
o-Carboranylalkoxy-1,3,5-Triazine Derivatives: Synthesis, Characterization, X-ray Structural Studies, and Biological Activity


Abstract
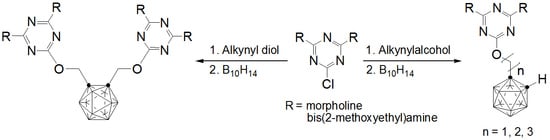
1. Introduction
2. Results and Discussion
2.1. Synthesis
2.2. X-ray Structural Studies on 5 and 6
2.3. Determination of IC50 and Incorporation of Boron into B16 Cells
3. Materials and Methods
3.1. General Considerations
3.2. Crystal Structure Determination
3.3. Cell Viability Assay (MTT Assay)
3.4. In Vitro Boron Incorporation into B16 Melanoma Cells
3.5. Synthesis of 4,4′-[(6-prop-2-ynylmethoxy)-1,3,5-triazine-2,4-diyl]dimorpholine (1)
3.6. Synthesis of 4,4′-[(6-but-2-ynylmethoxy)-1,3,5-triazine-2,4-diyl]dimorpholine (2)
3.7. Synthesis of 4,4′-[(6-pent-2-ynylmethoxy)-1,3,5-triazine-2,4-diyl]dimorpholine (3)
3.8. Synthesis of 1,4-bis(4,6-dimorpholino-1,3,5-triazin-2yloxy)but-2-yne (4)
3.9. Synthesis of N2,N2,N4,N4-tetrakis(2-methoxyethyl)-6-(propynyloxy)-1,3,5-triazine-2,4-diamine (9)
3.10. Synthesis of N2,N2,N4,N4-tetrakis(2-methoxyethyl)-6-(butynyloxy)-1,3,5-triazine-2,4-diamine (10)
3.11. Synthesis of N2,N2,N4,N4-tetrakis(2-methoxyethyl)-6-(pentynyloxy)-1,3,5-triazine-2,4-diamine (11)
3.12. Synthesis of 6,6’-[but-2-yne-1,4-diylbis(oxy)]bis[N2,N2,N4,N4-tetrakis(2-methoxyethyl)-1,3,5-triazine-2,4-diamine] (12)
3.13. Synthesis of 4,4′-[6-(o-carboranylmethoxy)-1,3,5-triazine-2,4-diyl]dimorpholine (5)
3.14. Synthesis of 4,4′-[6-(o-carboranylethoxy)-1,3,5-triazine-2,4-diyl]dimorpholine (6)
3.15. Synthesis of 4,4′-[6-(o-carboranylpropoxy)-1,3,5-triazine-2,4-diyl]dimorpholine (7)
3.16. Synthesis of 1,2-bis[(4,6-dimorpholino-1,3,5-triazin-2-yloxy)methyl]-o-carborane (8)
3.17. (6-o-carboranylmethoxy)-N2,N2,N4,N4-tetrakis(2-methoxyethyl)-1,3,5-triazine-2,4-diamine (13)
3.18. (6-o-carboranylethoxy)-N2,N2,N4,N4-tetrakis(2-methoxyethyl)-1,3,5-triazine-2,4-diamine (14)
3.19. (6-o-carboranylpropoxy)-N2,N2,N4,N4-tetrakis(2-methoxyethyl)-1,3,5-triazine-2,4-diamine (15)
3.20. 6,6’-[1,2-o-carboranylbis(methylene)bis(oxy)]bis[N2,N2,N4,N4-tetrakis(2-methoxyethyl)-1,3,5-triazine-2,4-diamine] (16)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
References
- Zhu, Y.; Hosmane, N.S. Nanostructured boron compounds for cancer therapy. Pure Appl. Chem. 2018, 90, 653–663. [Google Scholar] [CrossRef]
- Satapathy, R.; Dash, B.P.; Mahanta, C.S.; Swain, B.R.; Jena, B.B.; Hosmane, N.S. Cylcoconjugates of polyhedral boron clusters. J. Organomet. Chem. 2015, 798, 13–23. [Google Scholar] [CrossRef]
- Hawthorne, M.F.; Lee, M.W. A critical assessment of boron target compounds for boron neutron capture therapy. J. Neuro-Oncol. 2003, 62, 33–45. [Google Scholar] [CrossRef]
- Hosmane, N.S. Boron and Gadolinium Neutron Capture Therapy for Cancer Treatment; World Scientific Publishing Co.: Singapore, 2012. [Google Scholar]
