All reactions in organic medium were performed in standard oven dried glassware under an inert atmosphere of nitrogen using freshly distilled solvents stored over molecular sieves. Solvents were deoxygenated when necessary by bubbling nitrogen through the solution. All reagents were used as supplied without prior purification and obtained from Sigma-Aldrich Chemical Co. (Toronto, ON, Canada) Reactions were monitored by analytical thin-layer chromatography (TLC) using silica gel 60 F254 pre-coated plates (Merck, Darmstadt, Germany) and compounds were visualized by 254 nm light and/or by dipping into a mixture of sulfuric acid and methanol in water or into a mixture of KMin.O4 and K2CO3 in water followed by gentle warming with a heat-gun. Purifications were performed by flash column chromatography using silica gel from Canadian Life Science (60 Å, 40–63 μm) (Peterborough, ON, Canada) with the indicated eluent. 1H-NMR, 13C-NMR and 31P-NMR spectra were recorded at 300 and/or 600 MHz, 75 and/or 150 MHz and 122 and/or 243 MHz, respectively, on a Bruker spectrometer (300 MHz and 600 MHz) (Milton, ON, Canada) and Varian spectrometer (600 MHz) (Milton, ON, Canada). All NMR spectra were measured at 25 °C in indicated deuterated solvents. Proton and carbon chemical shifts (δ) are reported in ppm and coupling constants (J) are reported in Hertz (Hz). The resonance multiplicity in the 1H-NMR spectra are described as “s” (singlet), “d” (doublet), “t” (triplet) and “m” (multiplet) and broad resonances are indicated by “broad.” Residual protic solvent of CDCl3 (1H, 7.27 ppm; 13C, 77.0 ppm (central resonance of the triplet)), D2O (1H, 4.80 ppm and 30.9 ppm for CH3 of acetone for 13C spectra), MeOD (1H, 3.30 ppm and 13C, 49.0 ppm), 85% H3PO4 was used as an external reference for 31P-NMR. Two-dimensional homonuclear correlation 1H-1H COSY and 1H-13C HSQC experiments were used to confirm NMR peak assignments. Letters are used to NMR assignment. Accurate mass measurements (HRMS) were performed on a LC-MSD-TOF instrument from Agilent Technologies (Santa Clara, CA, USA) in positive electrospray mode. Either protonated molecular ions [M + nH]n+ or adducts [M + nX]n+ (X = Na, K, NH4) were used for empirical formula confirmation. Light Scattering-Multiangle Laser Light Scattering (LS-MALLS) detection with performances verified with polystyrene 100 kDa and 2000 kDa were used to determine the number-average molecular weight (Min.) and polydispersity index (MW/Min.). Calculations were performed with Zimm Plot (model). MALDI-TOF MS data were acquired on a Bruker Microflex LRF system (Bruker Daltonics, Billerica, MA, USA) equipped with a Compass 3.1 software platform. Acquisitions were performed in positive ion mode. Reflector mode was utilized for samples below 5 kDa and linear mode for samples above 5 kDa. Samples were dissolved in water (1 mg/mL) and mixed with 10 volumes of α-cyano-4-hydroxycinnamic acid matrix prepared at 10 mg/mL with 50% ACN/H2O 0.1% TFA. A volume of 2 μL was deposited on the target plate and dried. Representative acquisition parameters were: ion source 1, 19.5 kV; ion source 2, 18.05 kV; lens, 7.0 V; pulse ion extraction, 240 ns; detector, 1.905 kV. Approximately 200 laser shot/spectra were obtained with a 60 Hz N2-cartridge-laser set at 337 nm and the laser intensity adjusted according to the signal intensity. Sugar monomers were synthesized following the typical procedures found in literatures with a slight modification as describe bellow.
