
Kinetic Characterization of Novel HIV-1 Entry Inhibitors: Discovery of a Relationship between Off-Rate and Potency


Abstract
Highlights:
- The HIV-1 entry process represents an extremely attractive but underexploited therapeutic intervention point.
- Novel compounds with HIV-1 entry inhibitory activity were designed via field-based computational methods, synthesized and characterized biologically.
- A surface plasmon resonance (SPR) interaction assay was established for the first time for this class of antivirals, allowing assessment of affinity and kinetic parameters.
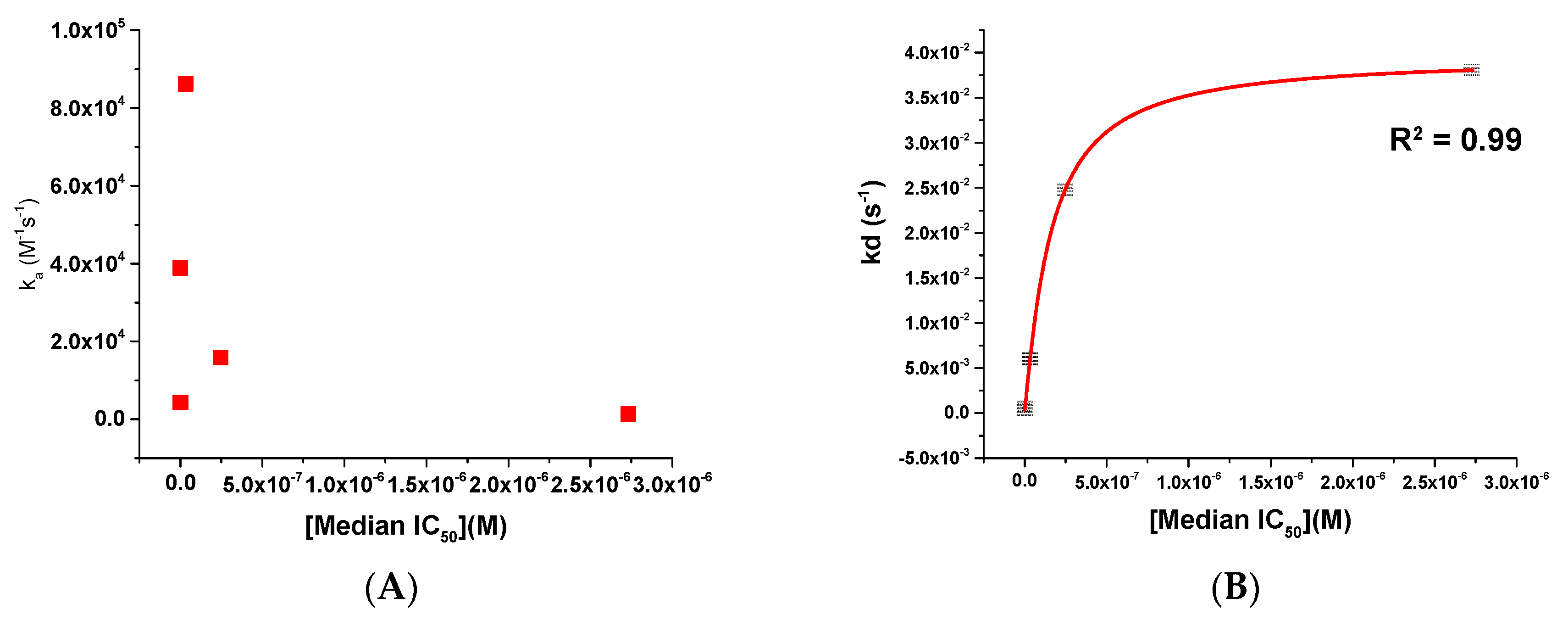
- The potency of a given entry inhibitor could be correlated with its dissociation rate parameter (kd) and its degree of electrostatic complementarity (EC) with its target, the Env complex.
1. Introduction
2. Results and Discussion
2.1. Design of Compounds SC16, SC18, and SC39
2.2. Potency Determination of Compounds SC11, SC16, SC18, and SC39 against Subtype B Env Pseudoviruses
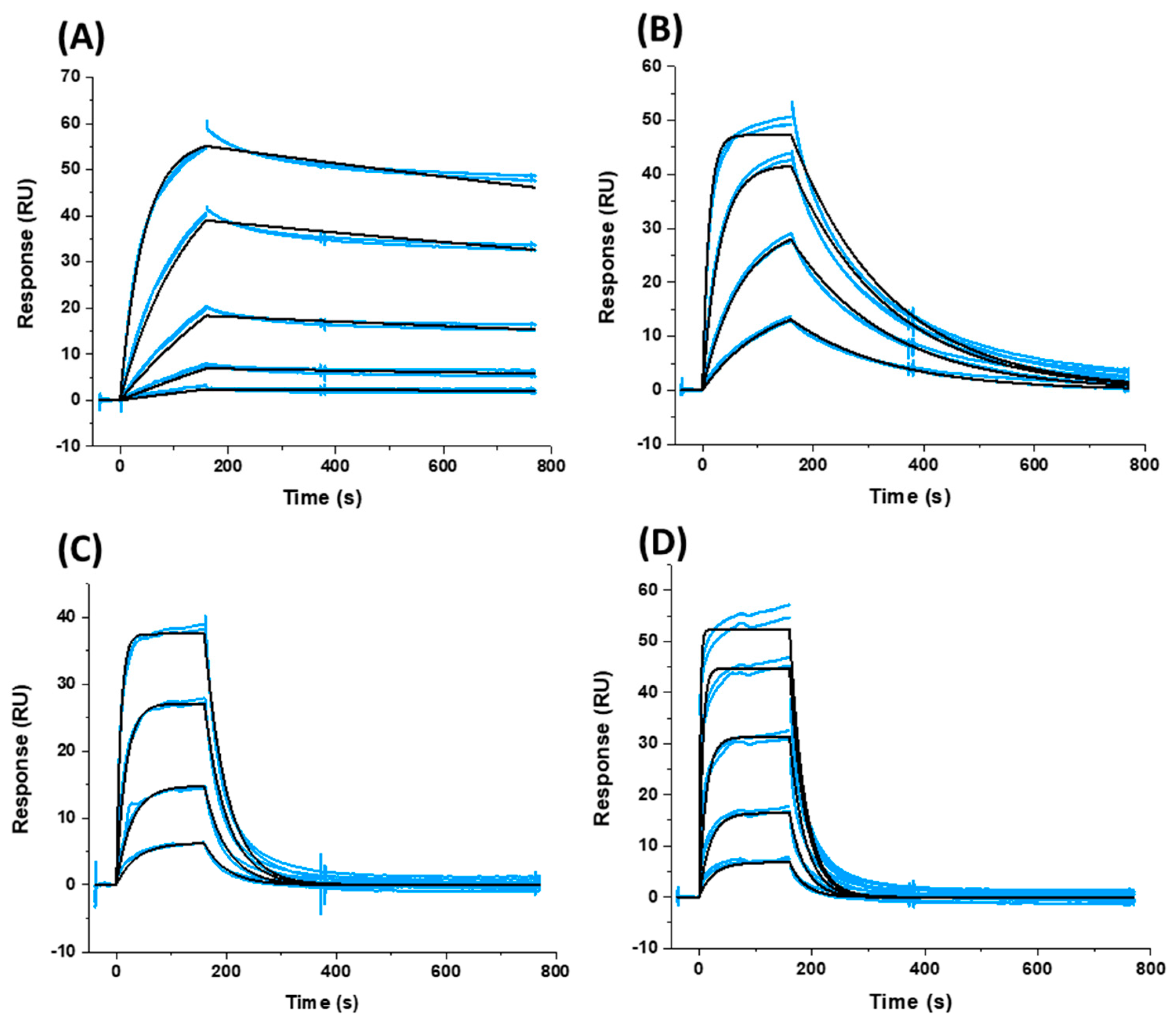
2.3. Affinity and Kinetic Characterization of SC11, SC16, SC18, and SC39 for Soluble Recombinant Env Trimer
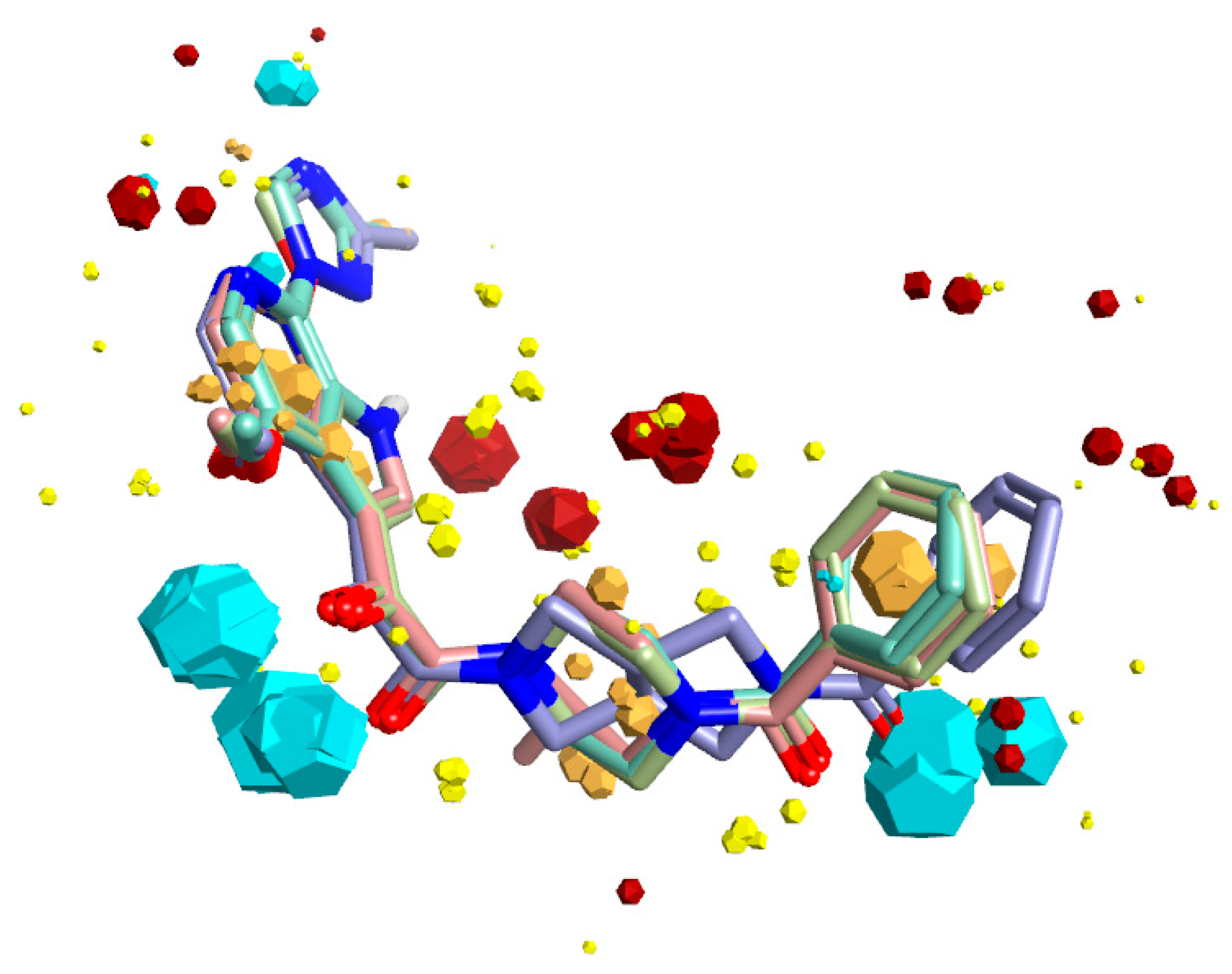
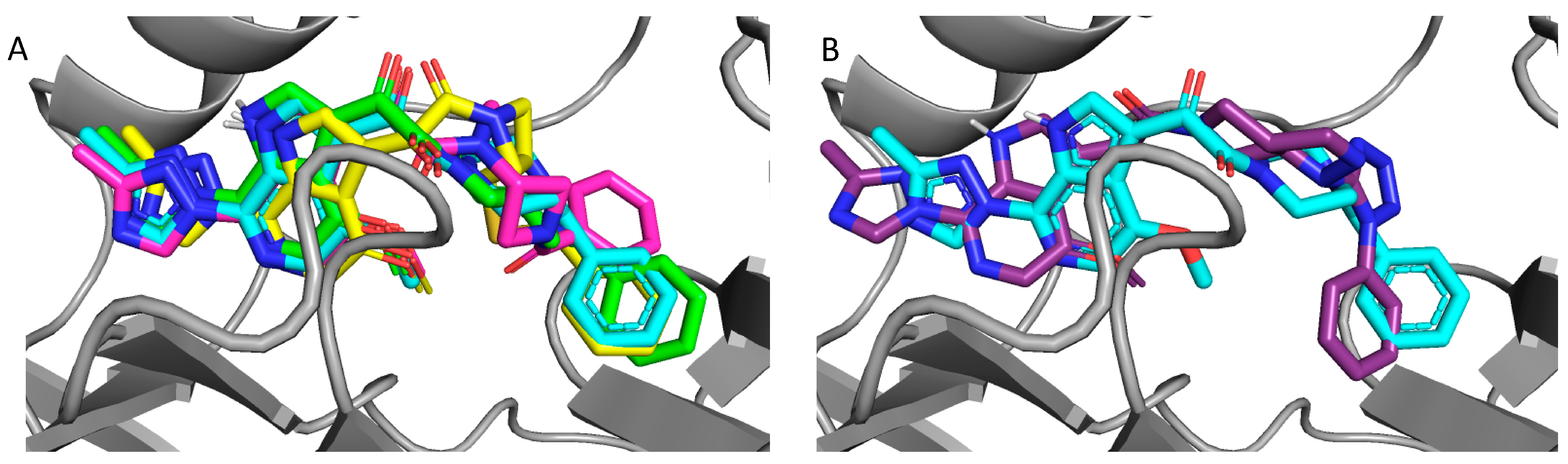