- Hosmane, N.S. Boron Science: New Technologies and Applications; CRC Press: Boca Raton, FL, USA, 2011. [Google Scholar]
- Azab, A.K.; Abu Ali, H.; Srebnik, M. Boron Neutron Capture Therapy. In Studies in Inorganic Chemistry; Abu Ali, H., Dembitsky, V.M., Srebnik, M., Eds.; Elsevier: New York, NY, USA, 2005; Chapter 5; pp. 337–366. [Google Scholar]
- Barth, R.F.; Coderre, J.A.; Vicente, M.G.H.; Blue, T.E. Boron neutron capture therapy of cancer: Current status and future prospects. Clin. Cancer Res. 2005, 11, 3987–4002. [Google Scholar] [CrossRef] [PubMed]
- Pisarev, M.A.; Dagrosa, M.A.; Juvenal, G.J. Boron neutron capture therapy in cancer: Past, present and future. Arq. Bras. Endocrinol. Metab. 2007, 51, 852–856. [Google Scholar] [CrossRef]
- Barth, R.F.; Vicente, M.G.H.; Harling, O.K.; Kiger, W.S., III; Riley, K.J.; Binns, P.J.; Wagner, F.M.; Suzuki, M.; Aihara, T.; Kato, I.; et al. Current status of boron neutron capture therapy of high grade gliomas and recurrent head and neck cancer. Radiat. Oncol. 2012, 7, 146. [Google Scholar] [CrossRef] [PubMed]
- Soloway, A.H.; Tjarks, W.; Barnum, B.A.; Rong, F.-G.; Barth, R.F.; Codogni, I.M.; Wilson, J.G. The chemistry of neutron capture therapy. Chem. Rev. 1998, 98, 1515–1562. [Google Scholar] [CrossRef] [PubMed]
- Hawthorne, M.F. New horizons for therapy based on the boron neutron capture reaction. Mol. Med. Today 1998, 4, 174–181. [Google Scholar] [CrossRef]
- Hawthorne, M.F. The role of chemistry in the development of boron neutron capture therapy of cancer. Angew. Chem. Int. Ed. Engl. 1993, 32, 950–984. [Google Scholar] [CrossRef]
- Fauchere, J.-L.; Leukart, O.; Eberie, A.; Schwyzer, R. The synthesis of [4-carboranylalanine, 5-leucine]-enkephalin (including an improved preparation of t-butoxycarbonyl-l-o-carboranylalanine, new derivatives of l-propargylglycine, and a note on melanotropic and opiate receptor binding characteristics. Helv. Chim. Acta 1979, 62, 1385–1395. [Google Scholar] [CrossRef]
- Leukart, O.; Caviezel, M.; Eberie, A.; Escher, E.; Tun-Kyi, A.; Schwyer, R. l-o-carboranylalanine, a boron analogue of phenylalanine. Helv. Chim. Acta 1976, 59, 2184–2187. [Google Scholar] [CrossRef]
- Valliant, J.F.; Guenther, K.J.; King, A.S.; Morel, P.; Schaffer, P.; Sogbein, O.O.; Stephenson, K.A. The medicinal chemistry of carboranes. Coord. Chem. Rev. 2002, 232, 173–230. [Google Scholar] [CrossRef]
- Lee, J.-D.; Lee, C.-H.; Nakamura, H.; Ko, J.; Kang, S.O. A convenient synthesis of the novel carboranyl-substituted tetrahydroisoquinolines: Application to the biologically active agent for BNCT. Tetrahedron Lett. 2002, 43, 5483–5486. [Google Scholar] [CrossRef]