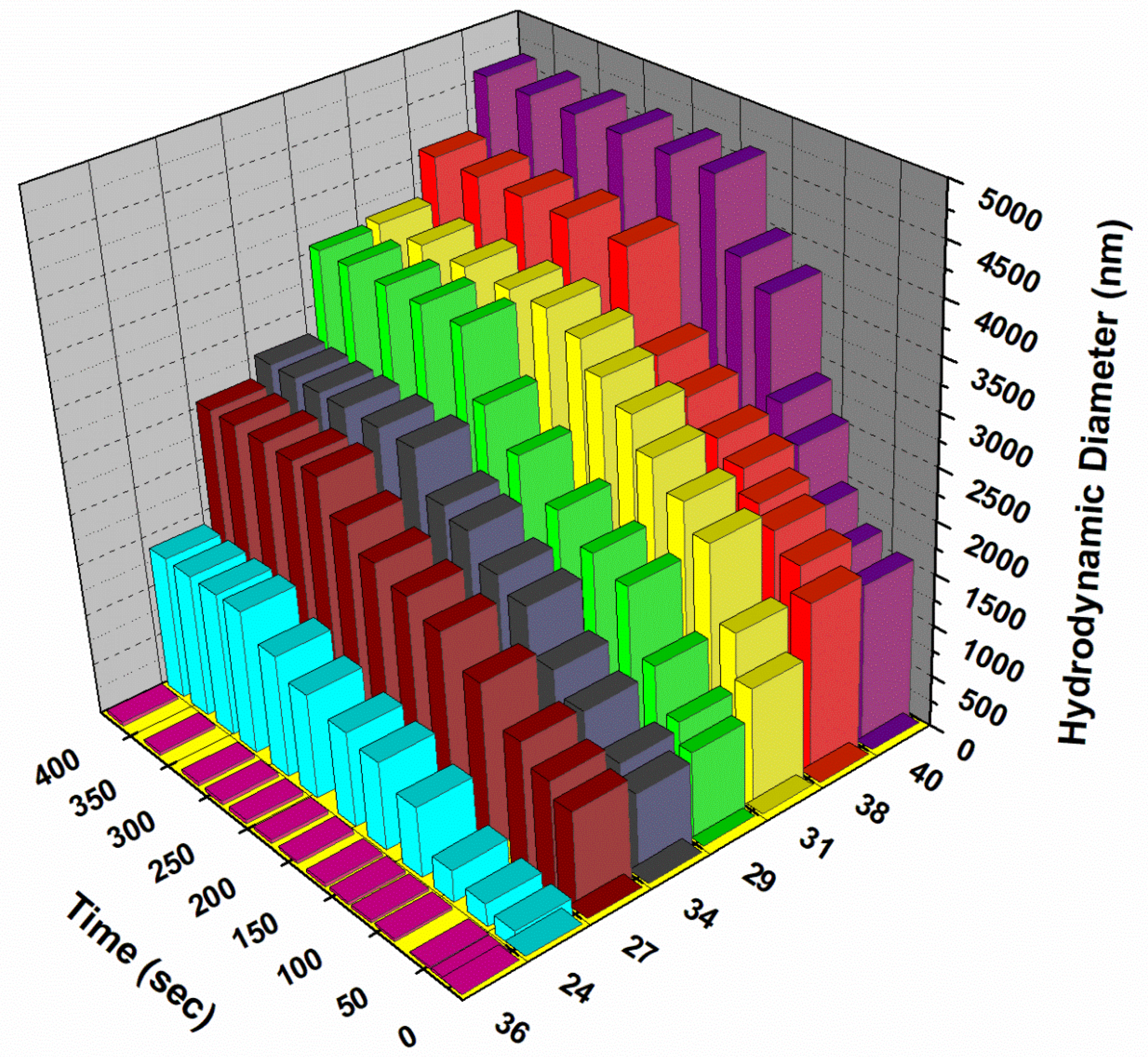
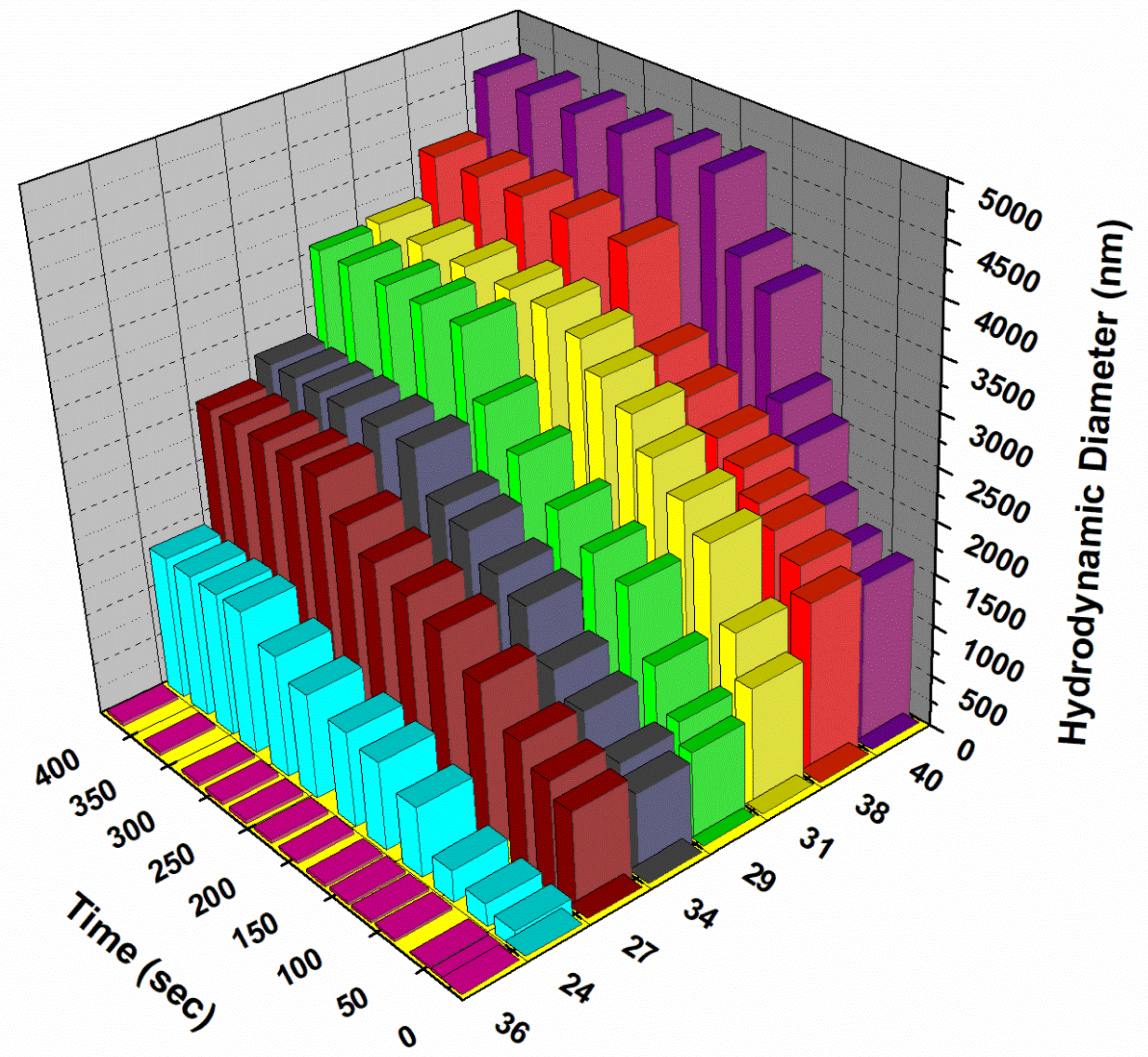
Dynamic Light Scattering
Particle size distribution (DLS) was measured in water with the help of Zetasizer Nano S90 from Malvern Instruments. Crosslinking studies were carried out in 1 mol/L phosphate buffered saline (PBS) for the plant lectin Concavalin A (Sigma-Aldrich).
General procedure for the Cu(I) azide-alkyne cycloaddition reaction (I) (CuAAc) [
28]. Solutions of alkyne (1 equiv.) and azide (1.5 equiv. per alkyne site) were prepared in minimum amount of THF. The resulting solutions were treated with an aqueous solution of CuSO
4 (0.3 equiv. per alkyne site) and sodium ascorbate solution in THF (0.35 equiv. per alkyne site). Biphasic mixture was then stirred at 50 °C for 2 h then left at room temperature until completion of the reaction as judged by the complete conversion of the limiting regents (alkyne). After completion of the reaction, EtOAc was added, then Na
2SO
4 was added to quench the reaction by regenerating the CuSO
4·5H
2O. The mixture was allowed to stir for 3 min then filtered before the solvent was removed under reduced pressure and the resulting viscous oil was subjected to silica gel column chromatography using the appropriate eluent system (0–10% MeOH in CH
2Cl
2) to afford the triazole products.
General procedure for the azide substitution (II) [
28]. To a solution of the appropriate bromo/tosyl derivatives (1 equiv.) in DMF was added NaN
3 (1.5 equiv. per bromide). The reaction mixtures were stirred at 80 °C until completion of reaction as judged by TLC. The excess solvent was next removed under reduced pressure with heating at 60 °C until dryness. The residues were dissolved in EtOAc and washed successively with water and brine. The organic layer was separated and dried on Na
2SO
4. The residue was subjected to silica gel flash chromatography to afford the desired azido derivative.
General Procedure for De-O-acetylation (III) (Zemplén reaction) [
28]. An acetylated glycocluster (0.1 mmol) was dissolved in dry MeOH (3 mL) and a solution of sodium methoxide (1 M in MeOH, 0.5 equiv.) was added. The reaction mixture was stirred at room temperature until the starting material disappeared. The solution was neutralized by the addition of a cationic ion-exchange resin (H
+), filtered and washed with MeOH and then the solvent was removed in vacuo. The residue was lyophilized to yield quantitatively the fully deprotected glycoclusters.