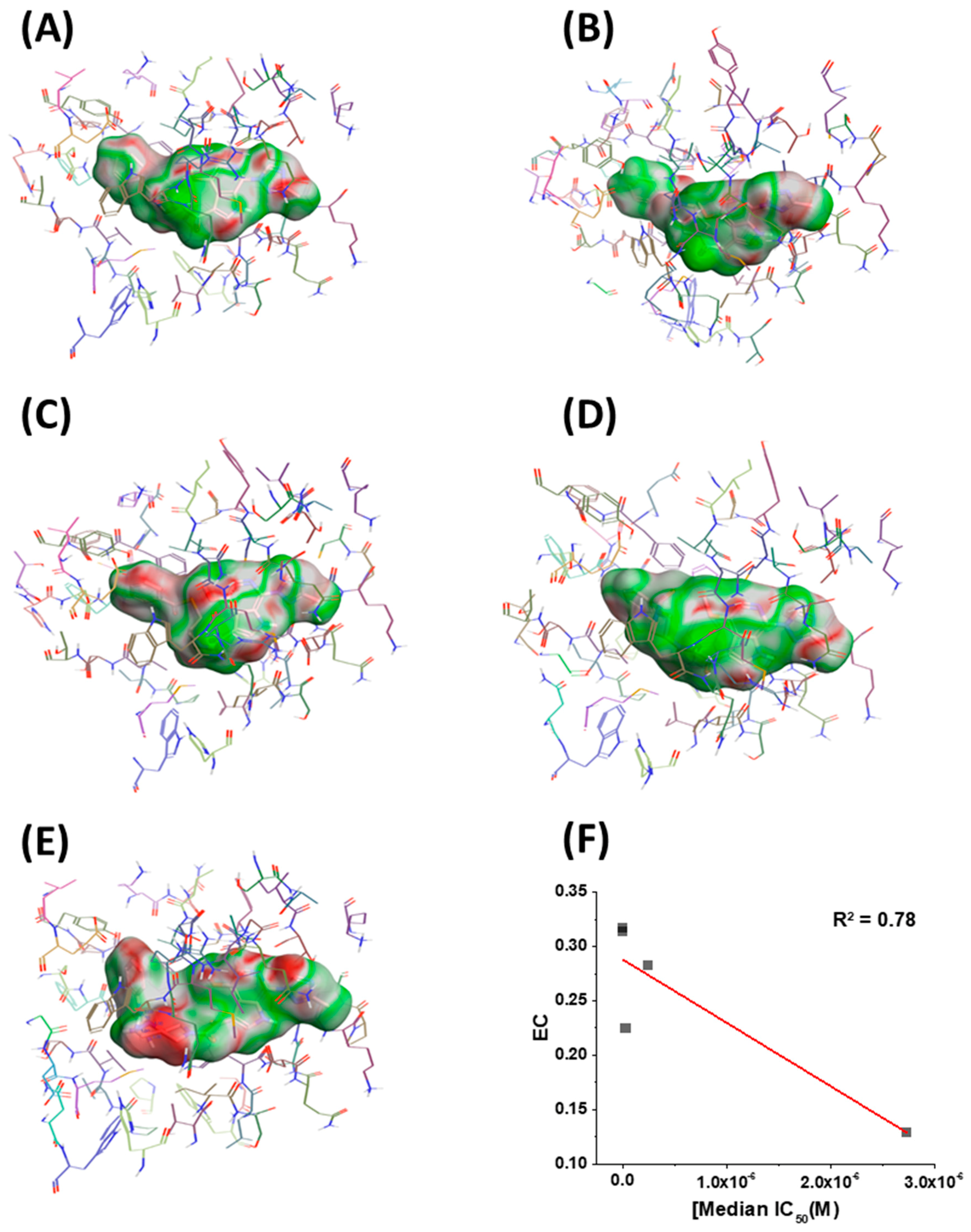
2.4. Molecular Modeling and Computational Analysis of Compounds of SC11, SC16, SC18, and SC39
3. Material and Methods
3.1. Compounds
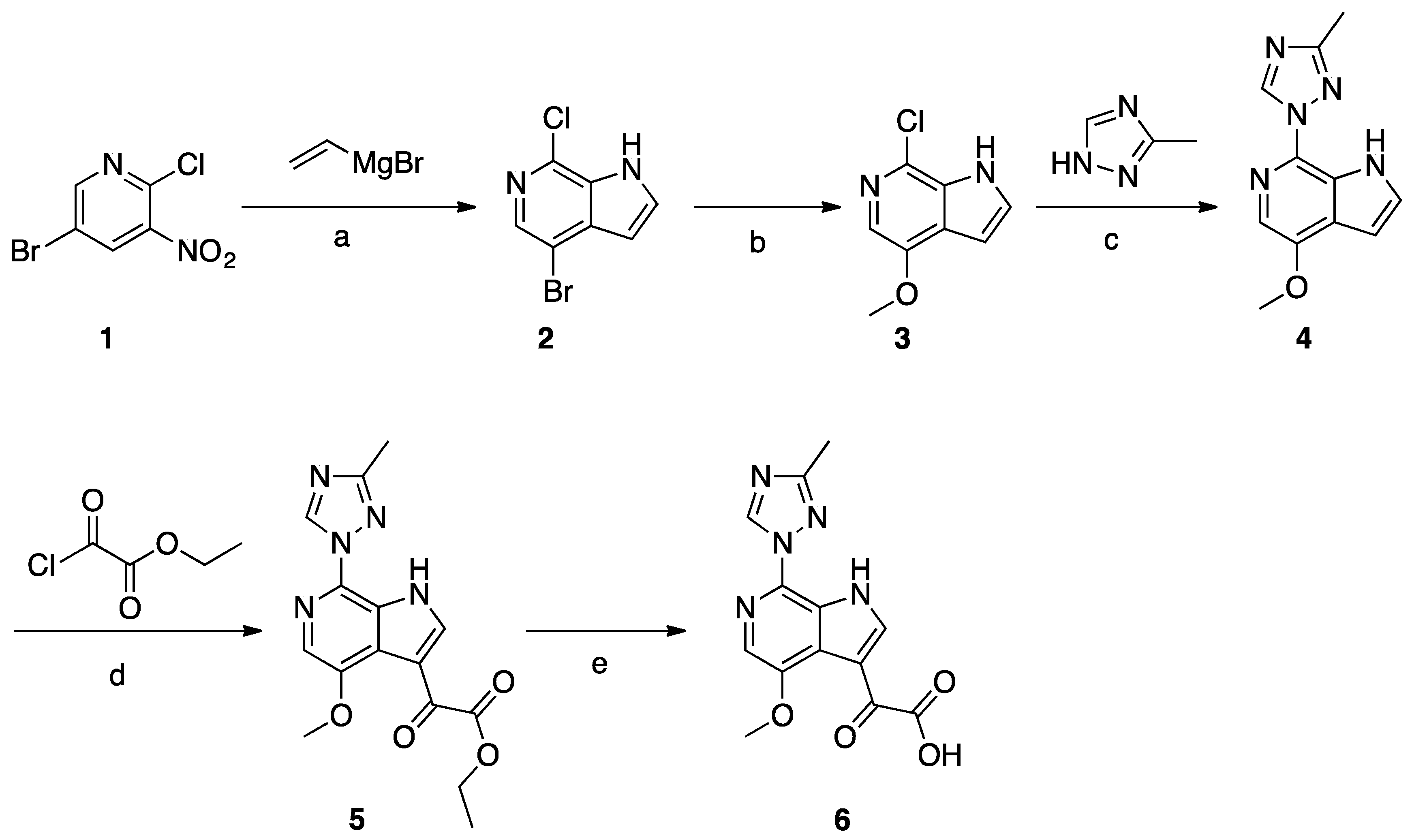
Synthesis of Common Intermediate 6 (Scheme 1)
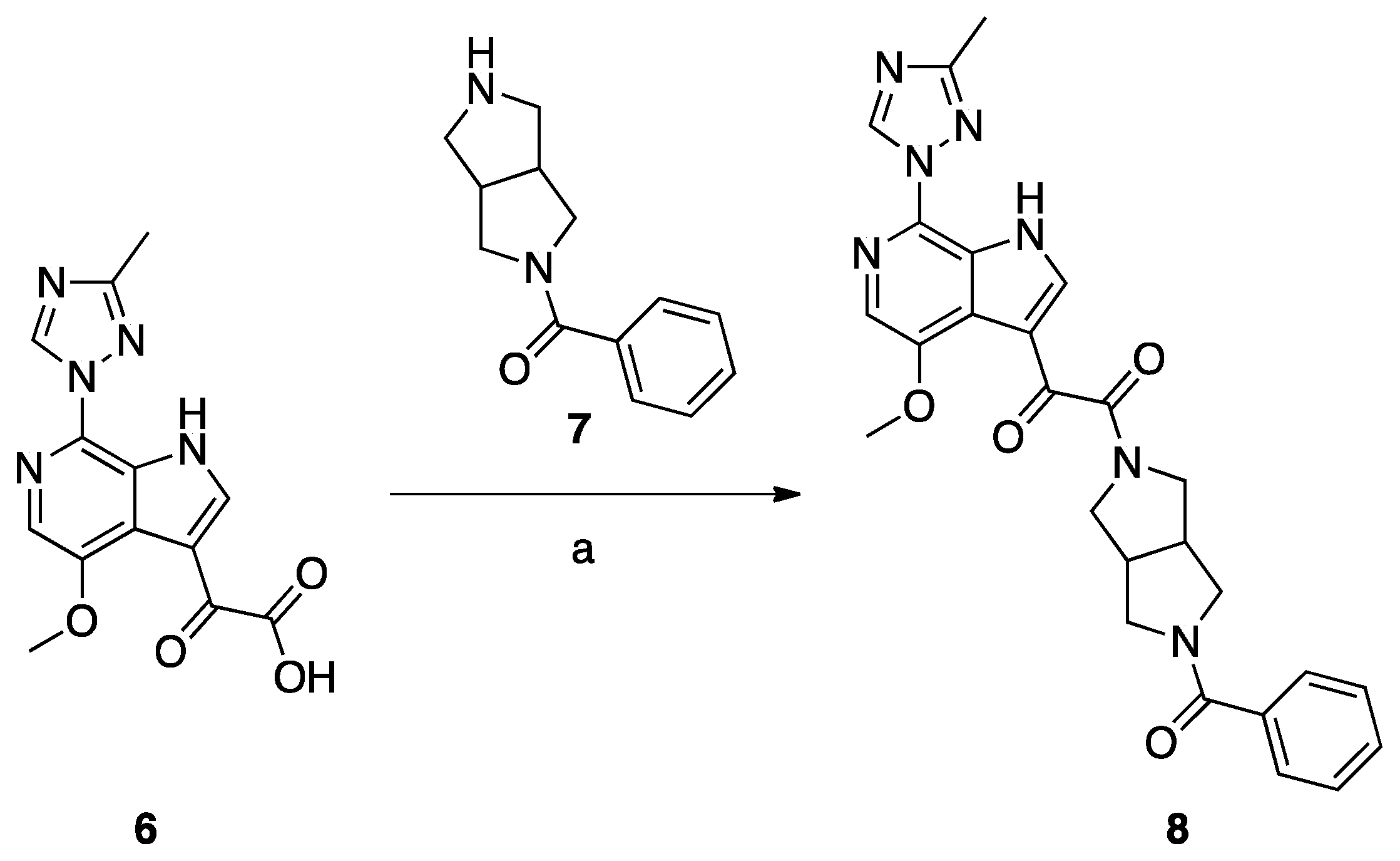
Synthesis of Compound 8 (SC11, Scheme 2)
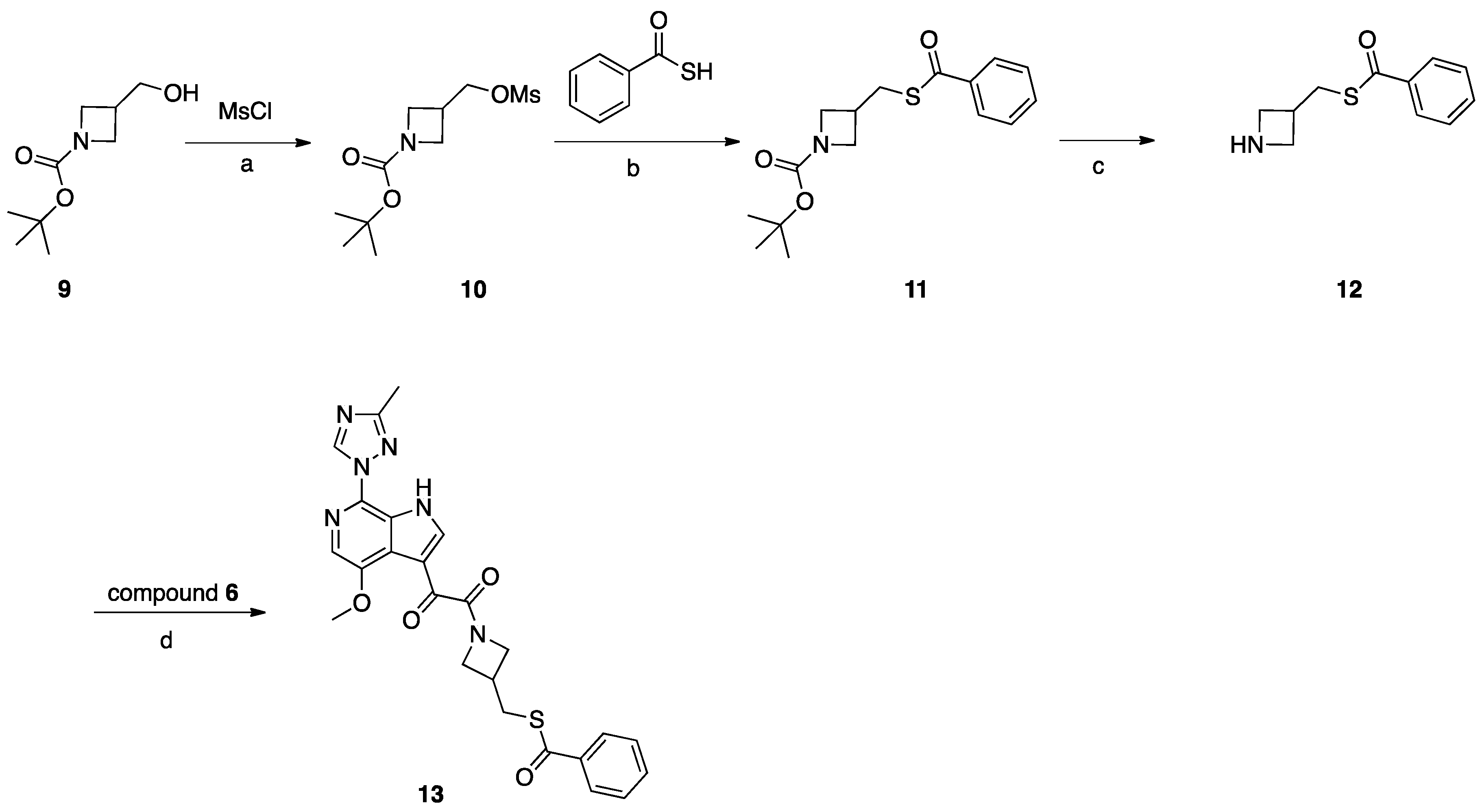
Synthesis of Compound 13 (SC16, Scheme 3)
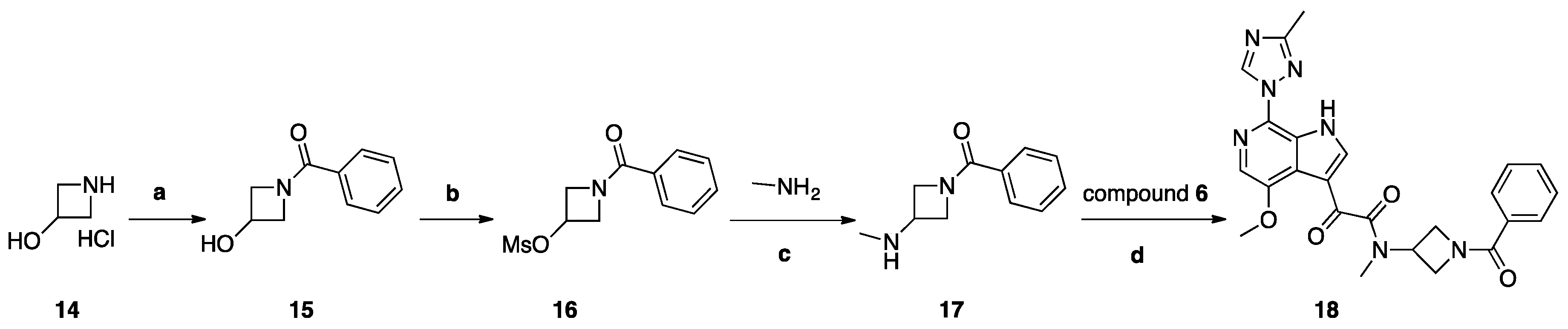
Synthesis of Compound 18 (SC18, Scheme 4)
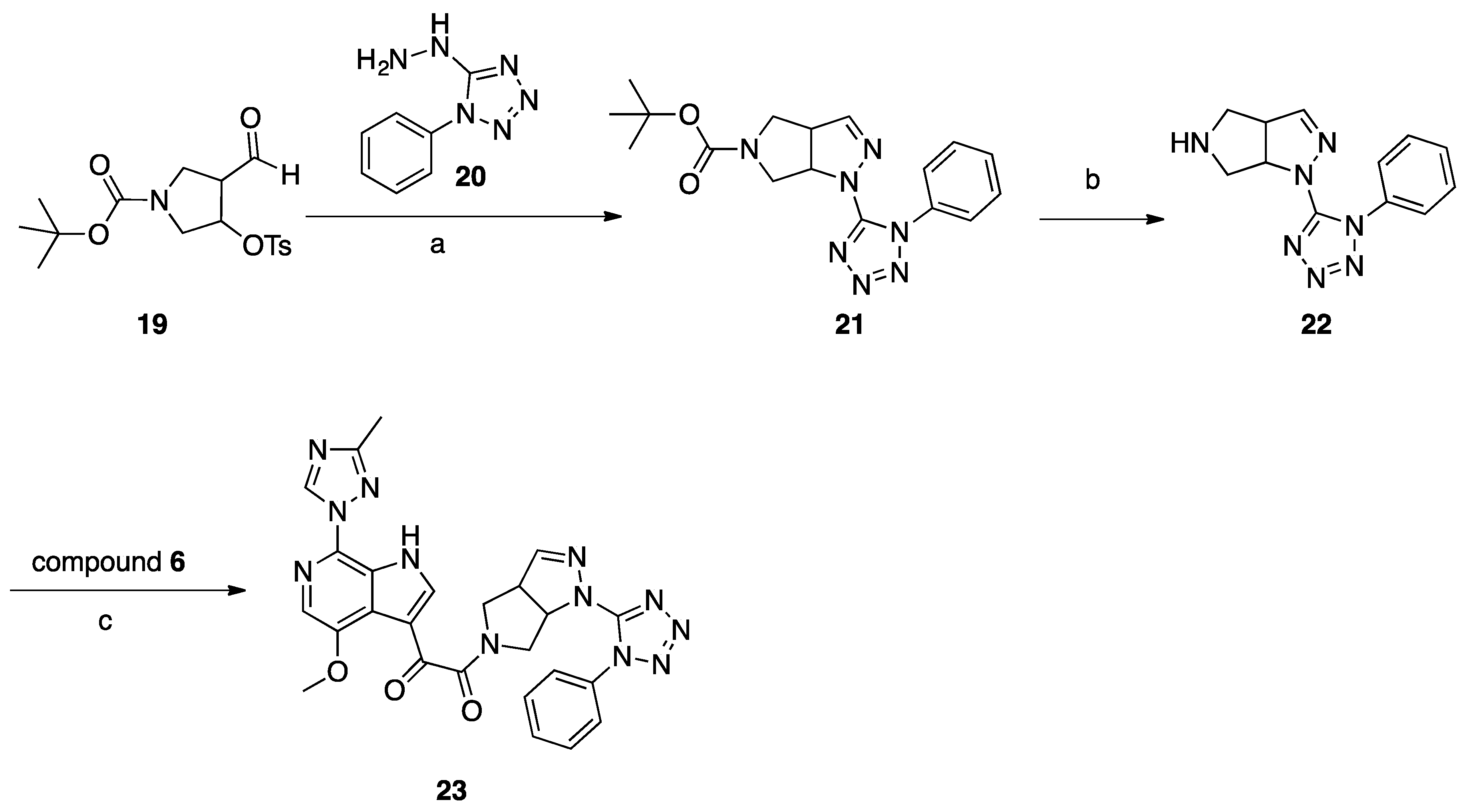
Synthesis of Compound 23 (SC39, Scheme 5)
3.2. Cells
3.3. Proteins
3.4. Production of Pseudotyped Viruses
3.5. ELISA-Based Quantification of p24 Content
3.6. Single-Round Infection Assay
3.7. Cellular Toxicity
3.8. SPR Direct Interaction Analysis
3.9. Molecular Modeling
3.9.1. Bioactive Conformation Hypothesis Prediction
3.9.2. Docking Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhao, Q.; Ma, L.; Jiang, S.; Lu, H.; Liu, S.; He, Y.; Strick, N.; Neamati, N.; Debnath, A.K. Identification of N-phenyl-N′-(2,2,6,6-tetramethyl-piperidin-4-yl)-oxalamides as a new class of HIV-1 entry inhibitors that prevent gp120 binding to CD4. Virology 2005, 339, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zhang, Z.; Wallace, O.B.; Deshpande, M.; Fang, H.; Yang, Z.; Zadjura, L.M.; Tweedie, D.L.; Huang, S.; Zhao, F.; et al. Discovery of 4-benzoyl-1-[(4-methoxy-1H-pyrrolo[2,3-b]pyridin-3-yl)oxoacetyl]-2-(R)-methylpiperazine (BMS-378806): A novel HIV-1 attachment inhibitor that interferes with CD4-gp120 interactions. J. Med. Chem. 2003, 46, 4236–4239. [Google Scholar] [CrossRef] [PubMed]
- Nettles, R.E.; Schurmann, D.; Zhu, L.; Stonier, M.; Huang, S.P.; Chang, I.; Chien, C.; Krystal, M.; Wind-Rotolo, M.; Ray, N.; et al. Pharmacodynamics, safety, and pharmacokinetics of BMS-663068, an oral HIV-1 attachment inhibitor in HIV-1-infected subjects. J. Infect. Dis. 2012, 206, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- Tuyishime, M.; Danish, M.; Princiotto, A.; Mankowski, M.K.; Lawrence, R.; Lombart, H.G.; Esikov, K.; Berniac, J.; Liang, K.; Ji, J.; et al. Discovery and optimization of novel small-molecule HIV-1 entry inhibitors using field-based virtual screening and bioisosteric replacement. Bioorg. Med. Chem. Lett. 2014, 24, 5439–5445. [Google Scholar] [CrossRef] [PubMed]
