Decision, 2005. Associated RED Fact Sheet Archived 2008-05-17 at the Wayback Machine. EPA. Available online: https://archive.epa.gov/pesticides/reregistration/web/html/24d_fs.html (accessed on 14 April 2018).
- Methyl Parathion. Available online: http://pmep.cce.cornell.edu/profiles/extoxnet/haloxyfop-methylparathion/methyl-parathion-ext.html#2 (accessed on 2 July 2018).
- Dicamba (Banvel) Herbicide Profile 10/83. Available online: http://pmep.cce.cornell.edu/profiles/herb-growthreg/dalapon-ethephon/dicamba/herb-prof-dicamba.html (accessed on 4 July 2018).
- CDC-Immediately Dangerous to Life or Health Concentrations (IDLH): Dimethyl-1,2-dibromo-2,2-dichlorethyl Phosphate (Naled)-NIOSH Publications and Products. Available online: https://www.cdc.gov/niosh/idlh/300765.html (accessed on 2 July 2018).
- Tsitsanou, K.E.; Thireou, T.; Drakou, C.E.; Koussis, K.; Keramioti, M.V.; Leonidas, D.D.; Eliopoulos, E.; Iatrou, K.; Zographos, S.E. Anopheles gambiae odorant binding protein crystal complex with the synthetic repellent DEET: Implications for structure-based design of novel mosquito repellents. Cell. Mol. Life Sci. 2012, 69, 283–297. [Google Scholar] [CrossRef] [PubMed]
- CDC-Immediately Dangerous to Life or Health Concentrations (IDLH): Parathion-NIOSH Publications and Products. Available online: https://www.cdc.gov/niosh/idlh/56382.html (accessed on 2 July 2018).
- Department of Pesticide Regulation, C. Summary of Toxicology Data-Sulfoxaflor. 2014. Available online: https://www.cdpr.ca.gov/docs/risk/toxsums/pdfs/6109.pdf (accessed on 5 June 2018).
- Čolović, M.B.; Krstić, D.Z.; Lazarević-Pasti, T.D.; Bondzić, AM.; Vasić, V.M. Acetylcholinesterase Inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol. 2013. [Google Scholar] [CrossRef]
- Li, Q.; Kong, X.; Xiao, Z.; Zhang, L.; Wang, F.; Zhang, H.; Li, Y.; Wang, Y. Structural determinants of imidacloprid-based nicotinic acetylcholine receptor inhibitors identified using 3D-QSAR, docking and molecular dynamics. J. Mol. Model. 2012, 18, 2279–2289. [Google Scholar] [CrossRef] [PubMed]
- Mohamadi, F.; Richards, N.G.J.; Guida, W.C.; Liskamp, R.; Lipton, M.; Caufield, C.; Chang, G.; Hendrickson, T.; Still, W.C. Macromodel: An integrated software system for modeling organic and bioorganic molecules using molecular mechanics. J. Comput. Chem. 1990, 11, 440–467. [Google Scholar] [CrossRef]
- Chang, G.; Guida, W.C.; Still, W.C. An internal-coordinate Monte Carlo method for searching conformational space. J. Am. Chem. Soc. 1989. [Google Scholar] [CrossRef]
- Allinger, N.L.; Yuh, Y.H.; Lii, J.H. Molecular mechanics. The MM3 force field for hydrocarbons. 1. J. Am. Chem. Soc. 1989, 111, 8551–8566. [Google Scholar] [CrossRef]
- Mladenović, M.; Patsilinakos, A.; Pirolli, A.; Sabatino, M.; Ragno, R. Understanding the molecular determinant of reversible human monoamine oxidase B inhibitors containing 2H-chromen-2-one core: Structure-based and ligand-based derived three-dimensional quantitative structure-activity relationships predictive models. J. Chem. Inf. Model. 2017. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.T.; Brozell, S.R.; Mukherjee, S.; Pettersen, E.F.; Meng, E.C.; Thomas, V.; Rizzo, R.C.; Case, D.A.; James, T.L.; Kuntz, I.D. DOCK 6: Combining techniques to model RNA-small molecule complexes. RNA 2009, 15, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, S.; Zhang, Y. Catalytic reaction mechanism of acetylcholinesterase determined by Born−Oppenheimer ab initio QM/MM molecular dynamics simulations. J. Phys. Chem. B 2010, 114, 8817–8825. [Google Scholar] [CrossRef] [PubMed]
