Linking Aromatic Hydroxy Metabolic Functionalization of Drug Molecules to Structure and Pharmacologic Activity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Objective of the Review
3. Methodology
- (a)
- Drugs that are metabolized by O-dealkylation or aromatic ring hydroxylation;
- (b)
- Availability of data regarding the pharmacologic activity of the major metabolite(s).
- The published literature;
- Drug manufacturers’ data sheets;
- Reference books on drug metabolism and activity of metabolites.
4. Review Strategy
- The chemical structure of the drug and metabolite(s), and the enzymes or isoenzymes involved;
- The status of the pharmacologic activity of the metabolite;
- The percentage concentration of the metabolite(s) with respect to the parent drug.
5. Aromatic Hydroxy (Arenolic) Metabolites
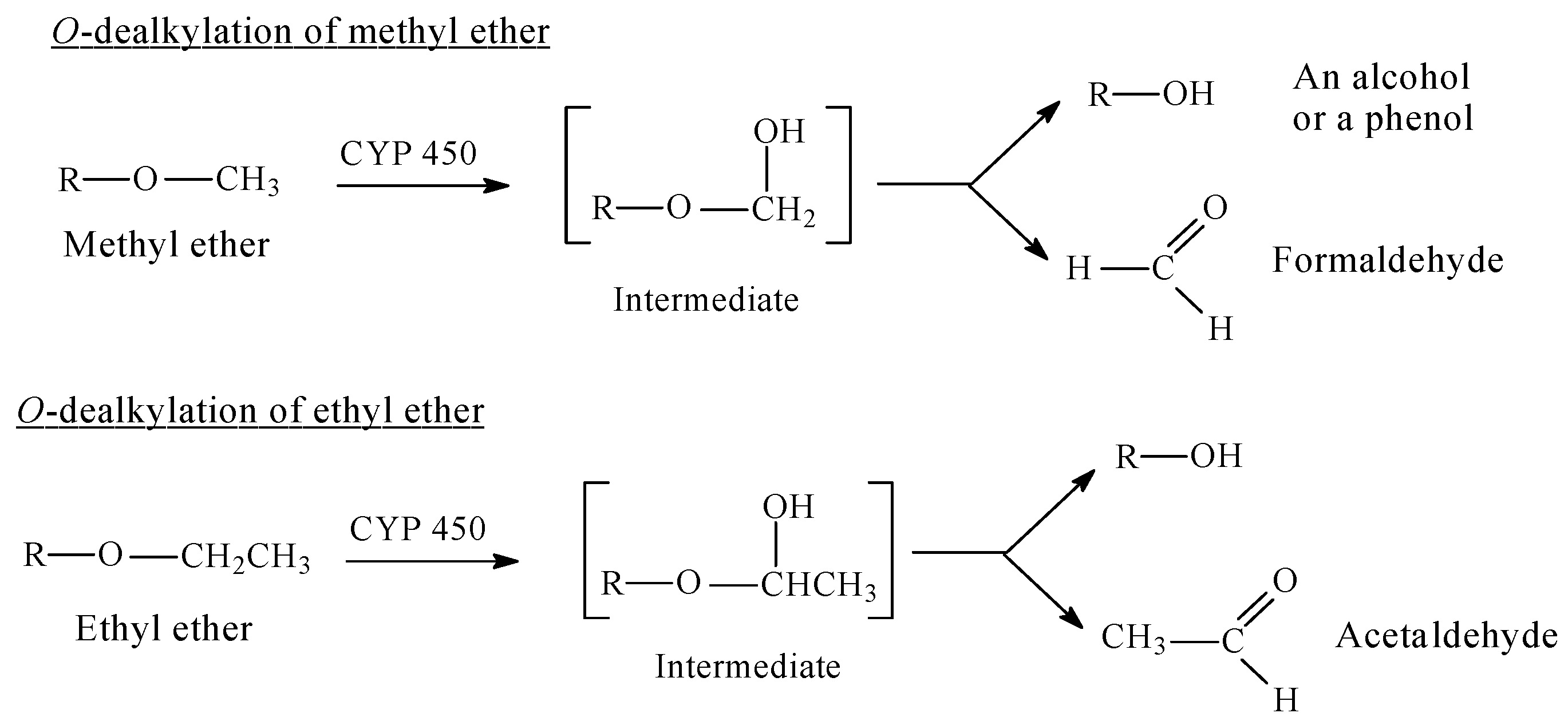
5.1. Arenolic Metabolites Resulting from the O-Dealkylation of Aralkoxy Groups
- (1)