- Tuyishime, M.; Mankowski, M.K.; Lawrence, R.; Ptak, R.G.; Cocklin, S. Core chemotype diversification in the HIV-1 entry inhibitor class using field-based bioisosteric replacement. Bioorg. Med. Chem. Lett. 2016, 26, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.F.; Blair, W.; Wang, T.; Spicer, T.; Guo, Q.; Zhou, N.; Gong, Y.F.; Wang, H.G.; Rose, R.; Yamanaka, G.; et al. A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc. Natl. Acad. Sci. USA 2003, 100, 11013–11018. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zadjura, L.M.; Marino, A.M.; D’Arienzo, C.J.; Malinowski, J.; Gesenberg, C.; Lin, P.F.; Colonno, R.J.; Wang, T.; Kadow, J.F.; et al. Utilization of in vitro Caco-2 permeability and liver microsomal half-life screens in discovering BMS-488043, a novel HIV-1 attachment inhibitor with improved pharmacokinetic properties. J. Pharm. Sci. 2010, 99, 2135–2152. [Google Scholar] [CrossRef] [PubMed]
- Nowicka-Sans, B.; Gong, Y.F.; McAuliffe, B.; Dicker, I.; Ho, H.T.; Zhou, N.; Eggers, B.; Lin, P.F.; Ray, N.; Wind-Rotolo, M.; et al. In vitro antiviral characteristics of HIV-1 attachment inhibitor BMS-626529, the active component of the prodrug BMS-663068. Antimicrob. Agents Chemother. 2012, 56, 3498–3507. [Google Scholar] [CrossRef] [PubMed]