- Lee, C.-H.; Jin, G.F.; Yoon, J.H.; Kim, S.H.; Lee, J.-D.; Nakamura, H.; Kang, S.O. Triazinyl architecture on bifunctional carboranyl templates for the production of potential neutron capture therapy agents: Synthesis and characterization of 1,3,5-triazinylcarborane derivatives. Synthesis 2008, 1193–1200. [Google Scholar] [CrossRef]
- Lee, C.-H.; Jin, G.F.; Yoon, J.H.; Jung, Y.U.; Lee, J.-D.; Cho, S.; Nakamura, H.; Kang, S.O. Synthesis and characterization of polar functional group substituted mono- and bis-(o-carboranyl)-1,3,5-triazine derivatives. Tetrahedron Lett. 2008, 49, 159–164. [Google Scholar] [CrossRef]
- Lee, C.-H.; Lim, H.-G.; Lee, J.-D.; Lee, Y.-J.; Ko, J.; Nakamura, H.; Kang, S.O. o-Carboranyl derivatives of 1,3,5-s-triazines: Structures, properties and in vitro activities. Appl. Organometal. Chem. 2003, 17, 539–548. [Google Scholar] [CrossRef]
- Lee, C.-H.; Oh, J.M.; Lee, J.-D.; Nakamura, H.; Ko, J.; Kang, S.O. Synthesis of THIQ derivatives as potential boron neutron capture therapy agents: N-functionalized o-carboranylmethyl benzopiperidines. Synlett 2006, 275–278. [Google Scholar] [CrossRef]
- Lee, C.-H.; Yang, I.D.; Lee, J.-D.; Nakamura, H.; Ko, J.; Kang, S.O. Synthesis of N-functionalized 3,4-o-carboranylenepiperidines as potential boron neutron capture therapy agents. Synlett 2004, 1799–1801. [Google Scholar] [CrossRef]
- Fujii, S.; Goto, T.; Ohta, K.; Hashimoto, Y.; Suzuki, T.; Ohta, S.; Endo, Y. Potent androgen antagonists based on carborane as a hydrophobic core structure. J. Med. Chem. 2005, 48, 4654–4662. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, R.C.; Campbell, S.R.; Fairchild, R.G.; Kisliuk, R.L.; Micca, P.L.; Queener, S.F.; Riordan, J.M.; Sedwick, W.D.; Waud, W.R.; Leung, A.K.W.; et al. Novel boron-containing, nonclassical antifolates: Synthesis and preliminary biological and structural evaluation. J. Med. Chem. 2007, 50, 3283–3289. [Google Scholar] [CrossRef] [PubMed]
- Azev, Y.; Slepukhina, I.; Gabel, D. Synthesis of boron-containing heterocyclic compounds. Appl. Radiat. Isotope 2004, 61, 1107–1110. [Google Scholar] [CrossRef] [PubMed]
- Ronchi, S.; Prosperi, D.; Compostella, F.; Panza, L. Synthesis of novel carborane-hybrids based on a triazine scaffold for boron neutron capture therapy. Synlett 2004, 1007–1010. [Google Scholar] [CrossRef]
- Lee, J.-D.; Cho, S.J.; Kim, S.H.; Chai, G.Y.; Lee, C.-H. New types of o-carboranyl heterocyclic compounds: Synthesis and characterization of morpholino and di(methoxyethyl)amino substituted 1,3,5-triazine derivatives. Bull. Korean Chem. Soc. 2012, 33, 3136–3138. [Google Scholar] [CrossRef]
- Zeng, T.; Dong, C.M.; Shu, X.G.; Li, J.S.; Huang, P.M. 2-Chloro-4,6-dimorpholino-1,3,5-triazine. Acta Crystallogr. Sect. E 2005, 61, o2211–o2212. [Google Scholar] [CrossRef]