- Enhancement of activity occurring in the opioid narcotic/analgesic drug, codeine, and its semisynthetic and synthetic congeners, all containing an aryl-methoxy group, and the analgesic/antipyretic drug, phenacetin, which contains an aryl-ethoxy group;
- (2)
- Retention of activity by venlafaxine, (a selective-serotonin-reuptake-inhibitor (SSRI) antidepressant, containing an aryl-methoxy group);
- (3)
- Attenuation or loss of activity by COX1/COX2-inhibitors, -naproxen, indomethacin, and nabumetone-each containing an aryl-methoxy group.
5.1.1. Metabolic Aralkoxy Group Cleavage Resulting in the Enhancement of Pharmacologic Activity: Opioids and Phenacetin
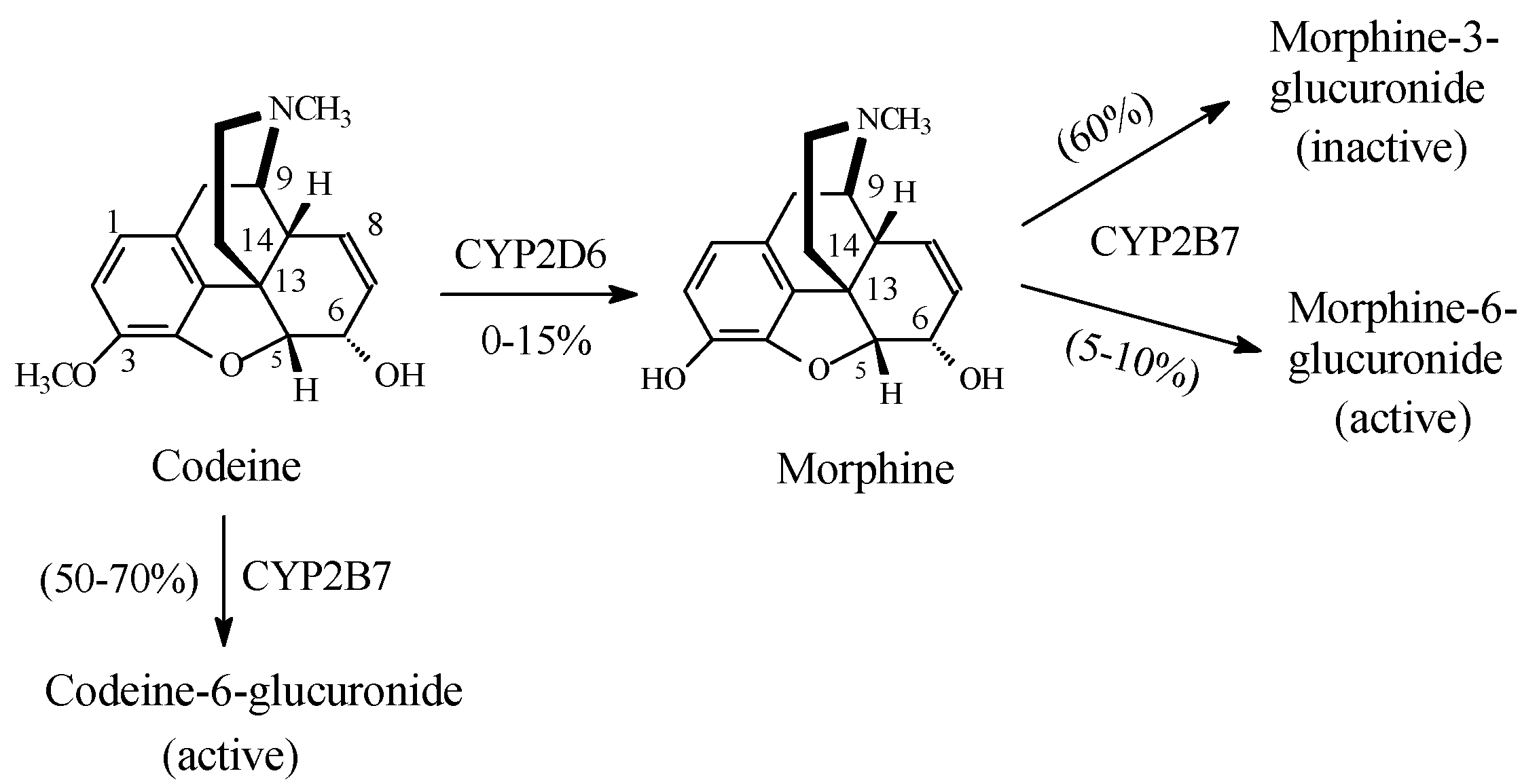
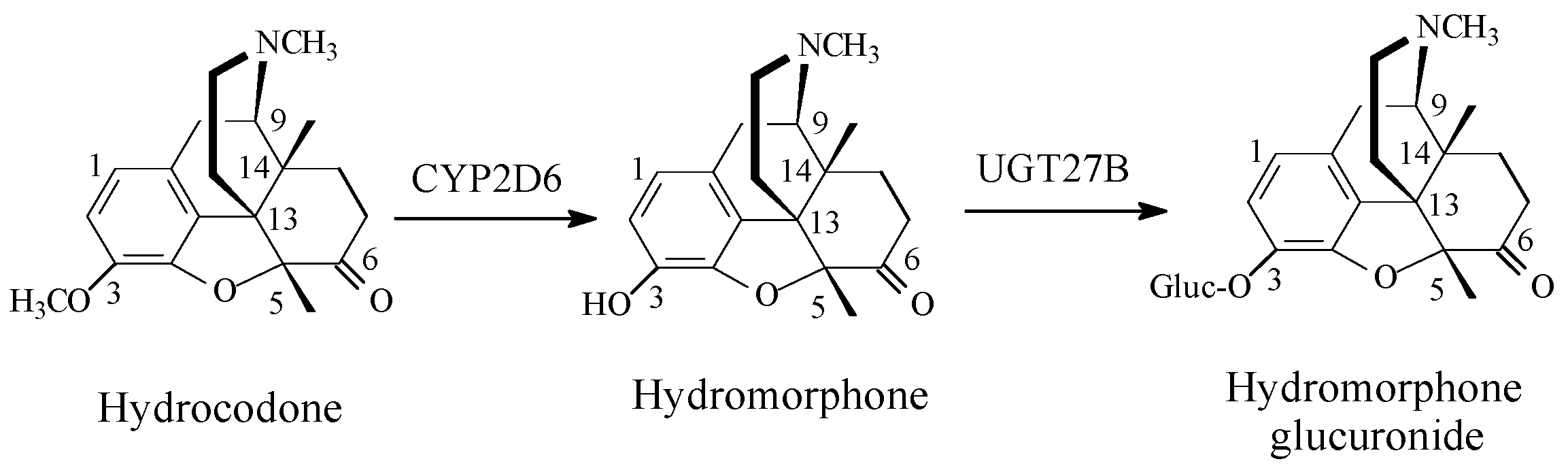
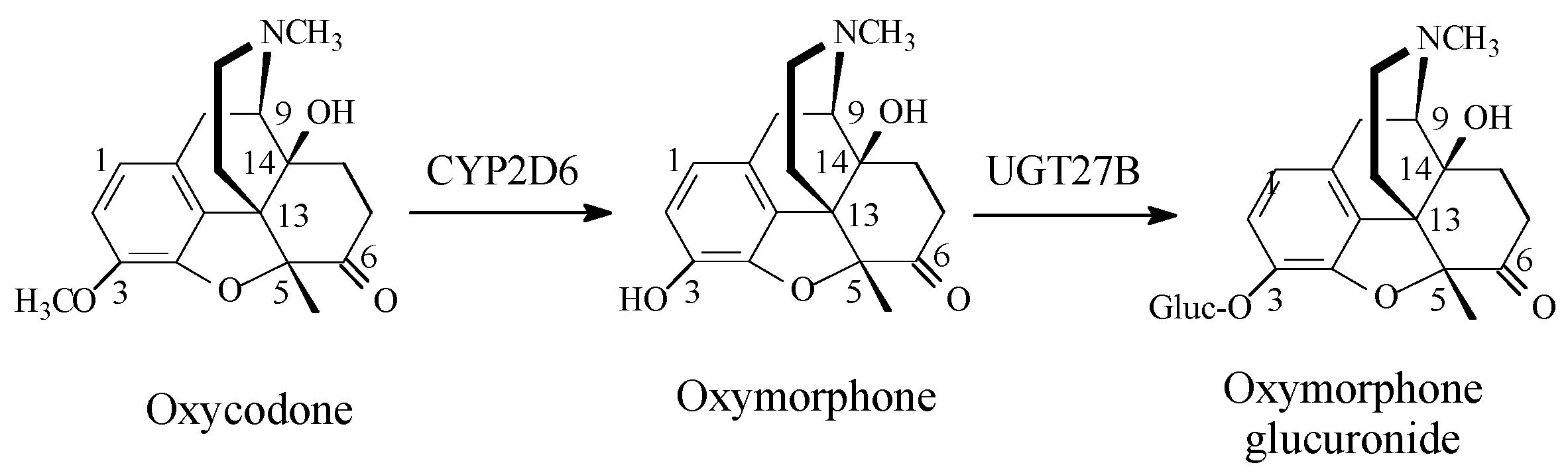
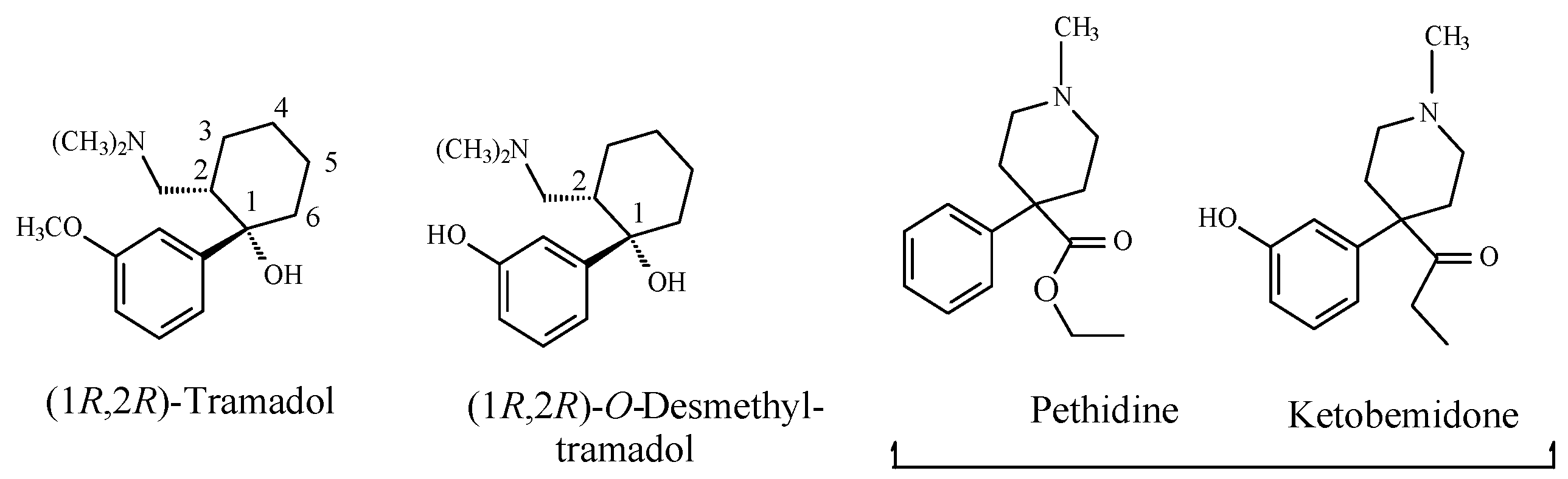
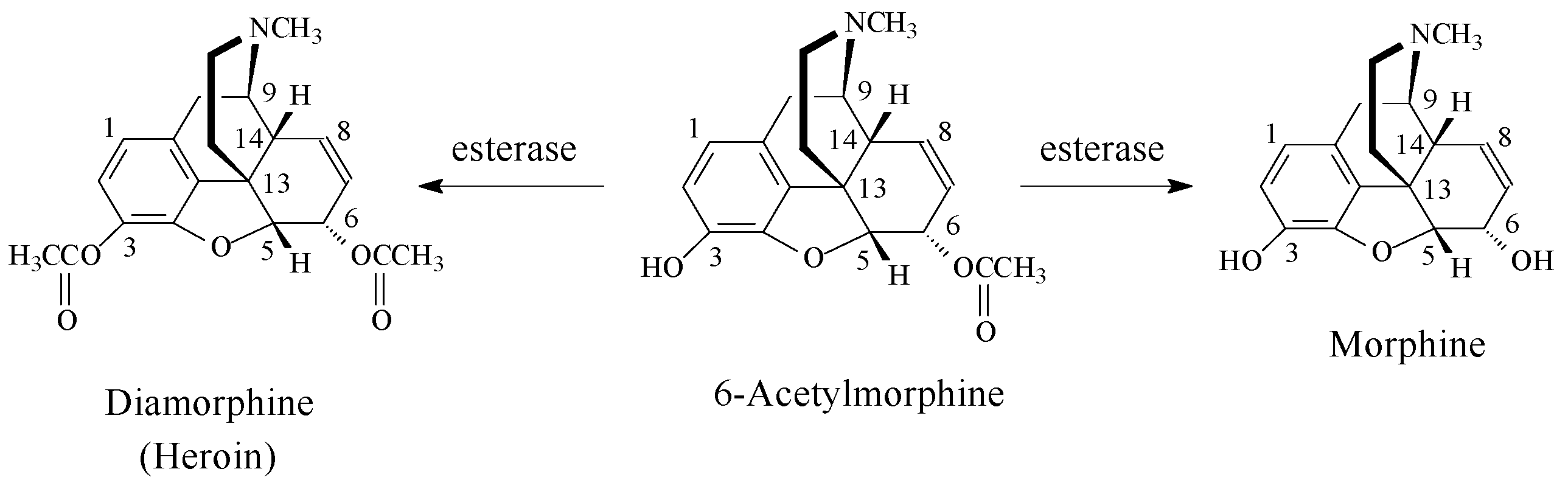
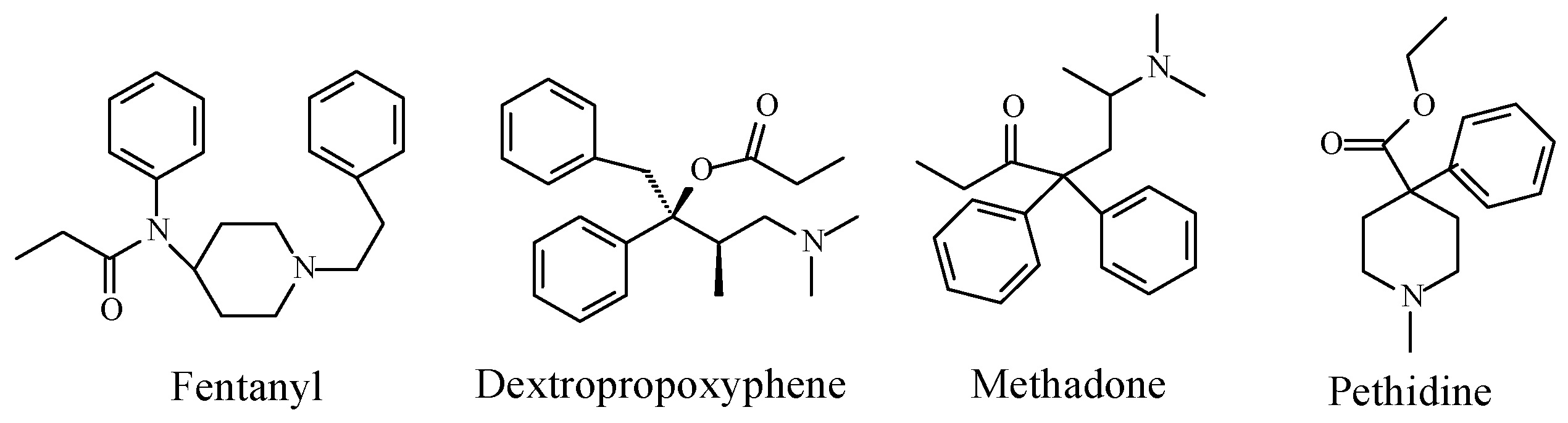
Opioids
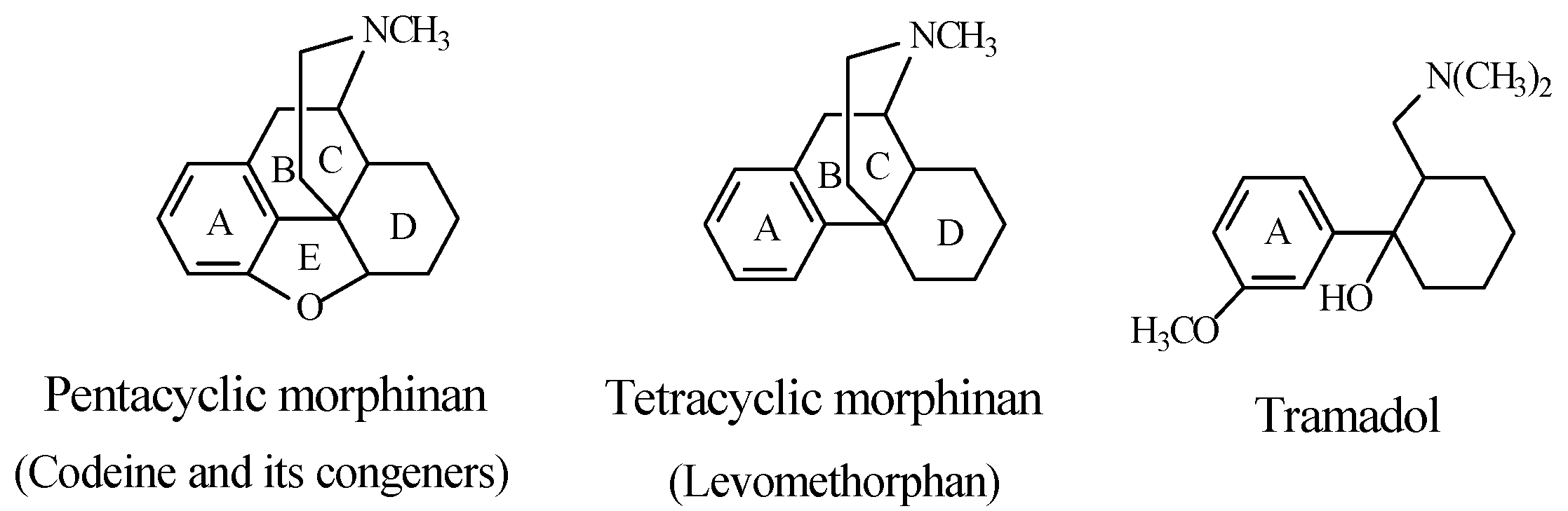
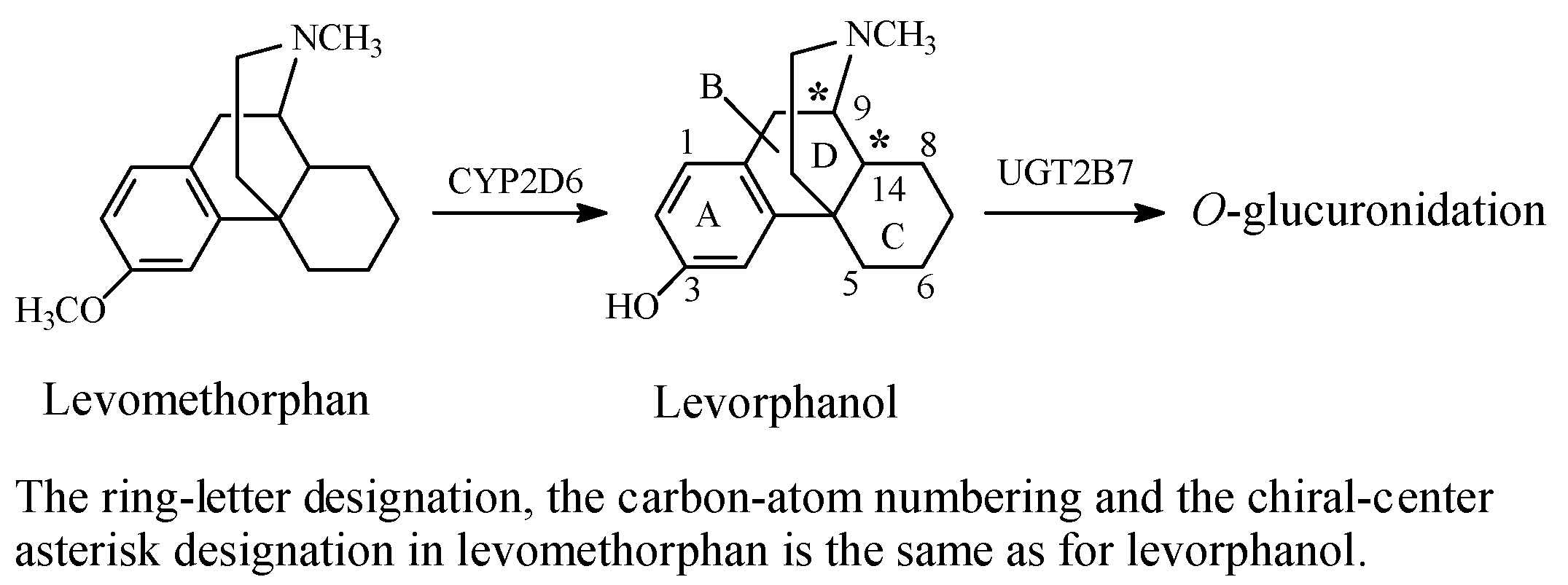
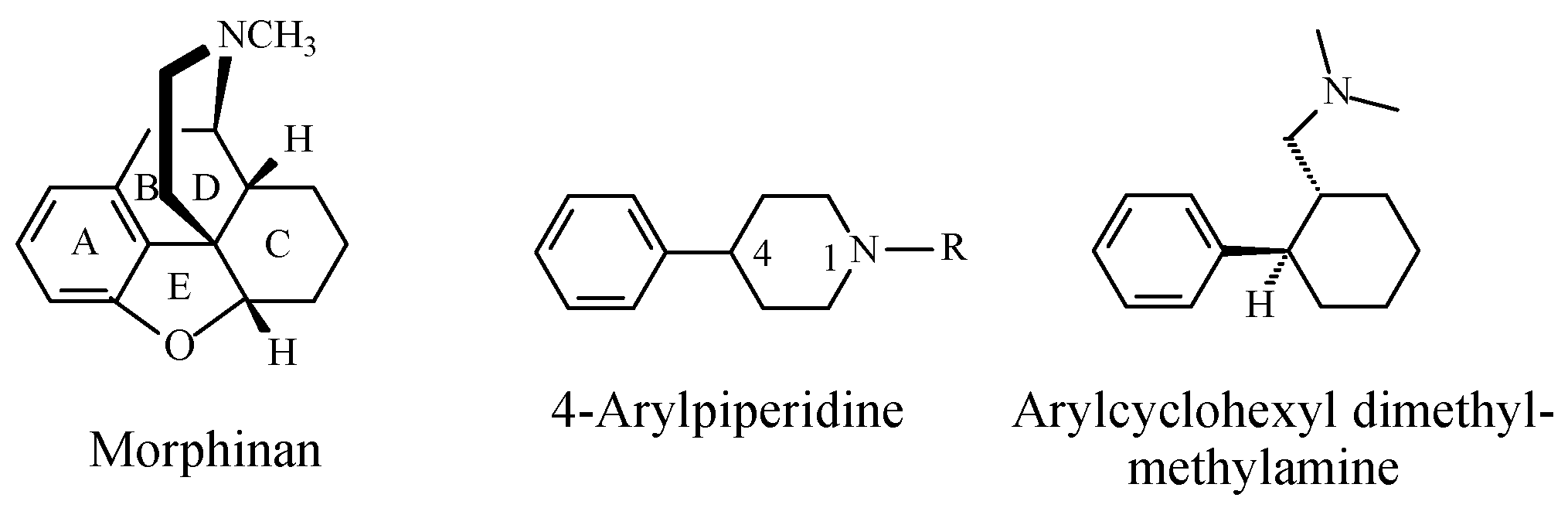