- Pugach, P.; Ozorowski, G.; Cupo, A.; Ringe, R.; Yasmeen, A.; de Val, N.; Derking, R.; Kim, H.J.; Korzun, J.; Golabek, M.; et al. A Native-Like SOSIP.664 Trimer Based on an HIV-1 Subtype B env Gene. J. Virol. 2015, 89, 3380–3395. [Google Scholar] [CrossRef] [PubMed]
- Pancera, M.; Lai, Y.T.; Bylund, T.; Druz, A.; Narpala, S.; O’Dell, S.; Schon, A.; Bailer, R.T.; Chuang, G.Y.; Geng, H.; et al. Crystal structures of trimeric HIV envelope with entry inhibitors BMS-378806 and BMS-626529. Nat. Chem. Biol. 2017, 13, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Bjorndal, A.; Deng, H.; Jansson, M.; Fiore, J.R.; Colognesi, C.; Karlsson, A.; Albert, J.; Scarlatti, G.; Littman, D.R.; Fenyo, E.M. Coreceptor usage of primary human immunodeficiency virus type 1 isolates varies according to biological phenotype. J. Virol. 1997, 71, 7478–7487. [Google Scholar] [PubMed]
- Zentner, I.; Sierra, L.J.; Fraser, A.K.; Maciunas, L.; Mankowski, M.K.; Vinnik, A.; Fedichev, P.; Ptak, R.G.; Martin-Garcia, J.; Cocklin, S. Identification of a small-molecule inhibitor of HIV-1 assembly that targets the phosphatidylinositol (4,5)-bisphosphate binding site of the HIV-1 matrix protein. ChemMedChem 2013, 8, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Kortagere, S.; Madani, N.; Mankowski, M.K.; Schon, A.; Zentner, I.; Swaminathan, G.; Princiotto, A.; Anthony, K.; Oza, A.; Sierra, L.J.; et al. Inhibiting early-stage events in HIV-1 replication by small-molecule targeting of the HIV-1 capsid. J. Virol. 