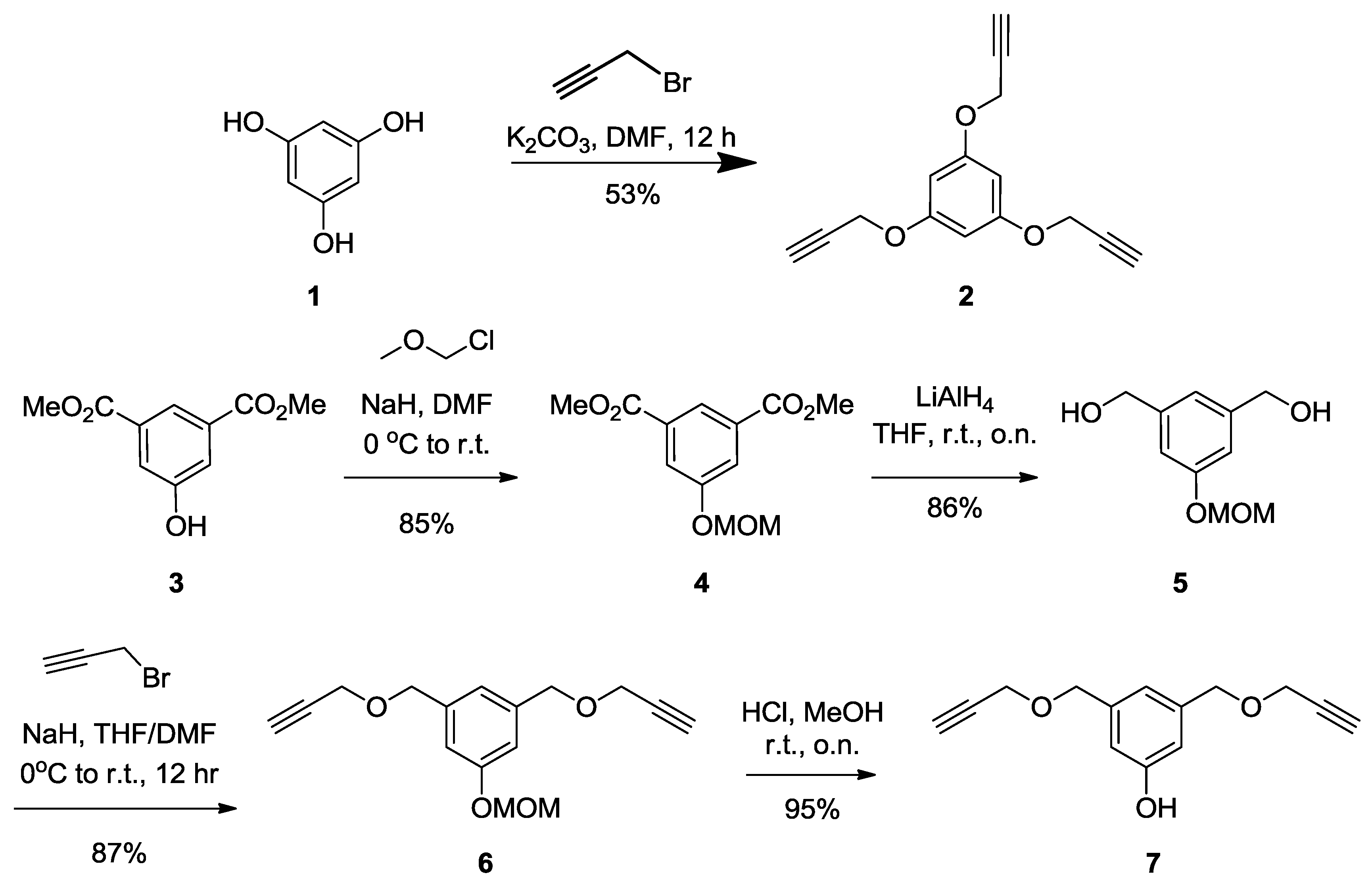
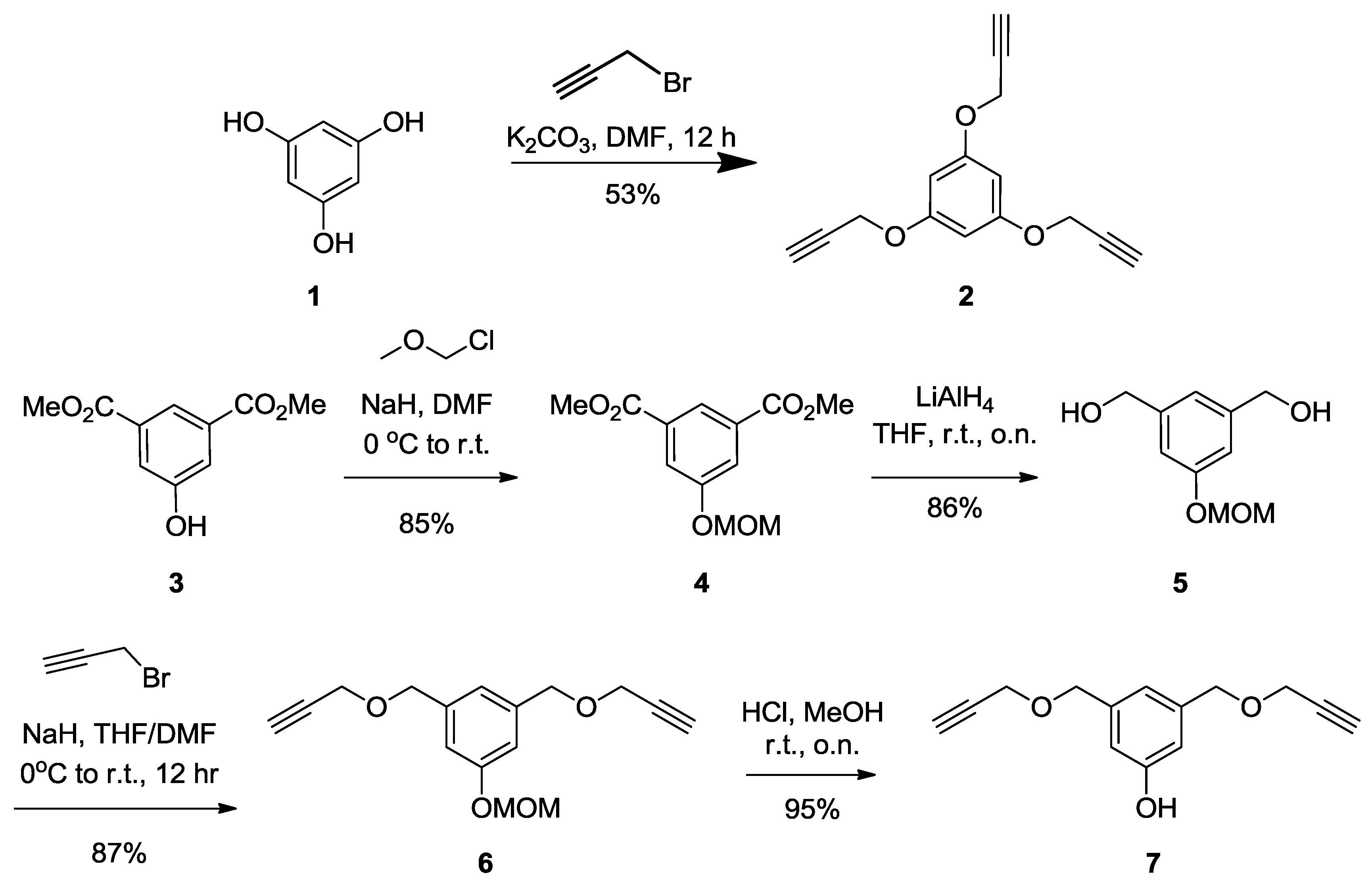
Synthesis of2.
Benzene-1,3,5-triol (
1) [
48] (2 g, 15.86 mmol) was dissolved in DMF (10 mL) followed by the addition of K
2CO
3 (3 g, 14.87 mmol). The mixture was reflux for 30 min, propargyl bromide (8.5 mL, 79.3 mmol, 5 equiv.) was added dropwise, then stirred over night at room temperature. The residue was dissolved in dichloromethane (DCM) and washed with water and brine. The organic layer was dried over Na
2SO
4, filtered, then concentered under vacuum. The crude product was purified by silica gel flash chromatography using 0–20% EtOAc in Hexane as eluents to afford compound
2 as a white powder (2 g, 53%).
Rf = 0.28 (EtOAc/Hexane, 1:4).
1H-NMR (300 MHz, CDCl
3) δ 6.27 (s, 3H, k), 4.65 (d,
J = 2.4 Hz, 6H, b), 2.53 (t,
J = 2.4 Hz, 3H, c).