Stereochemistry of the Morphinan Opioids
Discussion of Opioids
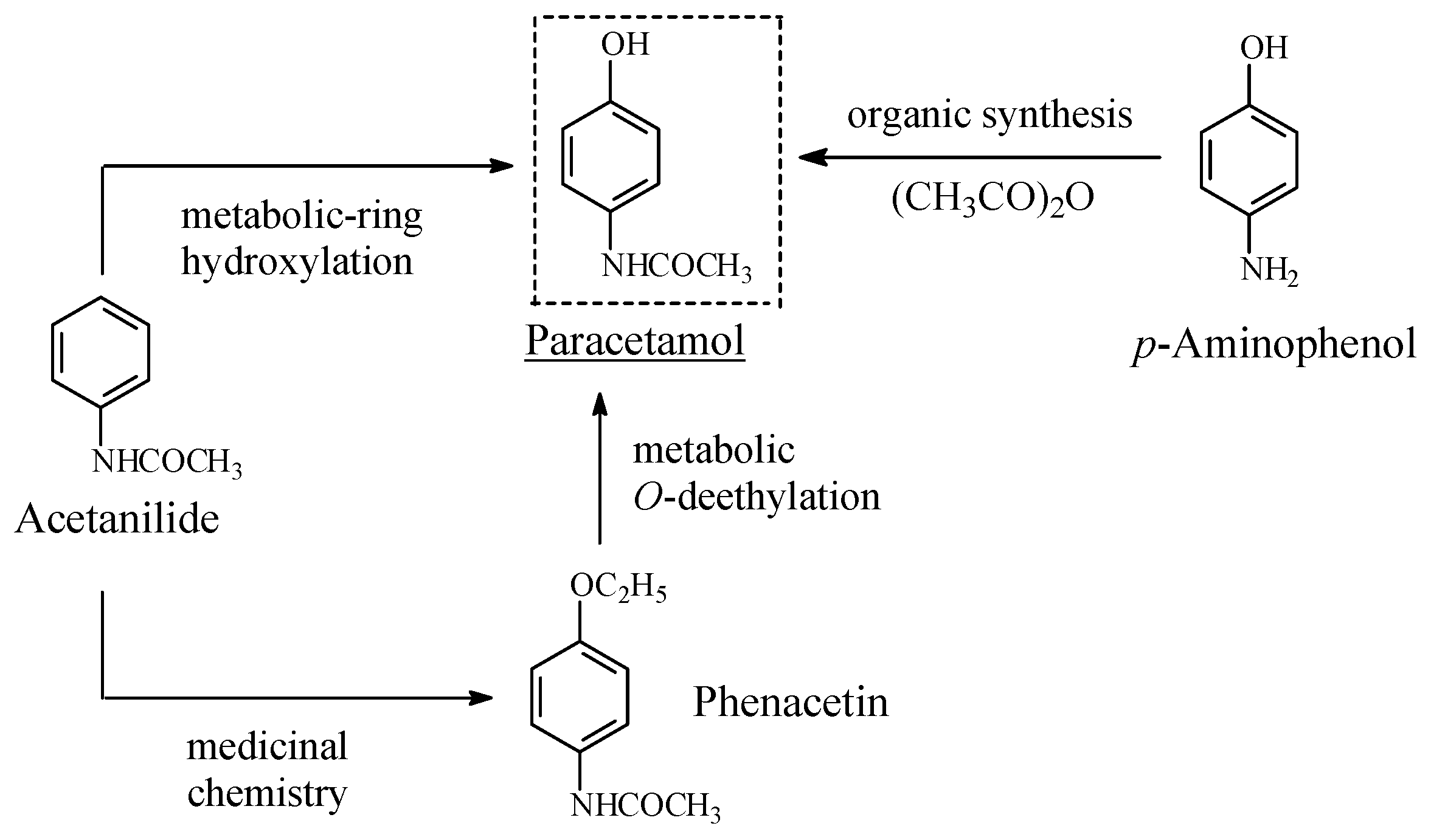
Acetanilide/Phenacetin/Paracetamol
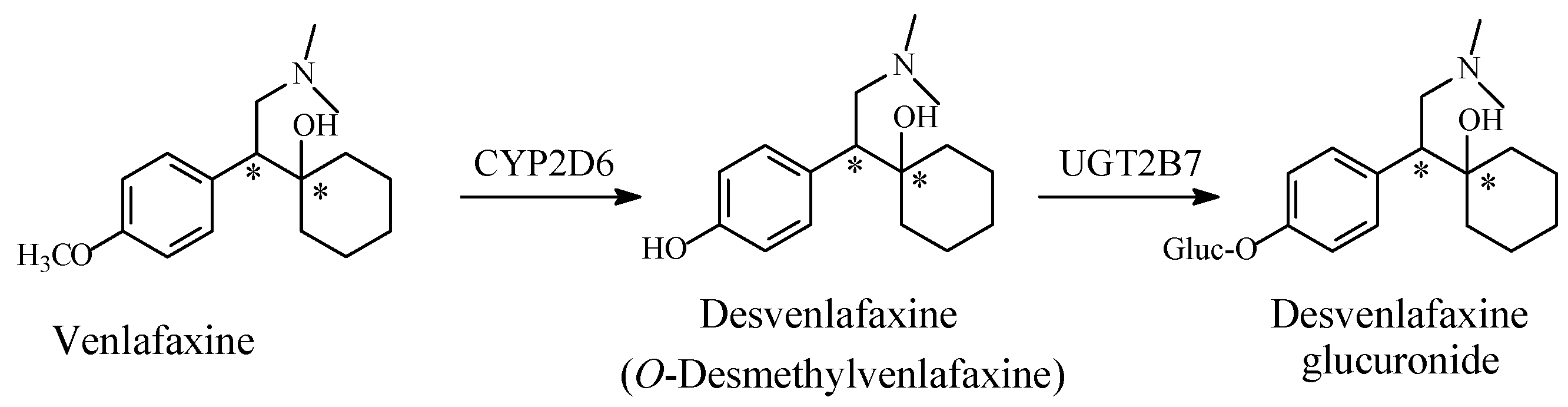
5.1.2. Metabolic O-Dealkylation of Aralkoxy Groups Resulting in Retaining Pharmacologic Activity: Venlafaxine/Desvenlafaxine
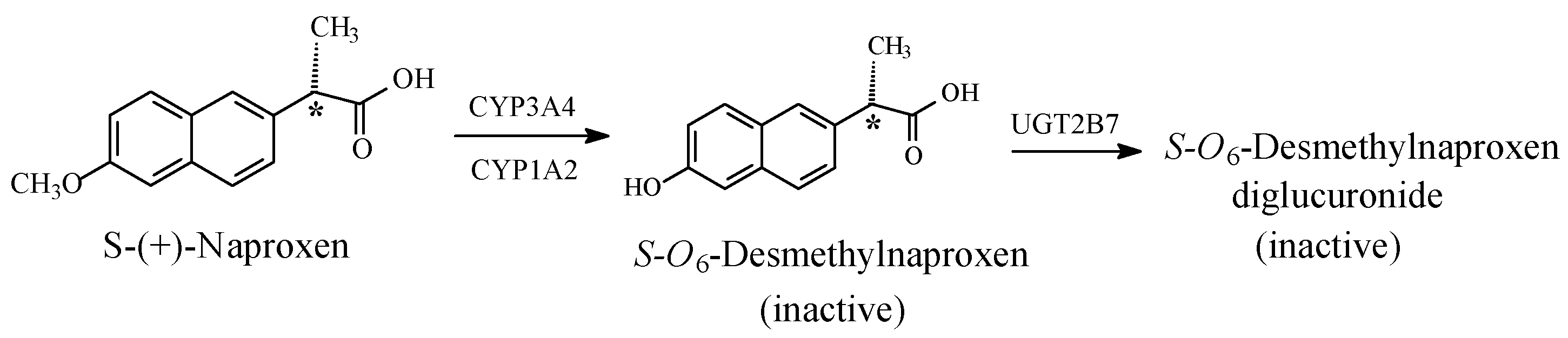
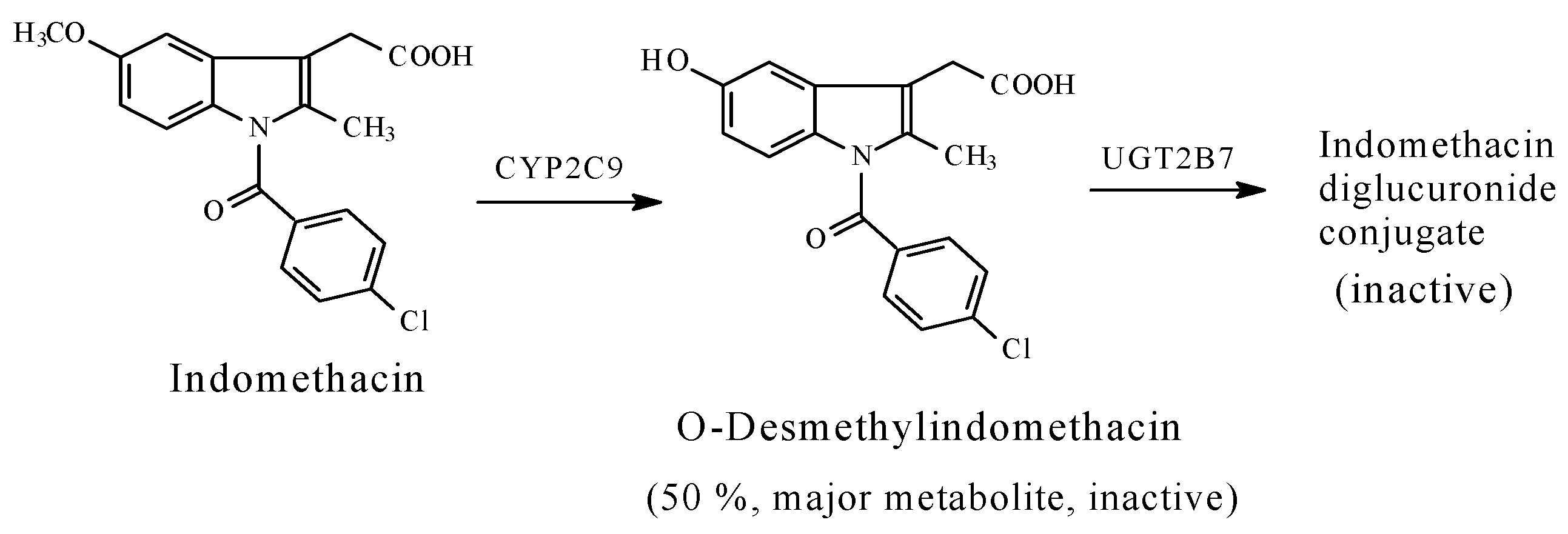
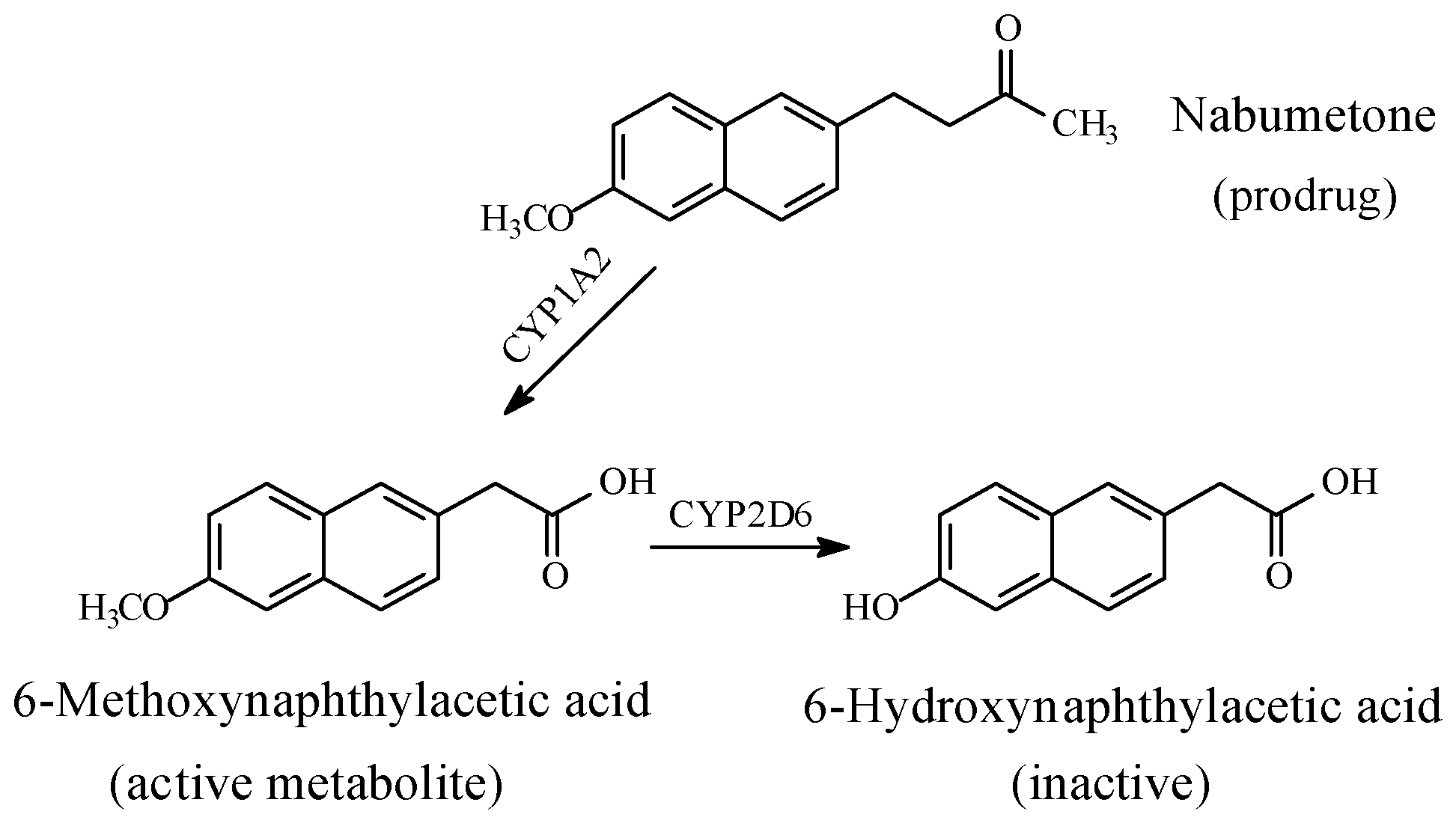
5.1.3. Metabolic O-Dealkylation of Aralkoxy Groups Resulting in the Attenuation or Loss of Pharmacologic Activity: NSAIDs (Naproxen, Indomethacin, and Nabumetone)
Stereochemistry of Arylalkanoic Acid NSAIDs
5.2. Overall Discussion
- (1)
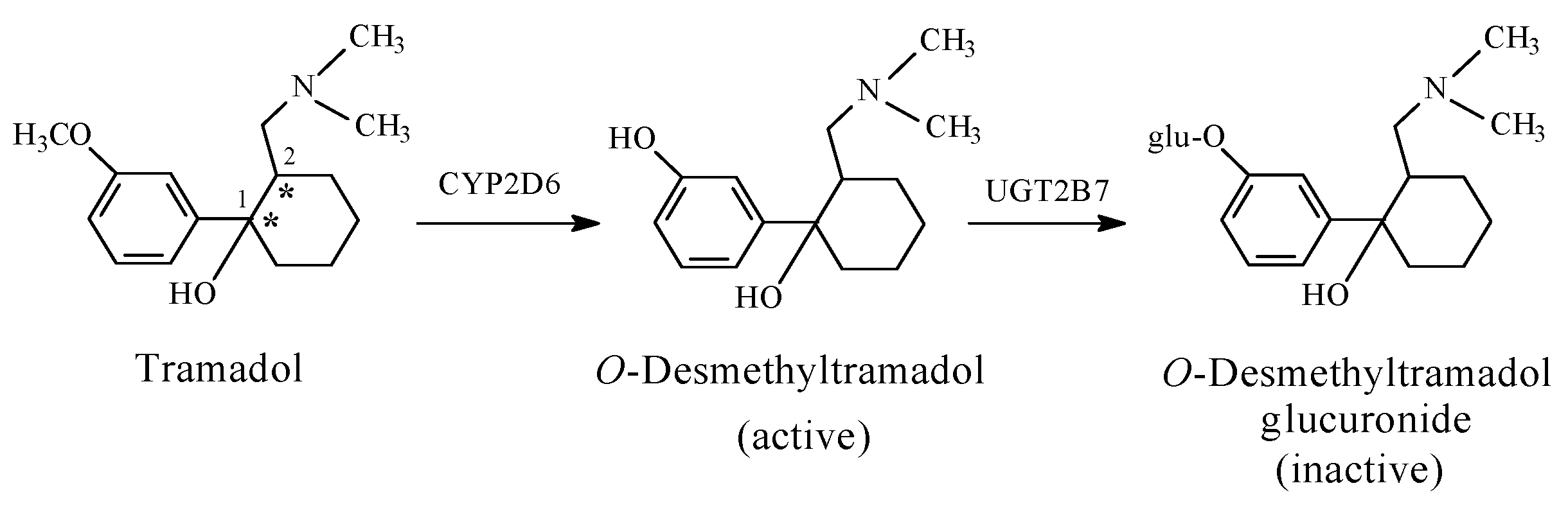
- Enhancement of activity is exhibited by the O-desmethyl morphinan opioids, O-desmethyltramadol and O-desethyl phenacetin;
- (2)
- Retention of activity is exhibited by O-desmethyl venlafaxine;
- (3)
- Significant attenuation or loss of activity is exhibited by O-desmethyl naproxen and O-desmethyl indomethacin.