2012, 86, 8472–8481. [Google Scholar] [CrossRef] [PubMed]
- Studier, F.W. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef] [PubMed]
- Connor, R.I.; Chen, B.K.; Choe, S.; Landau, N.R. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 1995, 206, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, W.; Schubert, D.; LaBonte, J.; Munson, L.; Gibson, S.; Scammell, J.; Ferrigno, P.; Sodroski, J. Species-specific, postentry barriers to primate immunodeficiency virus infection. J. Virol. 1999, 73, 10020–10028. [Google Scholar] [PubMed]
- Marcon, L.; Choe, H.; Martin, K.A.; Farzan, M.; Ponath, P.D.; Wu, L.; Newman, W.; Gerard, N.; Gerard, C.; Sodroski, J. Utilization of C-C chemokine receptor 5 by the envelope glycoproteins of a pathogenic simian immunodeficiency virus, SIVmac239. J. Virol. 1997, 71, 2522–2527. [Google Scholar] [PubMed]
- Cocklin, S.; Gopi, H.; Querido, B.; Nimmagadda, M.; Kuriakose, S.; Cicala, C.; Ajith, S.; Baxter, S.; Arthos, J.; Martin-Garcia, J.; et al. Broad-spectrum anti-human immunodeficiency virus (HIV) potential of a peptide HIV type 1 entry inhibitor. J. Virol. 