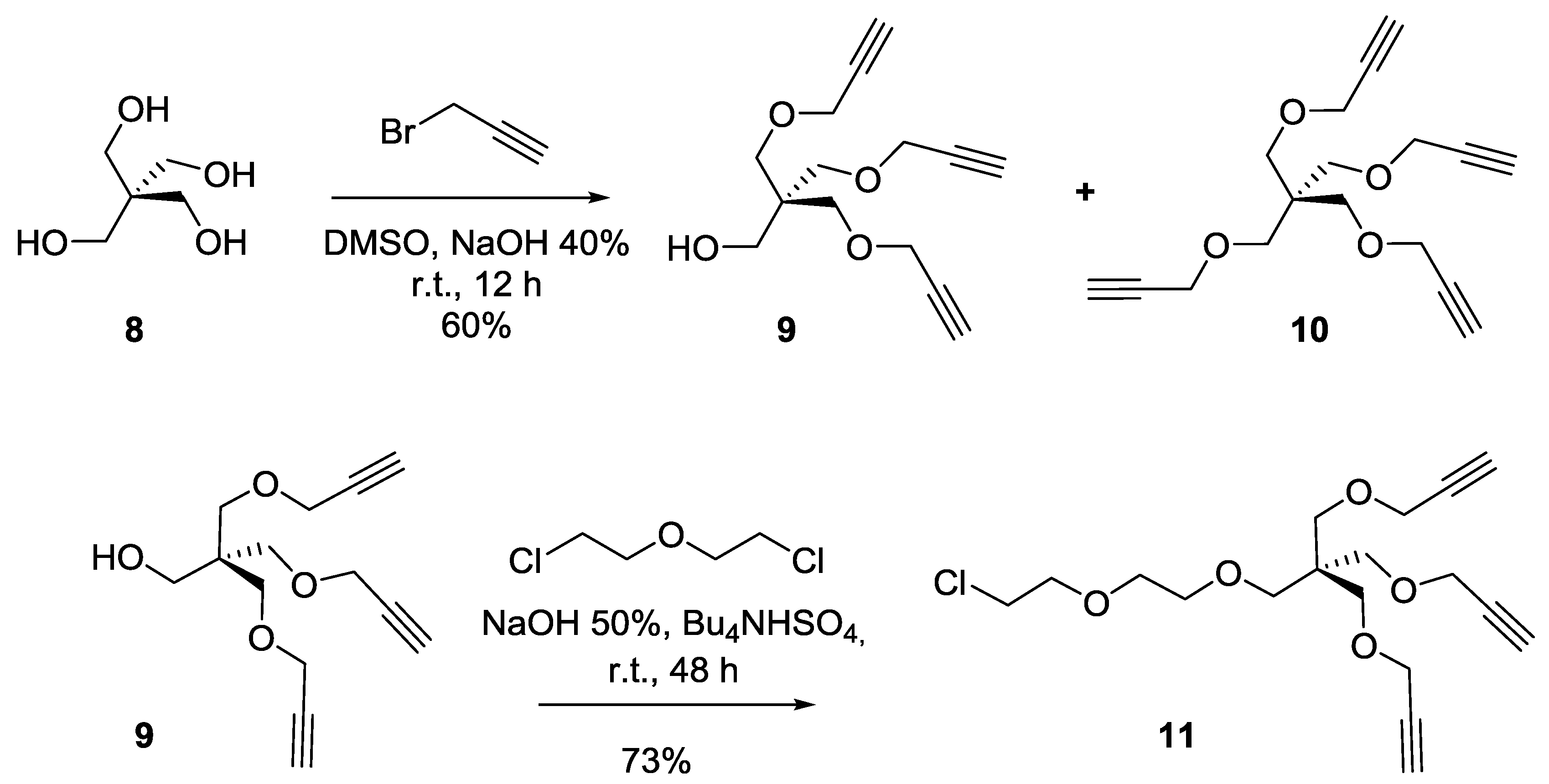
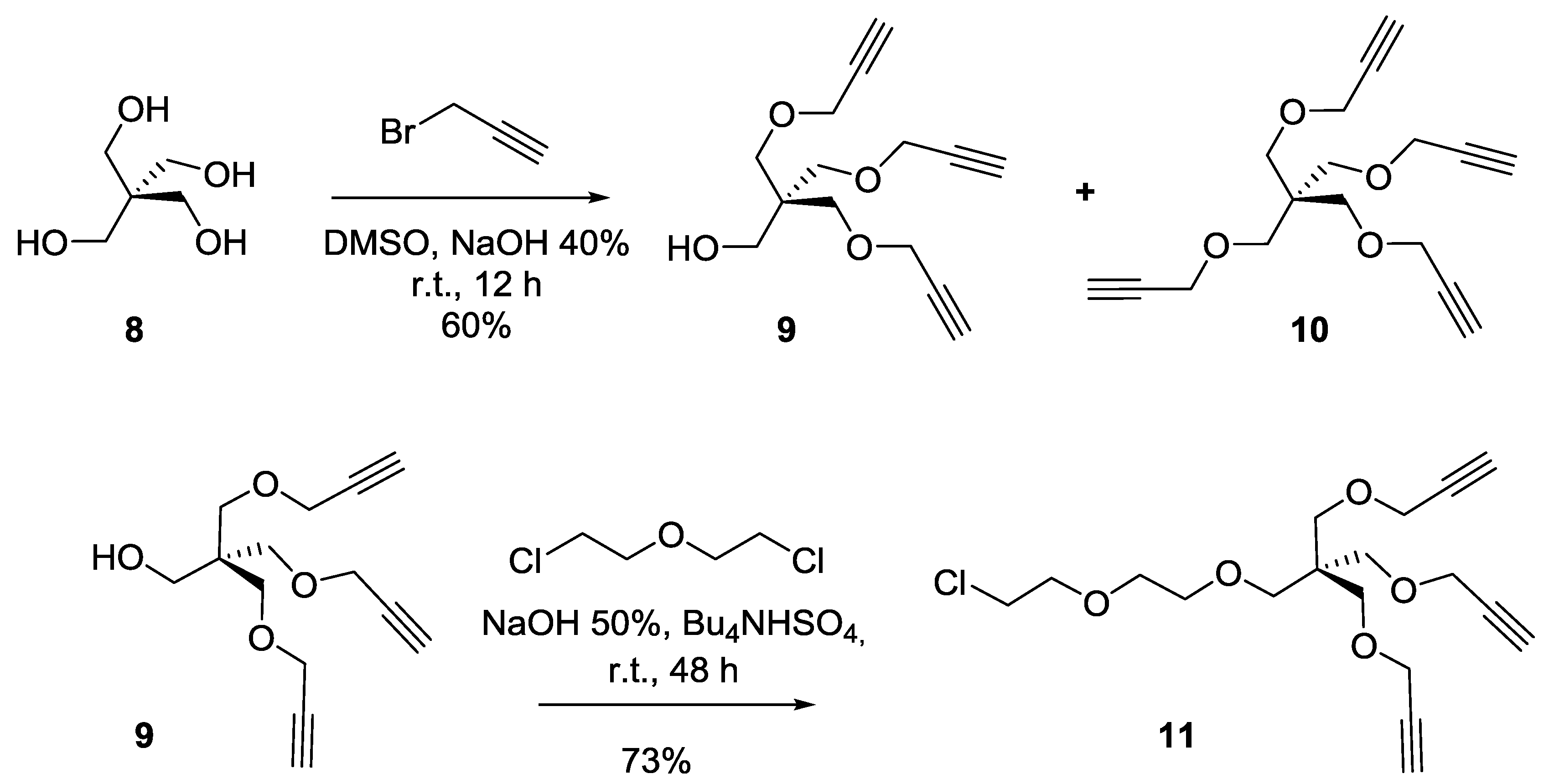
Synthesis of compound9. To a solution of pentaerythritol
8 (2 g, 14.7 mmol) in dimethylsulfoxide (DMSO, 15 mL) were added an aqueous solution of NaOH (40 wt%, 10 mL). The solution was kept under magnetic stirring at room temperature for 30 min Propargyl bromide (97%, 12 mL, 135.18 mmol) was then added and the solution was kept at room temperature for an additional 24 h. Ethyl ether was added to the reaction mixture then washed with water. The organic layer was dried over Na
2SO
4. The crude product was purified by silica gel flash chromatography using 0–20% EtOAc in Hexane as eluents to afford compound
9 as a colorless oil (2.2 g, 60%).
Rf = 0.25 (EtOAc/Hexane, 1:4). Compound
10 was obtained as a white powder (1.2 g, 28%).
Rf = 0.35 (EtOAc/Hexane, 1:4). Tetrapropargylpentaerythritol
10 [
49].
1H-NMR (300 MHz, CDCl
3) δ 4.08 (d,
J = 2.3 Hz, 8H, OC
H2CCH), 3.48 (s, 8H, C
H2OCH
2CCH), 2.42 (d,
J = 2.2 Hz, 4H, OCH
2CC
H).
Tripropargylpentaerythritol9 [
49].
1H-NMR (300 MHz, CDCl
3) δ 4.13 (d,
J = 2.4 Hz, 6H, OC
H2CCH), 3.69 (s, 2H, C
H2OH), 3.56 (s, 6H, CH
2OC
H2CCH), 2.42 (t,
J = 2.4 Hz, 3H, CC
H).