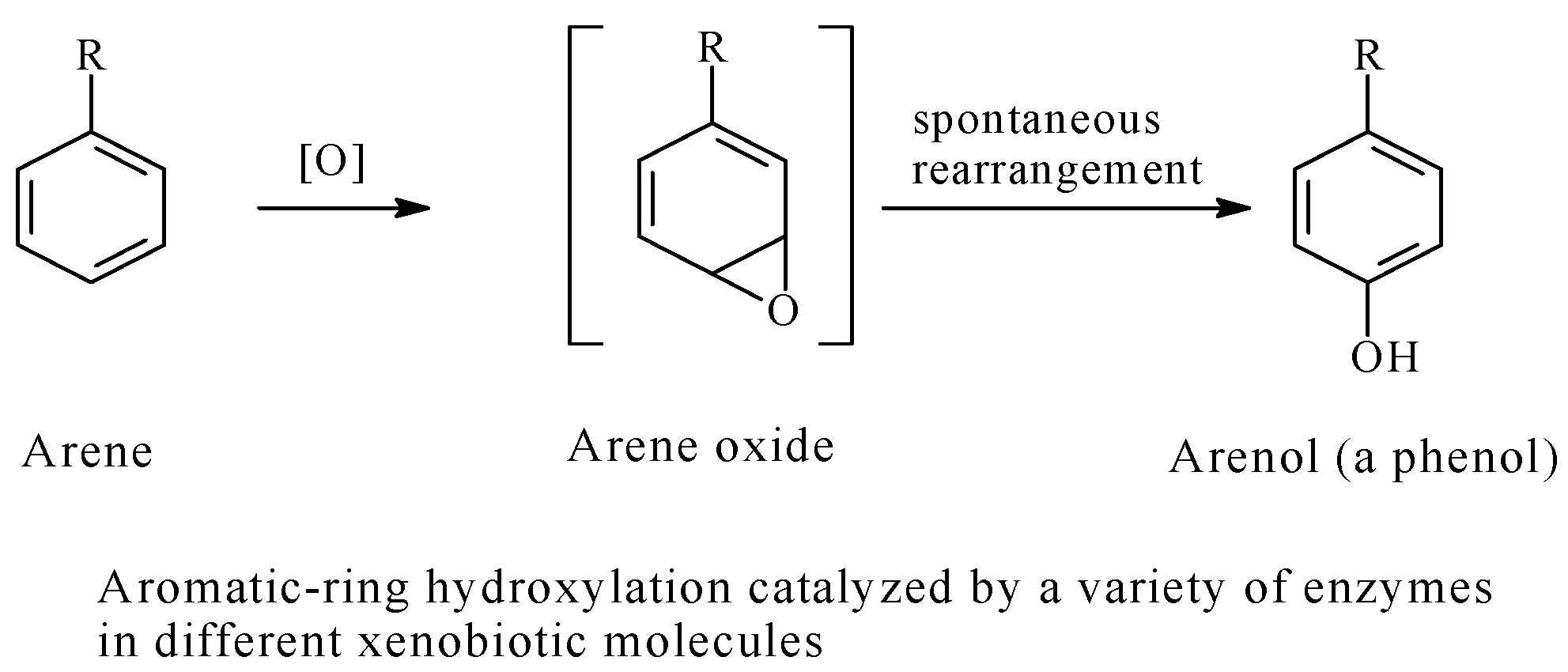
5.3. Arenolic Metabolites Resulting from Aromatic Ring Hydroxylation
- (a)
- The least substituted aromatic ring will be favorably oxidized, especially at the least hindered carbon atom;
- (b)
- The activated ring (i.e., the ring bearing an electron-donating group such as an alkyl) will be better oxidized;
- (c)
- Ring-deactivating groups (generally groups with negative inductive effects such as halo and nitro groups) discourage ring hydroxylation;
- (d)
- Being the farthest from steric effects in di-substituted benzene rings, the para position is the favored site of hydroxylation;
- (e)
- If two aromatic rings in a drug molecule have the same chemical environment, hydroxylation will occur in only one of them;
- (f)
- When the parent drug contains an aromatic hydroxy group, further metabolic hydroxylation is generally not favored even if there is more than one aromatic ring.