2007, 81, 3645–3648. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Choe, S.; Walker, R.; Di Marzio, P.; Morgan, D.O.; Landau, N.R. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J. Virol. 1995, 69, 6705–6711. [Google Scholar] [PubMed]
- Goodsell, D.S.; Morris, G.M.; Olson, A.J. Automated docking of flexible ligands: Applications of AutoDock. J. Mol. Recognit. 1996, 9, 1–5. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Huey, R.; Olson, A.J. Distributed automated docking of flexible ligands to proteins: Parallel applications of AutoDock 2.4. J. Comput. Aid Mol. Des. 1996, 10, 293–304. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 JR-CSF (μM) | IC50 B41 (μM) | IC50 AMLV (μM) | Median IC50 (μM) | CC50 (μM) |
|---|---|---|---|---|---|
| BMS-626529 | 0.00006 ± 0.000005 | 0.00005 ± 0.000006 | N.A. | 0.000055 | >50 |
Compound 8 (SC11) | 0.0006 ± 0.0001 | 0.002 ± 0.0002 | N.A. | 0.001 | |
Compound 13 (SC16) | 0.029 ± 0.008 | 0.037 ± 0.005 | N.A. | 0.033 | |
Compound 18 (SC18) | 0.132 ± 0.004 | 0.360 ± 0.012 | N.A. | 0.246 | |
Compound 23 (SC39) | 0.143 ± 0.052 | 5.32 ± 0.26 | N.A. | 2.73 |
| Compound | ka (M−1 s−1) | kd (s−1) | KD |
|---|---|---|---|
| BMS-626529 * | 3.89 × 104 | 5.9 × 10−4 | 15.2 nM |