- Xing, H.; Wang, J.; Li, J.; Fan, Z.; Wang, M.; Xu, S. Effects of atrazine and chlorpyrifos on acetylcholinesterase and Carboxylesterase in brain and muscle of common carp. Environ. Toxicol. Pharmacol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Jin-Clark, Y.; Lydy, M.J.; Zhu, K.Y. Effects of atrazine and cyanazine on chlorpyrifos toxicity in Chironomus tentans (Diptera: Chironomidae). Environ. Toxicol. Chem. 2002. [Google Scholar] [CrossRef]
- Rosenfeld, C.A.; Sultatos, L.G. Concentration-dependent kinetics of acetylcholinesterase inhibition by the organophosphate paraoxon. Toxicol. Sci. 2006. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006. [Google Scholar] [CrossRef] [PubMed]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A second generation force field for the simulation of sroteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996. [Google Scholar] [CrossRef]
- Arsic, B.; Aguilar, J.A.; Bryce, R.A.; Barber, J. Conformational study of tylosin A in water and full assignments of 1H and 13C spectra of tylosin A in D2O and tylosin B in CDCl3. Magn. Reson. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Clark Still, W.; Tempczyk, A.; Hawley, R.C.; Hendrickson, T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 1990. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Betz, R.M.; Botello-Smith, W.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; et al. AMBER 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Ragno, R.; Ballante, F.; Pirolli, A.; Wickersham, R.B.; Patsilinakos, A.; Hesse, S.; Perspicace, E.; Kirsch, G. Vascular endothelial growth factor receptor-2 (VEGFR-2) inhibitors: Development and validation of predictive 3-D QSAR models through extensive ligand- and structure-based approaches. J. Comput. Aided Mol. Des. 2015. [Google Scholar] [CrossRef] [PubMed]
- Bursulaya, B.D.; Totrov, M.; Abagyan, R.; Brooks, C.L. Comparative study of several algorithms for flexible ligand docking. J. Comput. Aided Mol. Des. 2003, 17, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Resource for Biocomputing, Visualization, and Informatics (RBVI). Available online: http://rbvi.ucsf.edu (accessed on 6 August 2018).
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the opls force field. J. Chem. Theory Comput. 2010. [Google Scholar] [CrossRef] [PubMed]
- Dennington, R.; Keith, T.; Millam, J. GaussView 2009. Available online: http://gaussian.com/glossary/g09/ (accessed on 13 March 2018).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pesticide | Structure | LD50 (mg/kg) | Ref. | Pesticide | Structure | LD50 (mg/kg) | Ref. |
|---|---|---|---|---|---|---|---|
| atrazine |  | 0.850 0.0689 | [20] | tebufenozide |  | 5000 405.40 | [29] |
| propazine |  | 3.18 0.258 | [21] | imidacloprid |  | 131 10.62 | [30] |
| simazine |  | 5 0.405 | [24] | acetamiprid |  | 18 14.92 | [31] |
| carbofuran |  | 2 0.162 | [25] | diuron |  | 500 40.54 | [22] |
| monocrotophos |  | 14 1.135 | [26] | monuron |  | 1700 437.84 | [23] |
| dimethoate |  | 60 4.86 | [27] | linuron |  | 2400 194.59 | [19] |
| carbaryl |  | 100 8.11 | [28] | acetylcholine |  |
| Pesticide | Structure | LD50 (mg/kg) | Ref. | Pesticide | Structure | LD50 (mg/kg) | Ref. |
|---|---|---|---|---|---|---|---|
| azamethiphos |  | 1040 84.324 | [44] | phosmet |  | 113 9.162 | [53] |
| azinphos-methyl |  | 7 0.567 | [54] | TCVP |  | 465 37.702 | [55] |
| chlorpyrifos |  | 2000 162.162 | [56] | terbufos |  | 1.6 0.129 | [57] |
| DDVP |  | 17 1.378 | [58] | methiocarb |  | 350 28.378 | [59] |
| diazinon |  | 66 5.351 | [60] | methomyl |  | 12 0.972 | [61] |
| fenitrothion |  | 50 40.540 | [62] | oxamyl |  | 5.4 0.437 | [63] |