- Varying the chemical classes of the drugs;
- Varying the pharmacologic class of the drugs, and accordingly, the type of drug-site-of-action interaction involved (i.e., the mechanism of action of the class of drugs);
- Varying the aromatic characteristics in both the number of rings and chemical environment.
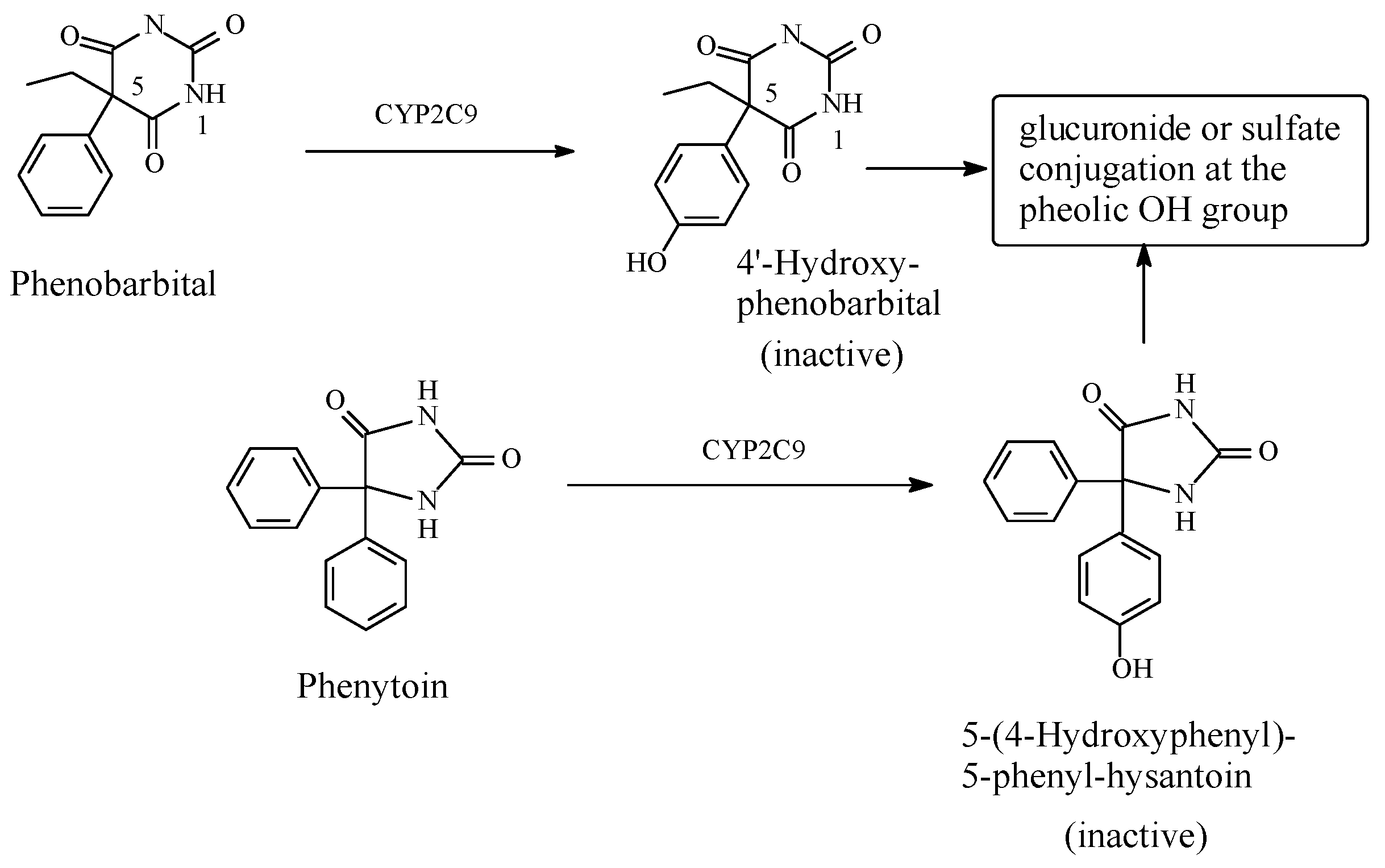
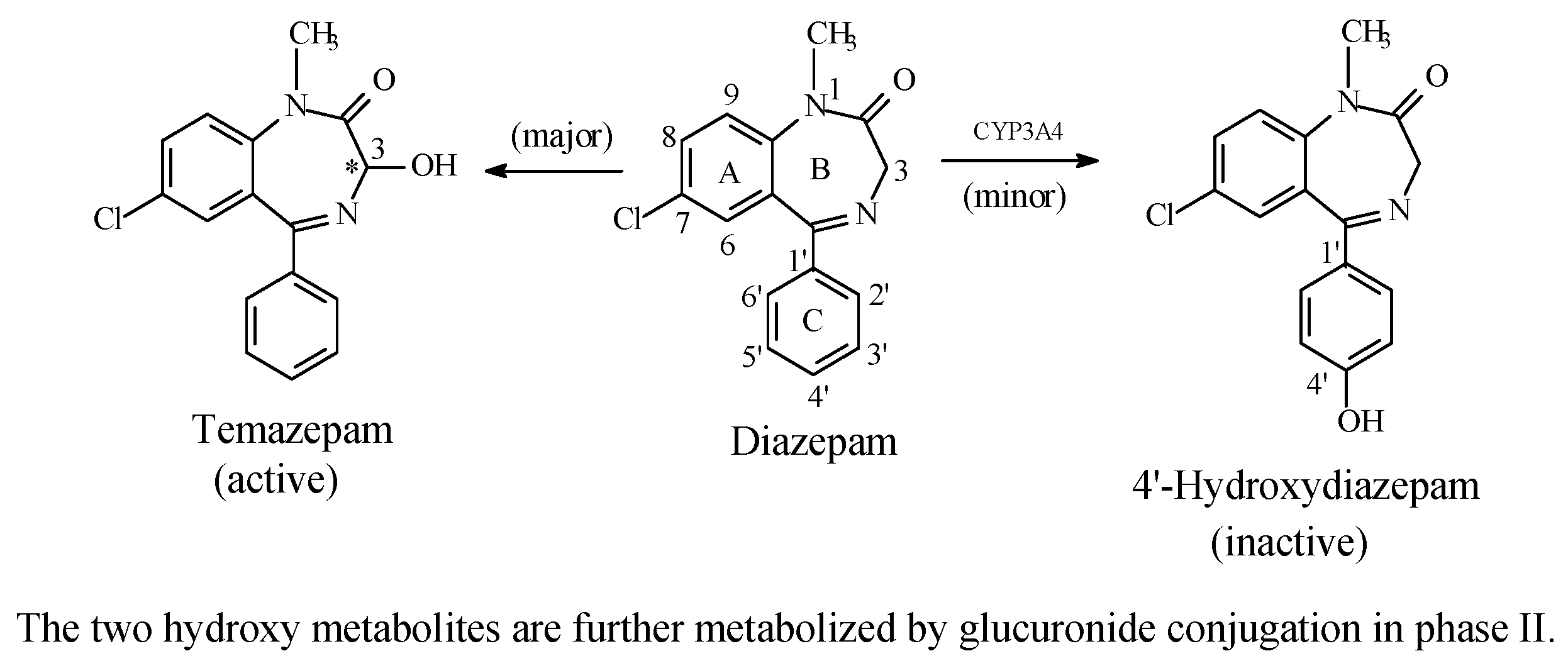
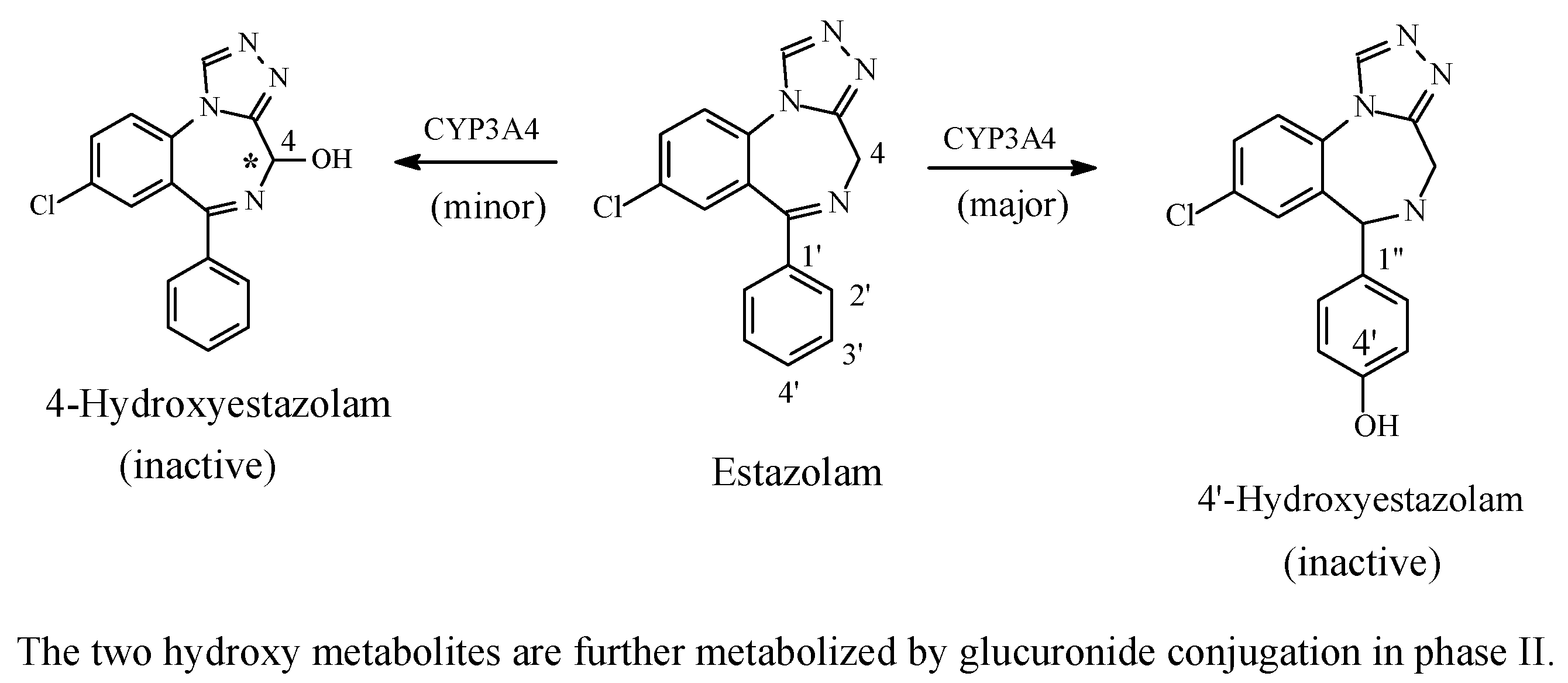
5.3.1. Metabolic Aromatic-Ring Hydroxylation Leading to Loss of Activity
Phenobarbital/Phenytoin
Benzodiazepines: Diazepam and Estazolam
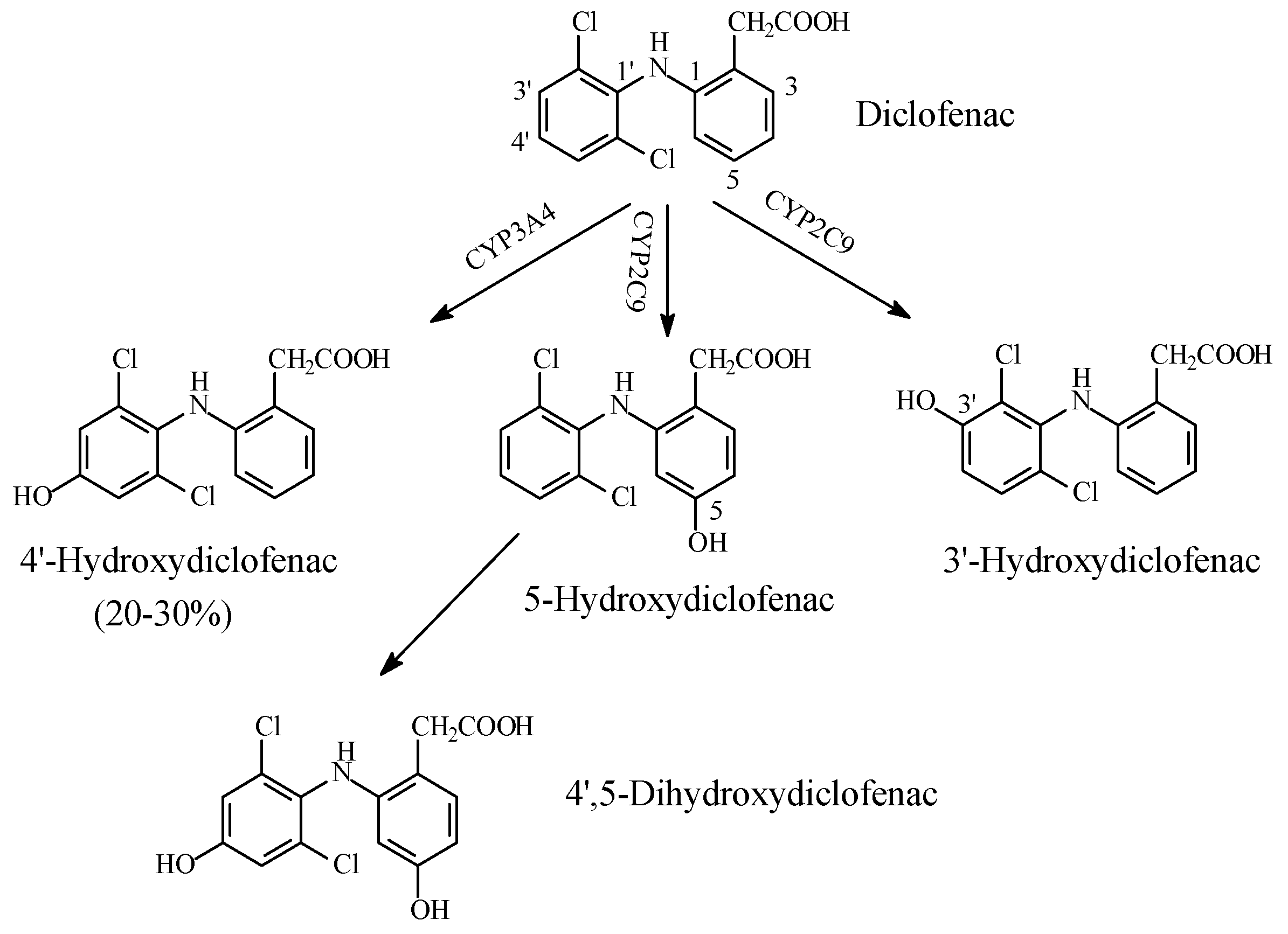
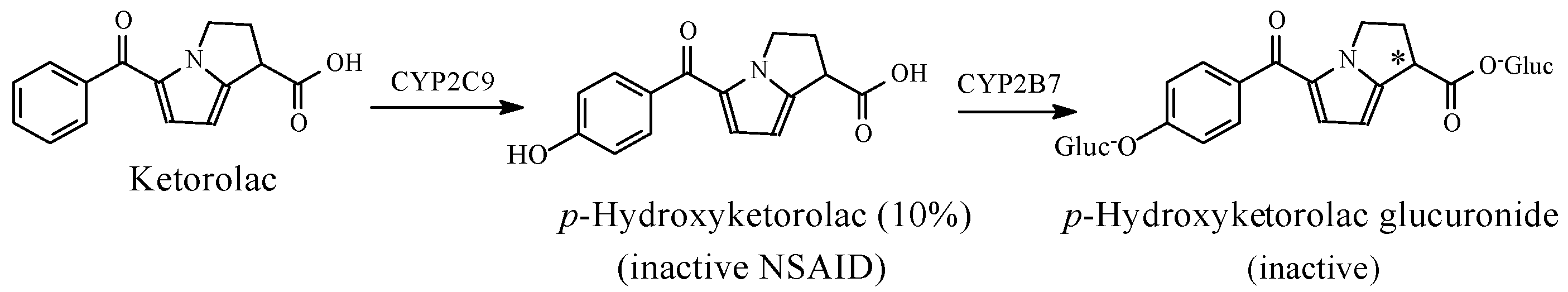
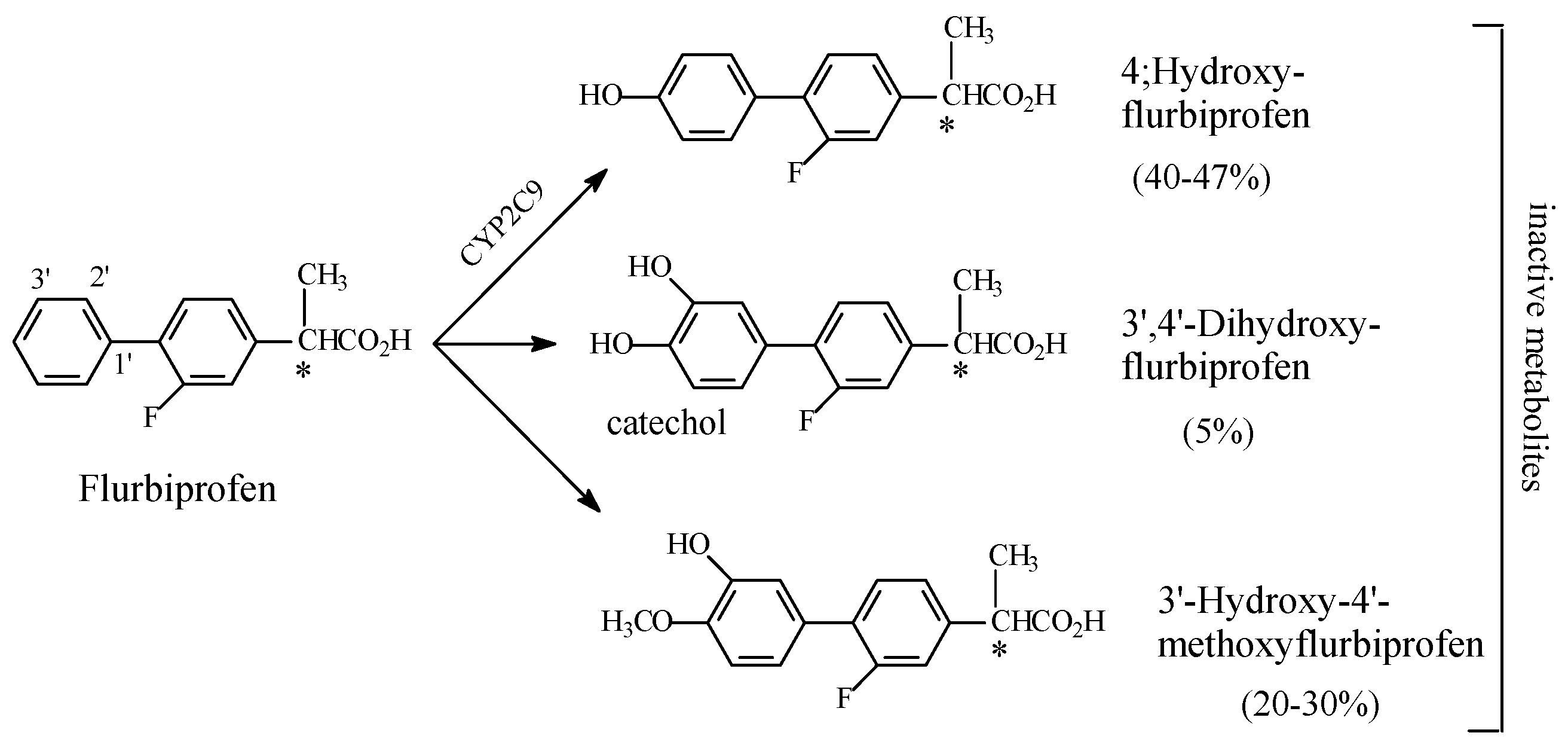
NSAIDs
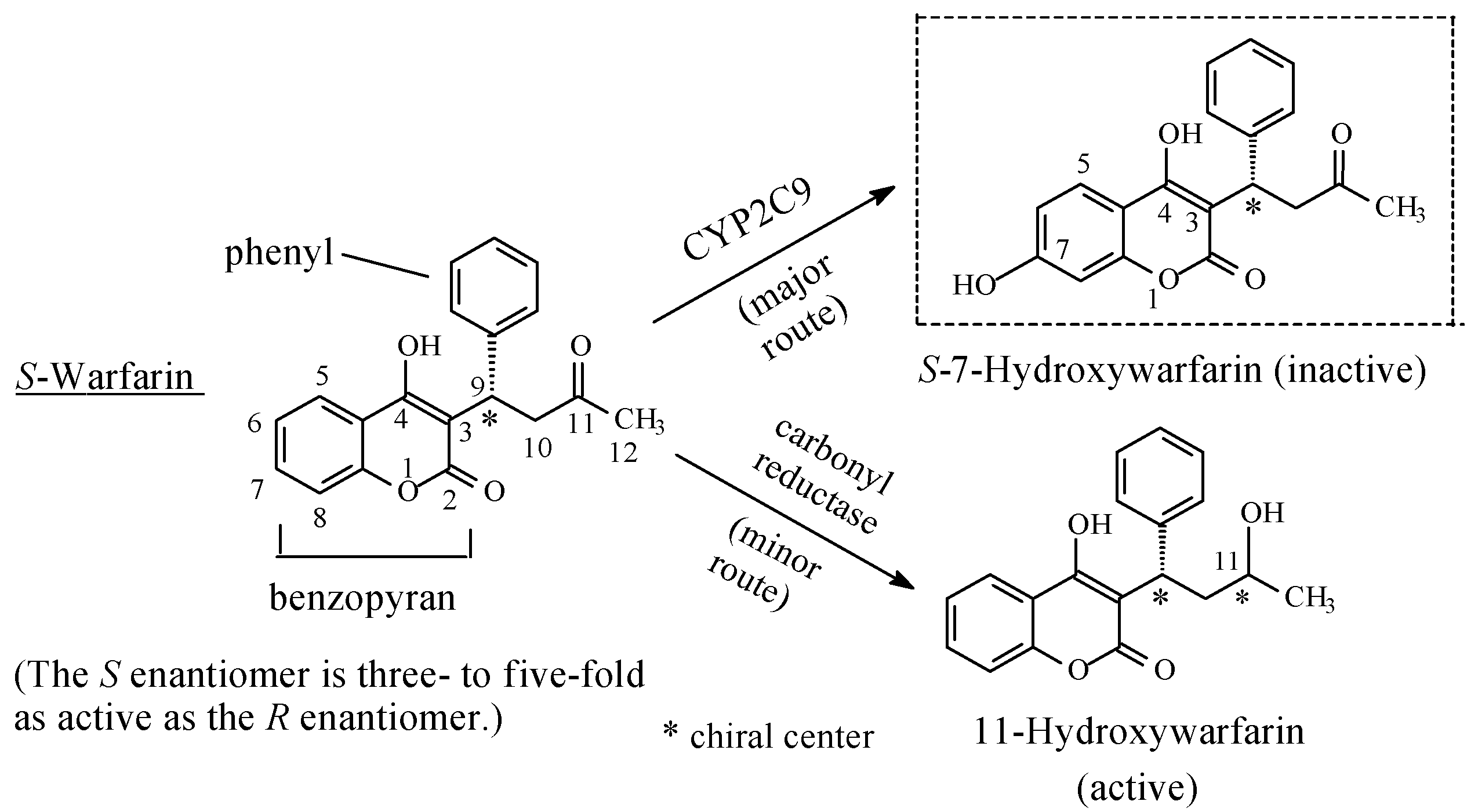
Miscellaneous: Warfarin