| SC11 | 4.33 × 103 | 2.87 × 10−4 | 66.3 nM |
| SC16 | 8.62 × 104 | 5.98 × 10−3 | 69.3 nM |
| SC18 | 1.59 × 104 | 2.48 × 10−2 | 1.56 μM |
| SC39 | 1.39 × 103 | 3.81 × 10−2 | 27.4 μM |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meuser, M.E.; Murphy, M.B.; Rashad, A.A.; Cocklin, S. Kinetic Characterization of Novel HIV-1 Entry Inhibitors: Discovery of a Relationship between Off-Rate and Potency. Molecules 2018, 23, 1940. https://doi.org/10.3390/molecules23081940
Meuser ME, Murphy MB, Rashad AA, Cocklin S. Kinetic Characterization of Novel HIV-1 Entry Inhibitors: Discovery of a Relationship between Off-Rate and Potency. Molecules. 2018; 23(8):1940. https://doi.org/10.3390/molecules23081940
Chicago/Turabian StyleMeuser, Megan E., Michael B. Murphy, Adel A. Rashad, and Simon Cocklin. 2018. "Kinetic Characterization of Novel HIV-1 Entry Inhibitors: Discovery of a Relationship between Off-Rate and Potency" Molecules 23, no. 8: 1940. https://doi.org/10.3390/molecules23081940
APA StyleMeuser, M. E., Murphy, M. B., Rashad, A. A., & Cocklin, S. (2018). Kinetic Characterization of Novel HIV-1 Entry Inhibitors: Discovery of a Relationship between Off-Rate and Potency. Molecules, 23(8), 1940. https://doi.org/10.3390/molecules23081940