| glyphosate |  | 5000 405.405 | [64] | DDT |  | 113 9.162 | [14] |
| malathion |  | 290 23.513 | [65] | 2,4-D |  | 639 51.810 | [66] |
| methyl parathion |  | 18 1.459 | [67] | dicamba |  | 75 61.378 | [68] |
| naled (dibrom) |  | 160 12.972 | [69] | DEET |  | 1.95 0.158 | [70] |
| parathion |  | 2 0.162 | [71] | sulfoxaflor |  | 750 60.810 | [72] |
| Pesticide | FF a | Eglob_minb | NGMS c | NF d | Pesticide | CAA f |
|---|---|---|---|---|---|---|
| (kJ/mol) | Alignment e | RMSD (Å) g | ||||
| simazine | MM3 | NA h | NA | NA |  | EC/MMFF |
 | AMBER94 | NA | NA | NA | 0.667 | |
| MMFF | −1022.18 | 136 | 2782 | |||
| MMFFs | NA | NA | NA | |||
| OPLSAA | −793.61 | 158 | 2306 | |||
| monocrotophos | MM3 | NA | NA | NA |  | EC/MMFF |
 | AMBER94 | NA | NA | NA | 0.620 | |
| MMFF | −307.36 | 241 | 62 | EC/MMFFs | ||
| MMFFs | −310.26 | 218 | 61 | 3.521 | ||
| OPLSAA | NA | NA | NA | |||
| dimethoate | MM3 | NA | NA | NA |  | EC/MMFF |
 | AMBER94 | NA | NA | NA | 0.148 | |
| MMFF | −383.68 | 340 | 44 | EC/MMFFs | ||
| MMFFs | −386.71 | 215 | 40 | 3.227 | ||
| OPLSAA | −228.66 | 998 | 15 | EC/OPLSAA | ||
| 2.746 | ||||||
| acetamiprid | MM3 | NA | NA | NA |  | EC/MMFF |
 | AMBER94 | NA | NA | NA | 0.783 | |
| MMFF | −31.45 | 1916 | 14 | EC/MMFFs | ||
| MMFFs | −31.29 | 1816 | 6 | 0.823 | ||
| OPLSAA | NA | NA | NA |
| Pesticide | FF a | Eglob_min b | NGMS c | NF d | Pesticide | FFDAA f |
|---|---|---|---|---|---|---|
| (kJ/mol) | Alignment e | RMSD (Å) g | ||||
| atrazine | MM3 | −1161.48 | 445 | 23 |  | MM3/MMFF |
 | AMBER94 | NA h | NA | NA | 0.892 | |
| MMFF | −1007.32 | 147 | 62 | MM3/MMFFs | ||
| MMFFs | −992.63 | 509 | 20 | 0.931 | ||
| OPLSAA | −783.57 | 364 | 19 | MMFF/MMFFS | ||
| 0.343 | ||||||
| carbofuran | MM3 | 29.60 | 3617 | 2 |  | MMFF/MMFFS |
 | AMBER94 | NA | NA | NA | 0.075 | |
| MMFF | 20.30 | 3307 | 2 | MMFF/OPLSAA | ||
| MMFFs | 15.92 | 3384 | 2 | 0.216 | ||
| OPLSAA | 73.27 | 5621 | 75 | MMFFs/OPLSAA | ||
| 0.239 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mladenović, M.; Arsić, B.B.; Stanković, N.; Mihović, N.; Ragno, R.; Regan, A.; Milićević, J.S.; Trtić-Petrović, T.M.; Micić, R. The Targeted Pesticides as Acetylcholinesterase Inhibitors: Comprehensive Cross-Organism Molecular Modelling Studies Performed to Anticipate the Pharmacology of Harmfulness to Humans In Vitro. Molecules 2018, 23, 2192. https://doi.org/10.3390/molecules23092192
Mladenović M, Arsić BB, Stanković N, Mihović N, Ragno R, Regan A, Milićević JS, Trtić-Petrović TM, Micić R. The Targeted Pesticides as Acetylcholinesterase Inhibitors: Comprehensive Cross-Organism Molecular Modelling Studies Performed to Anticipate the Pharmacology of Harmfulness to Humans In Vitro. Molecules. 2018; 23(9):2192. https://doi.org/10.3390/molecules23092192
Chicago/Turabian StyleMladenović, Milan, Biljana B. Arsić, Nevena Stanković, Nezrina Mihović, Rino Ragno, Andrew Regan, Jelena S. Milićević, Tatjana M. Trtić-Petrović, and Ružica Micić. 2018. "The Targeted Pesticides as Acetylcholinesterase Inhibitors: Comprehensive Cross-Organism Molecular Modelling Studies Performed to Anticipate the Pharmacology of Harmfulness to Humans In Vitro" Molecules 23, no. 9: 2192. https://doi.org/10.3390/molecules23092192
APA StyleMladenović, M., Arsić, B. B., Stanković, N., Mihović, N., Ragno, R., Regan, A., Milićević, J. S., Trtić-Petrović, T. M., & Micić, R. (2018). The Targeted Pesticides as Acetylcholinesterase Inhibitors: Comprehensive Cross-Organism Molecular Modelling Studies Performed to Anticipate the Pharmacology of Harmfulness to Humans In Vitro. Molecules, 23(9), 2192. https://doi.org/10.3390/molecules23092192