5.3.2. Metabolic Aromatic-Ring Hydroxylation Resulting in Attenuation of Pharmacologic Activity
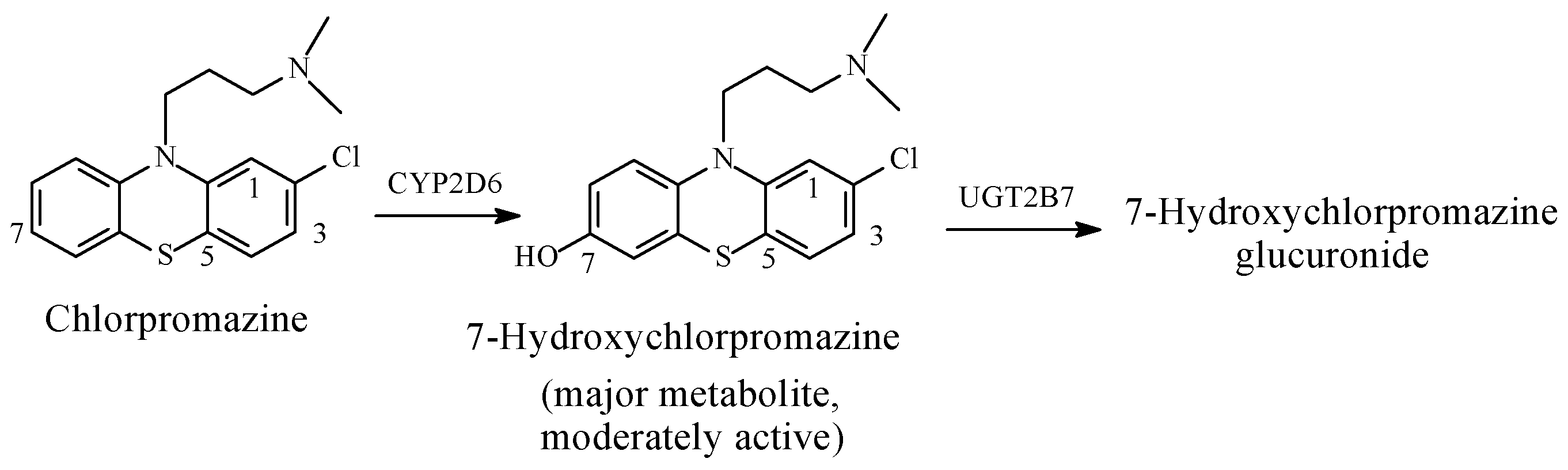
Chlorpromazine
5.3.3. Aromatic-Ring Hydroxylation Resulting in Parent-Drug Equiactive Metabolites
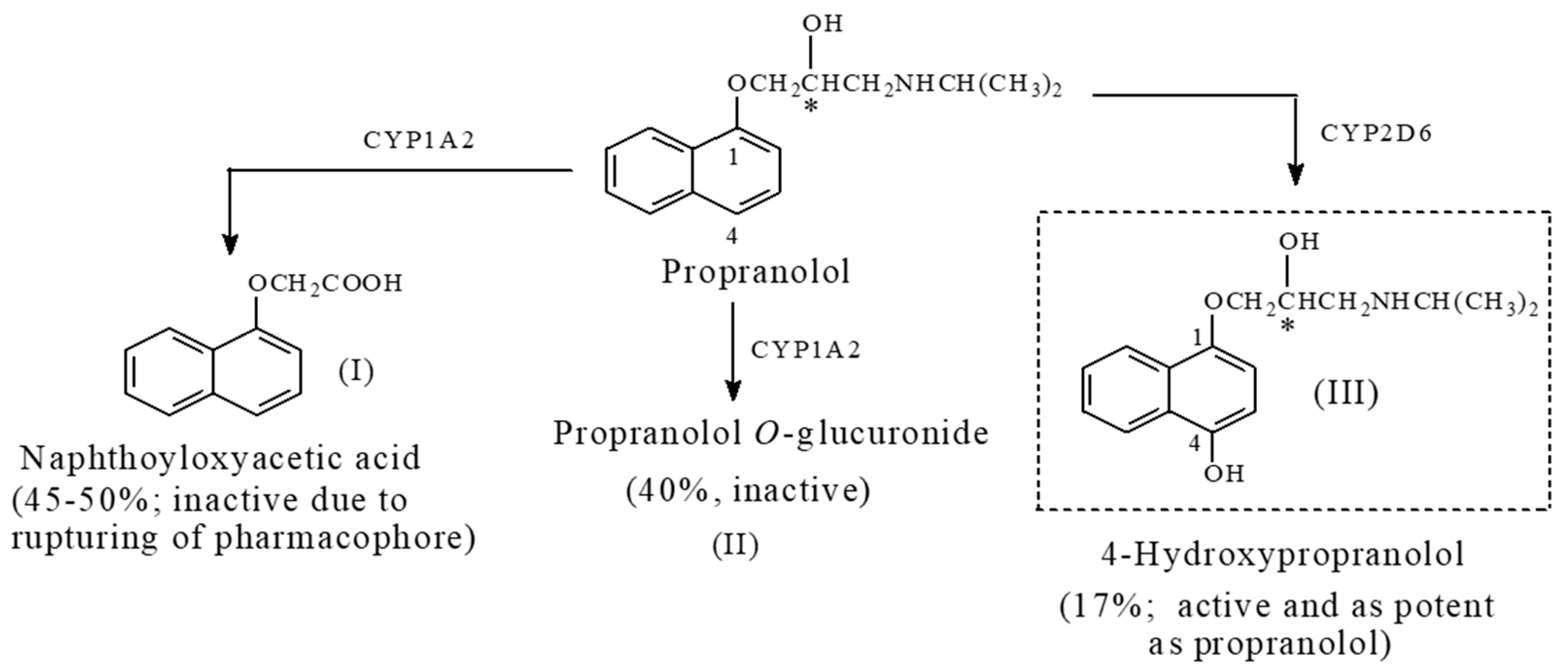
Propranolol
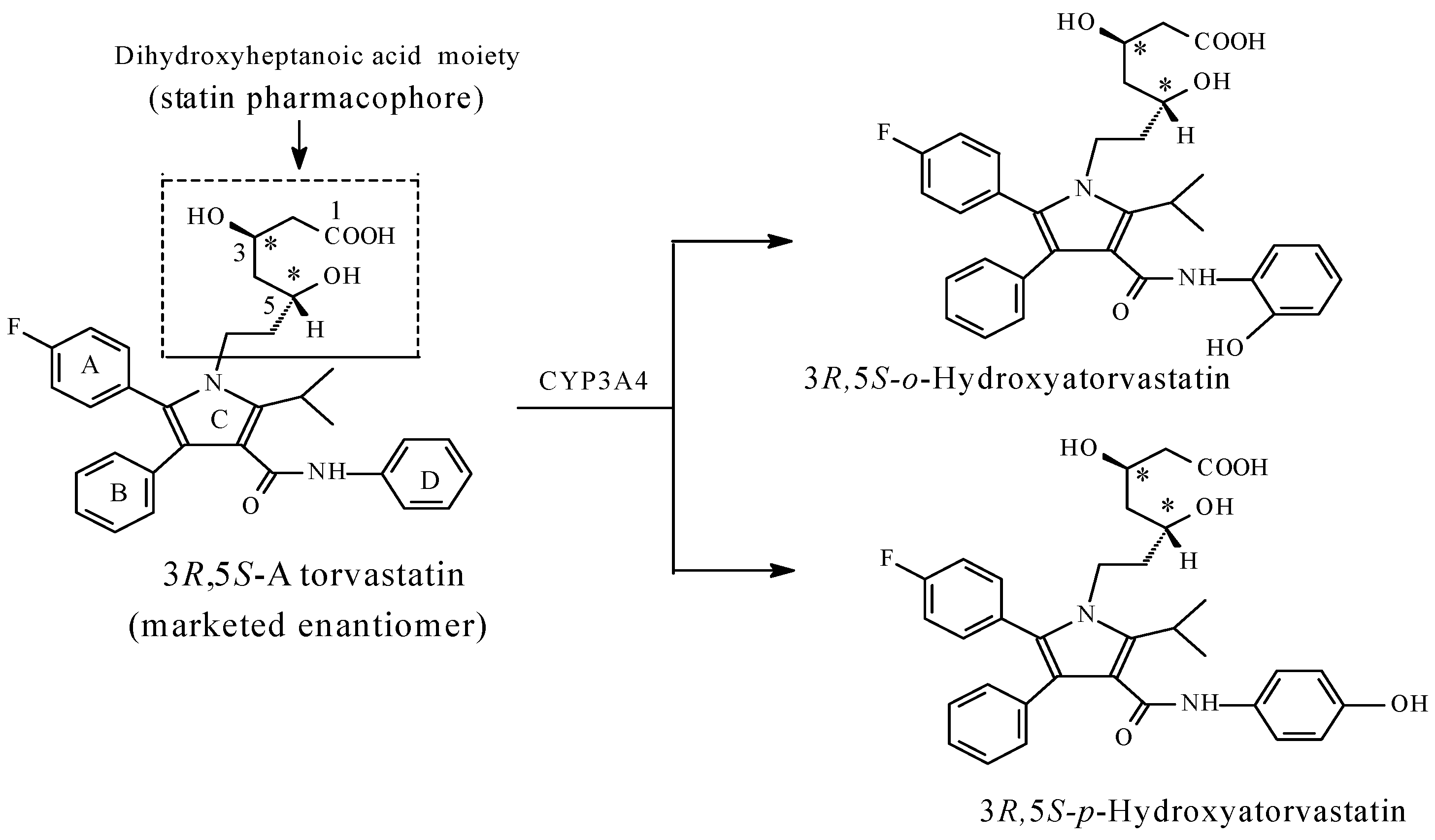
Atorvastatin
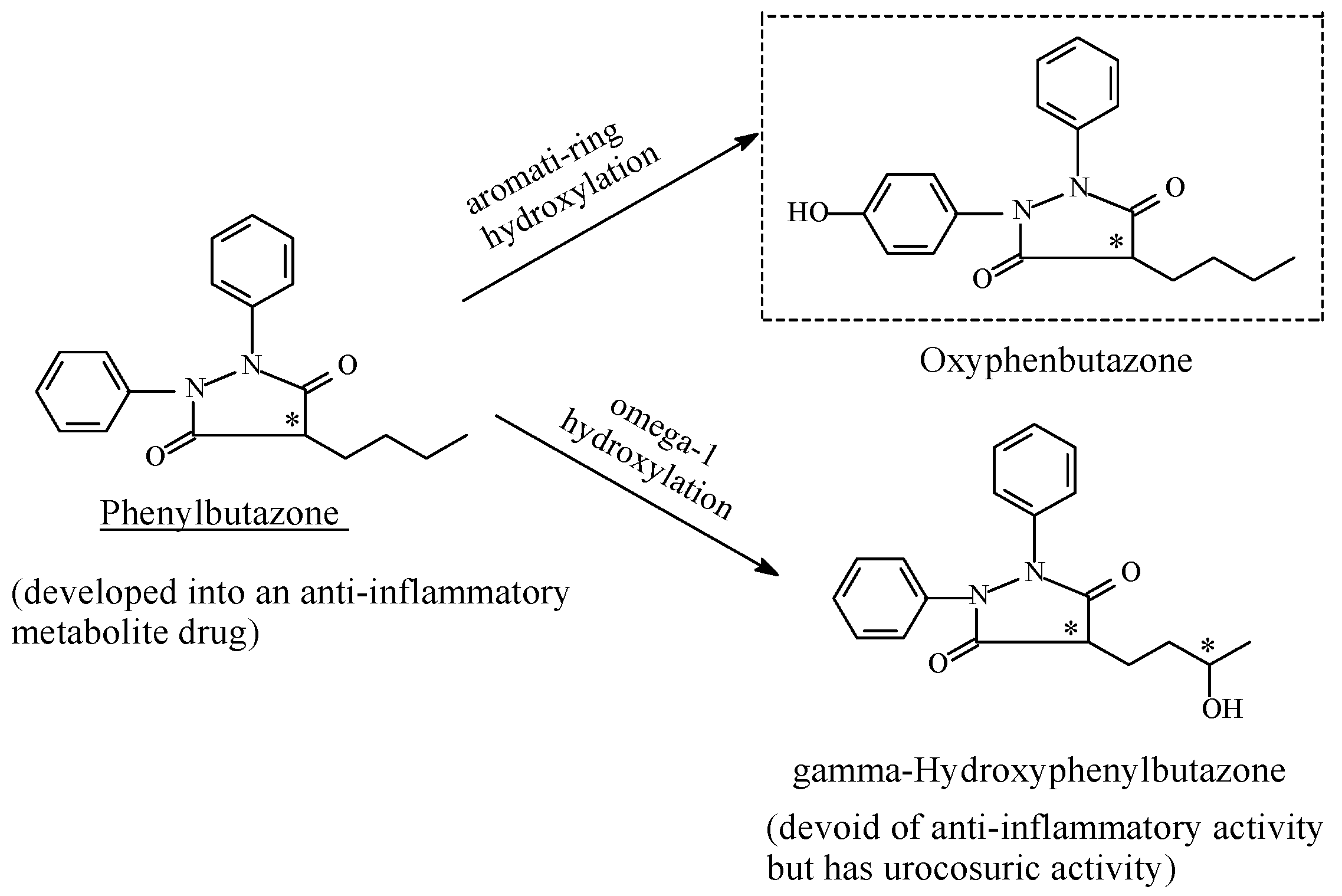
Phenylbutazone
5.4. Discussion of Metabolic Aromatic-Ring Hydroxylation
6. Racemic Drugs versus Enantiopure Drugs
Glucuronide Conjugation: Prevalence and Effect on Pharmacologic Activity
7. Conclusions
Funding
Conflicts of Interest
References
- The Importance of Stereochemistry in Drug Action and Disposition. Available online: https://www.researchgate.net/publication/21709119_The_Importance_of_Stereochemistry_in_DrugAction_and_Disposition (accessed on 25 July 2018).