- Dong, J.Y.; Huang, P.M. 4,6-Dimorpholino-N-(2,4,4-trimethylpentan-2-yl)-1,3,5-triazin-2-amine. Acta Crystallogr. Sect. E 2007, 63, o4028. [Google Scholar] [CrossRef]
- Davidson, M.G.; Hibbert, T.G.; Howard, J.A.K.; Mackinnon, A.; Wade, K. Definitive crystal structures of ortho-, meta- and para-carboranes: Supramolecular structures directed solely by C–H∙O hydrogen bonding to hmpa (hmpa = hexamethylphosphoramide). Chem. Commun. 1996, 2285–2286. [Google Scholar] [CrossRef]
- Lee, J.-D.; Lee, Y.-J.; Jeong, H.-J.; Lee, J.S.; Lee, C.-H.; Ko, J.; Kang, S.O. Practical synthesis of amonoethyl-o-carboranes. Organometallics 2003, 22, 445–449. [Google Scholar] [CrossRef]
- Welch, A.J.; Venkatasubramanian, U.; Rosair, G.M.; Ellis, D.; Donohoe, D.J. Crystal engineering with heteroboranes. I. 1-Carboxy-1,2-dicarba-closo-dodecaborane(11). Acta Crystallogr. Sect. C 2001, 57, 1295–1296. [Google Scholar] [CrossRef]
- Wu, Y.; Carroll, P.J.; Kang, S.O.; Quintana, W. Synthesis, characterization, and reactivity of isocyanato dicarbaboranes obtained from o-carborane. Inorg. Chem. 1997, 36, 4753–4761. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Wang, Y.; Miao, J.; Li, Y.; Zhang, Z. Synthesis and characterization of carboranyl Schiff base compounds from 1-amino-o-carborane. J. Organomet. Chem. 2015, 798, 182–188. [Google Scholar] [CrossRef]
- SMART and SAINT; Bruker Analytical X-ray Division: Madison, WI, USA, 2002.
- Sheldrick, G.M. SHELXTL-PLUS Software Package; Bruker Analytical X-ray Division: Madison, WI, USA, 2002. [Google Scholar]
Sample Availability: Samples of the compounds 5–8 and 13–16 are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identification Code | cnu1002 | cnu1001 |
|---|---|---|
| Empirical formula | C14 H29 B10 N5 O3 | C15 H30 B10 N5 O3 |
| Formula weight | 423.52 | 436.54 |
| Temperature | 293(2) K | 293(2) K |
| Wavelength | 0.71073 A | 0.71073 A |
| Crystal system, space group | Triclinic, P-1 | Triclinic, P-1 |
| Unit cell dimensions | a = 7.03880(10) Å, α = 87.1180(10)° b = 9.7116(2) Å, β = 88.4920(10)° c = 16.9533(3) Å, γ = 74.5480(10)° | a = 9.7505(3) Å, α = 88.224(2)° b = 11.1591(4) Å, β = 74.390(2)° c = 11.9630(4) Å,γ = 67.088(2)° |
| Volume | 1115.49(3) Å−3 | 1150.74(7) Å−3 |
| Z, Dcalc | 2, 1.261 g/cm3 | 2, 1.260 g/cm3 |
| m | 0.079 mm−1 | 0.079 mm−1 |
| F(000) | 444 | 458 |
| Crystal size | 0.24 × 0.20 × 0.15 mm | 0.26 × 0.22 × 0.19 mm |
| θ range for data collection | 1.20 to 28.14º | 1.77 to 28.34° |
| Limiting indices | −9 ≤ h ≤ 9, −10 ≤ k ≤ 12, −21 ≤ l ≤ 21 | −13 ≤ h ≤ 13, −14 ≤ k ≤ 14, −15 ≤ l ≤ 15 |
| Reflections collected/unique | 16295/5176 [R(int) = 0.0292] | 31151/5711 [R(int) = 0.0465] |
| Completeness to θ = 25.96 | 94.9% | 99.8% |
| Refinement method | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 5176/0/289 | 5711/0/299 |