- Williams, D.A. Drug Metabolism. In Foye’s Principles of Medicinal Chemistry, 7th ed.; Lemke, T.L., Williams, D.A., Roche, V.F., Zito, S.W., Eds.; Wolters Kluwer and Lippincott Williams and Wilkins: London, UK, 2013; p. 135. ISBN 978-1-60913-345-0. [Google Scholar]
- Cahill, C.M.; Taylor, A.M.W.; Cook, C.; Ong, E.; Morón, J.A.; Evans, C.J. Does the kappa opioid receptor system contribute to pain aversion? Front. Pharmmacol. 2014, 5, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.A.; Roche, V.E.; Roche, E.B. Central Analgesics, Opioid receptors. In Foye’s Principles of Medicinal Chemistry, 7th ed.; Lemke, T.L., Williams, D.A., Roche, V.F., Zito, S.W., Eds.; Wolters Kluwer and Lippincott Williams and Wilkins: London, UK, 2013; pp. 666–670. ISBN 978-1-60913-345-0. [Google Scholar]
- Satoh, M.; Minami, M. Molecular pharmacology of opioid receptors. Pharmacol. Ther. 1995, 68, 343–364. [Google Scholar] [CrossRef]
- The Opium Alkaloids. UNDOC, Creation Date 1 January 1953. pp. 13–14. Available online: https://www.unodc.org/unodc/en/data-and-analysis/bulletin/bulletin_1953-01-01_3_page005.html (accessed on 3 May 2018).
- Nogrady, T. Medicinal Chemistry: A Biochemical Approach, 2nd ed.; Oxford University Press: Oxford, UK, 1988; p. 316. ISBN 0-19-505368-0. [Google Scholar]
- Mikus, G.; Weiss, J. Influence of CYP2D6 on Opioid Kinetics, Metabolism and Response. Curr. Pharmacogenom. Pers. Med. 2005, 3, 43–52. [Google Scholar] [CrossRef]
- Codeine Metabolism Pathway. Pharmacokinetics. PharmGKB. Available online: http://www.pharmgkb.org/patheay/PA146123006 (accessed on 20 November 2017).
- Thompson, C.M.; Wojno, H.; Greiner, E.; May, E.L.; Rice, K.C.; Selley, D.E. Activation of G-Proteins by Morphine and Codeine Congeners: Insights to the Relevance of O- and N-Demethylated Metabolites at μ- and δ-Opioid Receptors. J. Pharmacol. Exp. Ther. 2004, 208, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Patrick, J.L. An Introduction to Medicinal Chemistry, 4th ed.; Oxford University Press: Oxford, UK, 2009; p. 634. ISBN 978-0-19-923447-9. [Google Scholar]
- Alder, T.K. The comparative potencies of codeine and its demethylated metabolites after intravenous injection in the mouse. J. Pharmacol. Exp. Ther. 1963, 140, 155–161. [Google Scholar]
- Findlay, J.W.A.; Jones, E.C.; Butz, R.F.; Welch, R.M. Plasma codeine and morphine concentrations after therapeutic oral doses of codeine-containing analgesics. Clin. Pharmacol. Ther. 1978, 24, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Vree, T.B.; van Dongen, R.T.; Koopman-Kimenai, P.M. Codeine analgesia is due to codeine-6-glucuronide, not morphine. Int. J. Clin. Pract. 2000, 54, 395–398. [Google Scholar] [PubMed]
- Vallejo, R.; Barkin, R.L.; Wang, V.C. Pharmacology of Opioids in the Treatment of Pain Syndromes. Pain Physician 2011, 14, E343–E360. [Google Scholar] [PubMed]
- Trescot, A.M.; Datta, S.; Lee, M. Opioid Pharmacology. Pain Physcian 2008, 11, S133–S153. [Google Scholar]
- Chen, Z.R.; Irvine, R.J.; Somogyi, A.A.; Bochner, F. Mu receptor binding of some commonly used opioids and their metabolites. Life Sci. 1991, 48, 2165–2171. [Google Scholar] [CrossRef]
- Howard, S.; Smith, M.D. Opioid Metabolism. Mayo Clin. Proc. 2009, 84, 613–624. [Google Scholar]
- Leppert, W. CYP2D6 in the Metabolism of Opioids for Mild to Moderate Pain. Pharmacology 2011, 87, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Ginsburg, D. Stereochemistry of Morphine, UNODC, Creation Date 1 January 1953. pp. 32–46. Available online: https://www.unodc.org/unodc/en/data-and-analysis/bulletin/bulletin_1953-01-01_4_page006.html (accessed on 3 May 2018).
- Beckett, A.H.; Anderson, P. The Determination of the Relative Configuration of Morphine, Levorphanol and Laevo-Phenazocine by Stereoselective Adsorbents. J. Pharm. Pharmacol. 1969, 12, 228T–236T. [Google Scholar] [CrossRef]
- Wong, B.Y.; Coulter, D.A.; Choi, D.W.; Prince, D.A. Dextrorphan and dextromethorphan, common antitussives, are antiepileptic and antagonize N-methyl-d-aspartate in brain slices. Neurosci. Lett. 1988, 85, 261–266. [Google Scholar] [CrossRef]
- Ground, S.; Sablotski, A. Clinical pharmacology of tramadol. Clin Pharmacol. Exp. Ther. 2004, 43, 879–923. [Google Scholar] [CrossRef]
- Williams, D.A. Central Analgesics. In Foye’s Principles of Medicinal Chemistry, 7th ed.; Lemke, T.L., Williams, D.A., Roche, V.F., Zito, S.W., Eds.; Wolters Kluwer and Lippincott Williams and Wilkins: London, UK, 2013; pp. 675–679. ISBN 978-1-60913-345-0. [Google Scholar]
- Mario, G. Tramadol Vs Tapentadol: A New Horizon in Pain Treatment? Am. J. Anim. Vet. Sci. 2012, 7, 7–11. [Google Scholar]
- Bergel, I.; Morrison, A.L. Pethidine ketobemidone. Q. Rev. Chem. Soc. 1948, 2, 349. [Google Scholar] [CrossRef]
- Beckett and Casy Model, Pharmacological Sciences How Drugs Act. Last Updated on 15 April 2017. Available online: https://www.pharmacologicalsciences.us/how-drugs-act/och3.html (accessed on 11 May 2017).
- Carrupt, P.A.; Testa, B.; Bechalany, A.; Tayar, N.; Descas, P.; Perrissoud, D. Morphine 6-glucuronide and morphine 3-glucuronide as molecular chameleons with unexpected lipophilicity. J. Med. Chem. 1991, 34, 1272–1275. [Google Scholar] [CrossRef] [PubMed]
- McCurdy, C.R. Development of opioid-receptor ligands. In Analogue-Based Drug Discovery; Fisher, J.C., Robin Ganellin, C.R., Eds.; WILEY-VCH Verlag GmbH and Co. KGaA: Weinheim, Germany, 2006; p. 263. ISBN 9783527608003. [Google Scholar]
- Paul, D.; Standifer, K.M.; Inturrisi, C.E.; Pasternak, G.W. Pharmacological characterization of morphine-6-glucuonide, a very potent morphine metabolite. J. Pharmacol. Exp. Ther. 1989, 251, 477–483. [Google Scholar] [PubMed]
- Vindenes, V.; Ripel, A.; Handal, M.; Boix, F.; Mørland, J. Interactions between morphine and the morphine-glucuronides measured by conditioned place preference and locomotor activity. Pharmacol. Biochem. Behav. 2009, 93, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Päivi, L.; Taina, S.; Olli, A.; Mika, K.; Katariina, V.; Moshe, F.; Risto, K. Glucuronidation of racemic O-desmethyltramadol, the active metabolite of tramadol. Eur. J. Pharm. Sci. 2010, 41, 523–530. [Google Scholar]
- Dixon, R.; Crews, T.; Inturrisi, C.; Foley, K. Levorphanol: Pharmacokinetics and steady state plasma concentrations in patients with pain. Res. Commun. Chem. Pathol. Pharmacol. 1983, 41, 3–17. [Google Scholar] [PubMed]
- Hubner, C.B.; Kornetsky, C. Heroin, 6-acetylmorphine and morphine effects on threshold for rewarding and aversive brain stimulation. J. Pharmacol. Exp. Ther. 1992, 260, 562–567. [Google Scholar] [PubMed]
- Inturrisi, C.E.; Schultz, M.; Shin, S.; Umans, J.G.; Angel, L.; Simon, E.J. Evidence from opiate binding studies that heroin acts through its metabolites. Life Sci. 1983, 1, 773–776. [Google Scholar] [CrossRef]
- Gudin, J.; Fudin, J.; Nalamachu, S. Levorphanol Use: Past, Present and Future. Postgrad. Med. 2016, 128, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Sahu, V.K.; Khan, A.K.R.; Singh, R.K.; Singh, P.P. Hydrophobic, Polar and Hydrogen Bonding Based Drug-Receptor Interactions of Tetrahydroimidazobenzodiazepinones. Am. J. Immunol. 2008, 4, 33–42. [Google Scholar] [CrossRef]
- Kalow, W.; Otton, S.V.; Kadar, D. Ethnic difference in drug metabolism: Debrisoquine 4-hydroxylation in Caucasians and Orientals. Can. J. Physiol. Pharmacol. 1980, 58, 1142–1144. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.I.; Radford, H. CYP2D6 Polymorphisms and Response to Codeine and Tramadol. Analg. Resusc. Curr. Res. 2016, 5, 1. [Google Scholar] [CrossRef]
- Yiannakopoulou, E. Pharmacogenomics and Opioid Analgesics: Clinical Implications. Int. J. Genom. 2015, 2015, 368979. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.; Hardy, J.R. The lipophilic opioids: Fentanil, sufetanil and remifentanil. In Opioids in Cancer Pain, 2nd ed.; Davis, M.P., Glare, P.A., Hardy, J., Quigley, C., Eds.; Oxford University Press: Oxford, UK, 2013; Chapter 11; ISBN 9780199236640. [Google Scholar]
- Prescott, L.F. Kinetics and metabolism of paracetamol and phenacetin. Br. J. Clin. Pharmacol. 1980, 2, 291S–298S. [Google Scholar] [CrossRef]
- Brodie, B.B.; Axelrod, J. The fate of acetanilide in man. J. Pharmacol. Exp. Ther. 1948, 94, 29–38. [Google Scholar] [PubMed]
- Aronoff, D.M.; Oates, J.A.; Boutaud, O. New insights into the mechanism of action of acetaminophen: Its clinical pharmacologic characteristics reflect its inhibition of the two prostaglandin H2 synthases. Clin. Pharmacol. Ther. 2006, 79, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Kingback, M. Genetic Influence on Enantiomeric Drug Disposition: Focus on Venlafaxine and Citalopram. Linköping University Medical Dissertations No. 1264, Linköping, Sweden, 2011; ISBN 978-1-7393-057-4. Available online: https://www.diva-portal.org/smash/get/diva2:458660/FULLTEXT01.pdf (accessed on 30 July 2018).
- Howell, S.R.; Husbands, G.E.; Scatina, J.A.; Sisenwine, S.F. Metabolic disposition of 14C-venlafaxine in mouse, rat, dog, rhesus monkey and man. Xenobiotica 1993, 23, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Preskornl, P.; Silman, H.; Isler, J.A.; Burczynski, M.E.; Ahmed, S.; Paul, J.; Nichols, A.I. Comparison of the pharmacokinetics of venlafaxine extended release and desvenlafaxine in extensive and poor cytochrome P450 2D6 metabolizers. J. Clin. Psychopharmacol. 2009, 29, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Sangkuhl, K.; Stingl, J.C.; Turpeinene, M.; Altman, R.B.; Kleina, T.E. Venlafaxine metabolic pathway. Pharmacogenet. Genom. 2014, 24, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Colvard, M.D. Key differences between Venlafaxine XR and Desvenlafaxine: An analysis of pharmacokinetic and clinical data. Ment. Health Clin. 2014, 4, 35–39. [Google Scholar] [CrossRef]
- Williams, D.A. Antidepressant Drugs. In Foye’s Principles of Medicinal Chemistry, 7th ed.; Lemke, T.L., Williams, D.A., Roche, V.F., Zito, S.W., Eds.; Wolters Kluwer and Lippincott Williams and Wilkins: London, UK, 2013; pp. 608–609. ISBN 978-1-60913-345-0. [Google Scholar]
- Cashman, J.N. The mechanism of action of NSAIDS in analgesia. Drug 1996, 52, 13–23. [Google Scholar] [CrossRef]
- Vance, J.R.; Botting, R.M. Mechanism of action of nonsteroidal anti-inflammatory drugs. Am. J. Med. 1998, 104, 2S–8S. [Google Scholar] [CrossRef]
- Vance, J.R. Introduction: Mechanism of action of NSAIDS. Br. J. Pharmacol. 1996, 35, 1–3. [Google Scholar]
- DeRuiter, J. Nonsteroidal Anti-Inflammatory Drugs (NSAIDS). Available online: www.auburn.edu/~deruija/nsaids_2002.pdf (accessed on 15 December 2017).
- Borne, R.; Levi, M.; Wilson, N. Nonsteroidal Anti-Inflammatory Drugs (metabolism of naproxen). In Foye’s Principles of Medicinal Chemistry, 7th ed.; Lemke, T.L., Williams, D.A., Roche, V.F., Zito, S.W., Eds.; Wolters Kluwer and Lippincott Williams and Wilkins: London, UK, 2013; p. 1014. ISBN 978-1-60913-345-0. [Google Scholar]
- Duggan, D.E.; Hogan, A.F.; Kuran, K.C.; McMahon, F.G. The metabolism of indomethacin in man. J. Pharmacol. Exp. Ther. 1972, 181, 563–575. [Google Scholar] [PubMed]
- Turpeinen, M.; Hofmann, U.; Klein, K.; Scwab, M.; Zanger, U.M. A predominate role of CYP1A2 for the metabolism of nabumetone to the active metabolite, 6-methoxy-2-naphthylacetic acid, in human liver microsomes. Drug Metab. Dispos. 2009, 37, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Wechter, W.J. Drug chirality: On the mechanism of R-aryl propionic acid class NSAIDs. Epimerization in humans and the clinical implications for the use of racemates. J. Clin. Pharmacol. 1996, 34, 1036–1042. [Google Scholar] [CrossRef]
- Duggan, K.C.; Walters, M.J.; Musee, J.; Harp, J.M.; Kiefer, J.R.; Oates, J.A.; Marnett, L.J. Molecular basis for cyclooxygenase inhibition by the nonsteroidal anti-inflammatory drug naproxen. J. Biol. Chem. 2010, 285, 34950–34959. [Google Scholar] [CrossRef] [PubMed]
- Helleberg, L. Clinical Pharmacokinetics of indomethacin. Clin. Pharmacokinet. 1981, 6, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Fura, A. Role of pharmacologically active metabolites in drug discovery and development. Drug Discov. Today 2006, 11, 133–142. [Google Scholar] [CrossRef]
- Morice, C.; Vermuth, C.G. Mechanism of Metabolic Aromatic-Ring Hydroxylation: The Practice of Medicinal Chemistry, 2nd ed.; Vermuth, C.G., Ed.; Academic Press (Elsevier): San Diego, CA, USA, 2003; pp. 243–263. ISBN 9780080497778. [Google Scholar]
- Sweety, M. Phase I Metabolism—Oxidative Reactions—Oxidation of Aromatic Compounds. Medicinal Chemistry Notes February 15, 2014 PharmaXchange Info. Available online: www://pharmaxchange.info/press/2014/02/phase-i-metabolism-oxidative-reactions-oxidation-of-aromatic-compounds/ (accessed on 15 March 2017).
- Dasgupta, A. 4-Hydroxyphenobarbital. In A Health Educators Guide to Understanding Drugs of Abuse Testing; Jones and Bartkett Publishers: Huston, TX, USA, 2010; p. 19. ISBN 978-0763765897. [Google Scholar]
- Lowry, W.T.; Garriott, J.C. Phenobarbital. In Forensic Toxicology: Controlled Substances and Dangerous Drugs, 17th ed.; Prenum Press: New York, NY, USA, 2012; pp. 347–348. ISBN 978-1-4684-3444-6. [Google Scholar]
- Butler, T.C. The metabolic hydroxylation of phenobarbital. J. Pharmacol. Exp. Ther. 1956, 116, 326–336. [Google Scholar] [PubMed]
- Veronese, M.E.; Doecke, C.J.; Mackenzie, P.I.; McManus, M.E.; Miners, J.O.; Ree, D.L.P.; Gasser, R.; Meyer, U.A.; Birkett, D.J. Site-directed mutation studies of human liver cytochrome P-450 isoenzymes in the CYP2 subfamily. Biochem. J. 1993, 289, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Moniri, N.H. Sedatives-Hypnotics. In Foye’s Principles of Medicinal Chemistry, 7th ed.; Lemke, T.L., Williams, D.A., Roche, V.F., Zito, S.W., Eds.; Wolters Kluwer and Lippincott Williams and Wilkins: London, UK, 2013; pp. 491–492. ISBN 978-1-60913-345-0. [Google Scholar]
- Rudolph, U.; Crestani, F.; Benke, D. Benzodiazepine actions mediated by specific gamma-aminobutyric acid(A) receptor subtypes. Nature 1999, 401, 796–800. [Google Scholar] [CrossRef] [PubMed]
- Kaye, A.M.; Bueno, F.R.; Kaye, A.D. Benzodiazepine Pharmacology and Central Nervous System–Mediated Effects. Ochsner J. 2013, 13, 214–223. [Google Scholar]
- Moniri, N.H. Pharmacokinetics and Metabolism of Benzodiazepines. In Foye’s Principles of Medicinal Chemistry, 7th ed.; Lemke, T.L., Williams, D.A., Roche, V.F., Zito, S.W., Eds.; Wolters Kluwer and Lippincott Williams and Wilkins: London, UK, 2013; pp. 494–495. ISBN 978-1-60913-345-0. [Google Scholar]
- Chwartz, M.A.; Koechlin, B.A.; Postma, E.; Palmer, S.; Krol, G. Metabolism of diazepam in rat, dog, and man. J. Pharmacol. Exp. Ther. 1965, 149, 423–435. [Google Scholar]
- Saito, K.; Kim, S.-H.; Sakai, N.; Ishizuka, M.; Fujita, S. Polymorphism in Diazepam Metabolism in Wistar Rats. J. Pharm. Sci. 2004, 93, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Miura, M.; Otani, K.; Ohkubo, T. Identification of human cytochrome P450 enzyme involved in the formation of 4-hydroyestazolam from estazolam. Xenobiotica 2005, 35, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Miura, M.; Otani, K.; Ohkubo, T. Identification of human cytochrome P450 enzymes involved in the formation of 4-hydroxyestazolam from estazolam. Xenobiotica 2004, 35, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Harrold, M.W.; Zavod, R.M. Basic Concepts in Medicinal Chemistry; eBook; Publisher: Amer Soc of Health-System Pharm; 2013; pp. 26–61. ISBN 978-1-58528-266-1. Available online: www.abebooks.co.uk/PassionFor Books (accessed on 13 August 2018).
- Tang, W.I. The metabolism of diclofenac—Enzymology and toxicology perspectives. Curr. Drug Metab. 2003, 4, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Borne, R.; Levi, M.; Wilson, N. Nonsteroidal Anti-Inflammatory Drugs. In Foye’s Principles of Medicinal Chemistry, 7th ed.; Lemke, T.L., Williams, D.A., Roche, V.F., Zito, S.W., Eds.; Wolters Kluwer and Lippincott Williams and Wilkins: London, UK, 2013; pp. 1009–1010. ISBN 978-1-60913-345-0. [Google Scholar]
- CHEBI:76227-(R)-Ketorolac. Available online: http://www.ebi.ac.uk/chebi/searchId.do;jsessionid=C2865A23BBB5839E5BD9FB55909ED68B?chebiId=CHEBI:76227 (accessed on 28 July 2018).
- Kauffman, R.E.; Lieh-Lai, M.W.; Uy, H.G.; Aravind, M.K. Enantiomer-selective pharmacokinetics and metabolism of ketorolac in children. Clin. Pharmacol. Ther. 1999, 65, 382–388. [Google Scholar] [CrossRef]
- Risdall, P.C.; Adams, S.S.; Crampton, E.L.; Marchant, B. The Disposition and Metabolism of Flurbiprofen in Several Species Including Man. Xenobiotica 1978, 8, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Kiss, L.E.; Soares-da-Silva, P. Medicinal Chemistry of Catechol O-Methyltransferase (COMT) Inhibitors and Their Therapeutic Utility. J. Med. Chem. 2014, 57, 8692–8717. [Google Scholar] [CrossRef] [PubMed]
- Akamine, Y.; Uno, T. Warfarin Enantiomers Pharmacokinetics by CYP2C19. Available online: www.intechopen.com (accessed on 30 May 2017).
- Rettie, A.E.; Korzekwa, K.L.; Kunze, K.L.; Lawrence, R.F.; Eddy, A.C.; Aoyama, H.; Gelboin, H.V.; Gonzalez, F.J.; Trager, W.F. Hydroxylation of warfarin by human cDNA expressed cytochrome P-450. A role forP4502C9 in the etiology of (S)-warfarin drug intoxications. Chem. Res. Toxicol. 1992, 5, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Kaminsky, L.S.; de Morais, S.M.F.; Faletto, D.A.; Dunbar, D.A.; Goldstein, J. Correlation of human P4502C substrate specificities with primary structure: Warfarin as a probe. Mol. Pharmacol. 1993, 43, 234–239. [Google Scholar] [PubMed]
- Kaminsky, L.S.; Zhang, Z.Y. Human P450 metabolism of warfarin. Pharmacol. Ther. 1997, 73, 67–74. [Google Scholar] [CrossRef]
- Trager, W.F.; Lewis, R.J. Mass spectral analysis in the identification of human metabolites of warfarin. J. Med. Chem. 1970, 13, 1196–1204. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Trager, W.F. Warfarin metabolism in man: identification of metabolites in urine. J. Clin. Investig. 1970, 49, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Beckett, A.H.; Beaven, M.A.; Robinson, A.F. Metabolism of chlorpromazine in humans. Biochem. Pharmacol. 1963, 12, 779–794. [Google Scholar] [CrossRef]
- Mackay, A.V.P.; Healy, A.F.; Baker, J. The relationship of plasma chlorpromazine to its 7-hydroxy and sulfoxide metabolites in a large population of chronic schizophrenics. Br. J. Clin. Pharmacol. 1974, 1, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, D.; Reid, E. Determination of Chlorpromazine and Its Sulfoxide and 7-Hydroxy Metabolites by Ion-Pair High Pressure Liquid Chromatography. Anal. Lett. 1981, 14, 1785–1805. [Google Scholar] [CrossRef]
- Goldenberg, H.; Fishman, V. Metabolism of chlorpromazine: V. Confirmation of position 7 as the major site of hydroxylation. Biochem. Biophys. Res. Commun. 1964, 14, 404–407. [Google Scholar] [CrossRef]
- Chen, B.M.; Herschel, M.; Aoba, A. Neuroleptic, antimuscarinic and antiadrenergic activity of chlorpromazine, thioridazine and their metabolites. Psychiatry Res. 1979, 1, 199–208. [Google Scholar] [CrossRef]
- Mehvar, R.; Brocks, R. Stereospecific Pharmacokinetics and Pharmacodynamics of Beta-Adrenergic Blockers in Humans. J. Pharm. Pharm. Sci. 2001, 4, 185–200. [Google Scholar] [PubMed]
- Paterson, J.W.; Conolly, M.F.; Dollery, C.T. The Pharmacodynamics and Metabolism of Propranolol in Man. Pharmacol. Clin. 1970, 2, 127–133. [Google Scholar] [CrossRef]
- Stoschitzky, K.; Egginger, G.; Zernig, G.; Klein, W.; Lindner, W. Stereoselective features of (R)- and (S)-atenolol: clincal pharmacological, pharmacokinetic, and radioligand binding studies. Chirality 1993, 5, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Harrold, M. Antihyperlipoproteinemics and Inhibitors of Cholesterol Biosynthesis. In Foye’s Principles of Medicinal Chemistry, 7th ed.; Lemke, T.L., Williams, D.A., Roche, V.F., Zito, S.W., Eds.; Wolters Kluwer and Lippincott Williams and Wilkins: London, UK, 2013; pp. 826–830. ISBN 978-1-60913-345-0. [Google Scholar]
- Drugbank: Atorvastatin (DB01076). Available online: https://www.drugbank.ca/drugs/DB01076 (accessed on 10 March 2018).
- Chemical Structures of Enantiopure Forms of Statins. Available online: https://www.researchgate.net/figure/Chemical-structures-of-enantiopure-forms-of-statins-Four-individual-enantiomers-of_fig7_281779178 (accessed on 26 July 2018).
- Lomas, S.; McCoy, M.; Behr, H. Enantiomeric Purity Analysis of the Drug Product Atorvastatin on Lux® Amylose-1 According to the United States Pharmacopeia (USP) Monograph 2263; Jacob, M., Ed.; Phenomenex, Inc.: Torrance, CA, USA; Phenomenex, Ltd.: Aschaffenburg, Germany, 2013. [Google Scholar]
- Portoghese, P.; Sultana, M.; Nagase, H.; Takemon, A.E. Application of the message-address concept in the design of highly potent and selective. Delta. Opioid receptor antagonists. J. Med. Chem. 1988, 31, 281–282. [Google Scholar] [PubMed]
- Yü, T.F.; Burns, J.J.; Paton, B.C.; Brodie, B.B. Phenylbutazone Metabolites: Antirheumatic, Sodium-Retaining andUricosuric Effects in Man. J. Pharmacol. Exp. Ther. 1958, 123, 63–69. [Google Scholar] [PubMed]
- Aarbakke, J.; Bakke, O.M.; Milde, E.J.; Davies, D.S. Disposition and oxidative metabolism of phenylbutazone in man. Eur. J. Clin. Pharmacol. 1977, 11, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Woolfe, G.; Deesi, L.; Uchleke, H.; Archer, S.; Harris, L.S.; Whitehouse, M.W.; Montgomery, J.A.Y. Progress in Drug Research; Jucker, E., Ed.; Springer: Basel, Switzerland; Stuttgart, Germany, 1965; Volume 8, p. 381. ISBN 978-3-0348-7133-4. [Google Scholar]
- Sriram, D.; Yogeeswar, P. Medicinal Chemistry, 2nd ed.; Pearson Publisher: Delhi, India, 2007; p. 361. ISBN 8131731448. [Google Scholar]
- Bagwell, E.E.; Williams, E.M. Further studies regarding the structure activity relationships of beta-adrenoceptor antagonists. Br. J. Pharmacol. 1973, 58, 686–692. [Google Scholar] [CrossRef]
- Hedberg, A.; Carlsso, E.; Fellenius, E.; Lundgren, B. Cardiostimulatory effects of prenalterol, a beta-1 adrenoceptor partial agonist, in vivo and in vitro. Naunyn Schmiedelberg’s Arch. Pharmacol. 1982, 318, 185–191. [Google Scholar] [CrossRef]
- Volpe, A.D. Application of Method Suitability for Drug Permeability Classification. APPS J. 2010, 12, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Maestri, E.; Maltoni, S.; Marata, A.M. Racemic Drugs and Enantiomers: Identifying the Really Useful Innovations. Available online: https://www.researchgate.net/publication/282879049_Racemic_drugs_and_enantiomers_identifying_the_really_useful_innovations (accessed on 1 August 2018).
- Hodgson, E. Introduction to Biotansformation (Metabolism). In Pesticide Biotransformation and Disposition; Hodgson, E., Ed.; Academic Press (Elsevier): Cambridge, MA, USA, 2012; Chapter 11; pp. 53–72. ISBN 9780123854810. [Google Scholar]





























© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Haj, B.M.; Ahmed, S.B.M.; Garawi, M.A.; Ali, H.S. Linking Aromatic Hydroxy Metabolic Functionalization of Drug Molecules to Structure and Pharmacologic Activity. Molecules 2018, 23, 2119. https://doi.org/10.3390/molecules23092119
El-Haj BM, Ahmed SBM, Garawi MA, Ali HS. Linking Aromatic Hydroxy Metabolic Functionalization of Drug Molecules to Structure and Pharmacologic Activity. Molecules. 2018; 23(9):2119. https://doi.org/10.3390/molecules23092119
Chicago/Turabian StyleEl-Haj, Babiker M., Samrein B. M. Ahmed, Mousa A. Garawi, and Heyam S. Ali. 2018. "Linking Aromatic Hydroxy Metabolic Functionalization of Drug Molecules to Structure and Pharmacologic Activity" Molecules 23, no. 9: 2119. https://doi.org/10.3390/molecules23092119
APA StyleEl-Haj, B. M., Ahmed, S. B. M., Garawi, M. A., & Ali, H. S. (2018). Linking Aromatic Hydroxy Metabolic Functionalization of Drug Molecules to Structure and Pharmacologic Activity. Molecules, 23(9), 2119. https://doi.org/10.3390/molecules23092119