| Goodness-of-fit on F2 | 1.055 | 1.124 |
| Final R indices [I > 2s (I)] | R1 = 0.0528, wR2 = 0.1335 | R1 = 0.0647, wR2 = 0.2064 |
| R indices (all data) | R1 = 0.0829, wR2 = 0.1540 | R1 = 0.0840, wR2 = 0.2238 |
| Extinction coefficient | 0.011(5) | |
| Largest diff. peak and hole | 0.205 and −0.272 e.Å−3 | 0.807 and −0.360 e.Å−3 |
| 5 | |||
|---|---|---|---|
| C1–C2 | 1.627(2) | N1–C14 | 1.317(2) |
| O1–C14 | 1.358(2) | N1–C15 | 1.352(2) |
| O1–C13 | 1.429(2) | N3–C14 | 1.318(2) |
| N4–C15 | 1.348(2) | N3–C16 | 1.355(2) |
| N5–C16 | 1.350(2) | N2–C15 | 1.339(2) |
| C1 C13 | 1.520(2) | N2–C16 | 1.335(2) |
| C14–O1–C13 | 118.8(1) | C13–C1–C2 | 119.7(1) |
| O1–C13–C1 | 109.4(1) | N1–C14–N3 | 129.4(1) |
| N1–C14–O1 | 111.7(1) | N3–C14–O1 | 118.9(1) |
| N2–C15–N1 | 125.4(1) | N2–C16–N3 | 125.5(1) |
| 6 | |||
| C1–C2 | 1.638(3) | O1–C14 | 1.444(2) |
| O1–C15 | 1.351(2) | C13–C14 | 1.504(3) |
| C1–C13 | 1.531(2) | N1–C15 | 1.324(2) |
| N1–C16 | 1.349(2) | N2–C16 | 1.342(2) |
| N2–C17 | 1.336(2) | N3–C15 | 1.311(2) |
| N3– C17 | 1.347(2) | ||
| C15–N3–C17 | 113.03(15) | C15–N1–C16 | 112.50(14) |
| C17–N2–C16 | 114.04(16) | C15–O1–C14 | 116.47(14) |
| C13–C1–C2 | 116.26(14) | C14–C13–C1 | 112.70(16) |
| O1–C14–C13 | 111.28(16) | N3–C15–N1 | 128.89(17) |
| N3–C15–O1 | 118.03(15) | N1–C15–O1 | 113.08(15) |
| Compound | Cytotoxicity IC50 (μM) a | |
|---|---|---|
| B16 | HeLa | |
| 5 | 14.6 ± 0.5 | 17.4 ± 2.0 |
| 6 | 13.9 ± 1.1 | 15.9 ± 3.3 |
| 7 | 20.9 ± 1.8 | 18.1 ± 0.7 |
| 8 | 18.5 ± 0.6 | 20.3 ± 3.5 |
| 13 | 17.0 ± 1.4 | 16.4 ± 1.9 |
| 14 | 13.1 ± 1.2 | 17.5 ± 1.4 |
| 15 | 27.9 ± 1.6 | 21.5 ± 0.9 |
| 16 | 28.6 ± 1.0 | 20.5 ± 1.4 |
| BPA | 44.9 ± 0.3 | n.d. b |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, G.F.; Ban, H.S.; Nakamura, H.; Lee, J.-D. o-Carboranylalkoxy-1,3,5-Triazine Derivatives: Synthesis, Characterization, X-ray Structural Studies, and Biological Activity. Molecules 2018, 23, 2194. https://doi.org/10.3390/molecules23092194
Jin GF, Ban HS, Nakamura H, Lee J-D. o-Carboranylalkoxy-1,3,5-Triazine Derivatives: Synthesis, Characterization, X-ray Structural Studies, and Biological Activity. Molecules. 2018; 23(9):2194. https://doi.org/10.3390/molecules23092194
Chicago/Turabian StyleJin, Guo Fan, Hyun Seung Ban, Hiroyuki Nakamura, and Jong-Dae Lee. 2018. "o-Carboranylalkoxy-1,3,5-Triazine Derivatives: Synthesis, Characterization, X-ray Structural Studies, and Biological Activity" Molecules 23, no. 9: 2194. https://doi.org/10.3390/molecules23092194
APA StyleJin, G. F., Ban, H. S., Nakamura, H., & Lee, J.-D. (2018). o-Carboranylalkoxy-1,3,5-Triazine Derivatives: Synthesis, Characterization, X-ray Structural Studies, and Biological Activity. Molecules, 23(9), 2194. https://doi.org/10.3390/molecules23092194