Synthesis of compound11. A solution of pentaerythritol propargyl ether (9) (2 g, 7.9 mmol, 1 equiv.), Bu3NHSO4 (5 g, 14.7 mmol, 1.8 equiv.), Bis(2-chloroethyl) ether (22 mL, 187.61 mmol) in NaOH 50% (30 mL), was stirred at room temperature for 24 h. DCM was added to the reaction mixture and the organic layer was washed successfully with water and brine, then dried over anhydrous sodium sulfate. The crude product was purified by silica gel flash chromatography using 0–20% EtOAc in Hexane as eluents to afford compound 11 as a colorless oil (2.1 g, 73%). Rf = 0.25 (EtOAc/Hexane, 1:4). 1H-NMR (300 MHz, CDCl3) δ 4.11 (d, J = 2.4 Hz, 6H, OCH2CCH), 3.76 (dd, J = 8.8, 3.2 Hz, 2H, CH2-Cl), 3.67–3.56 (m, 6H, OCH2-CH2), 3.52 (s, 6H, CH2OCH2CCH), 3.46 (s, 2H, CH2OCH2), 2.40 (t, J = 2.4 Hz, 3H, CCH).
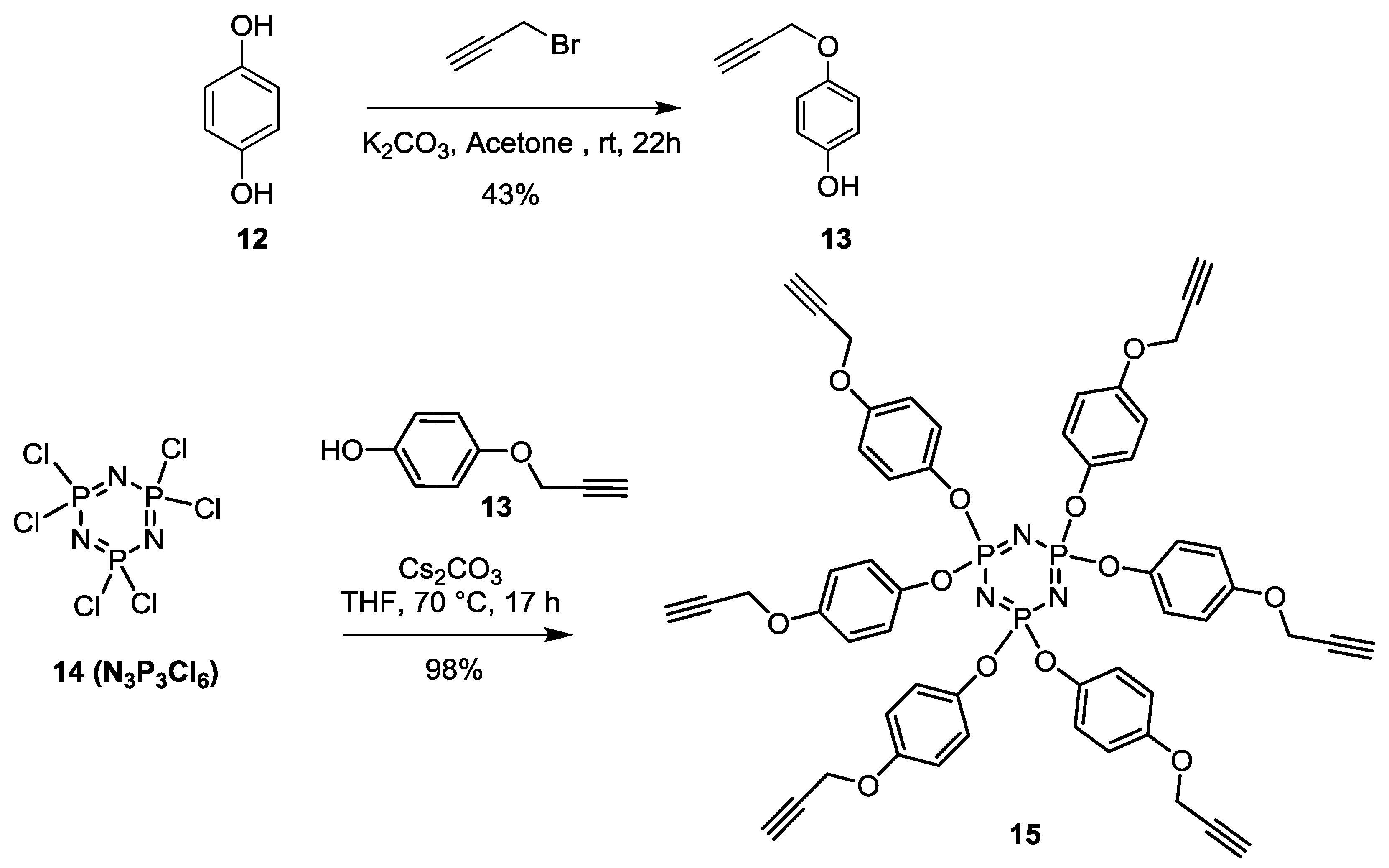
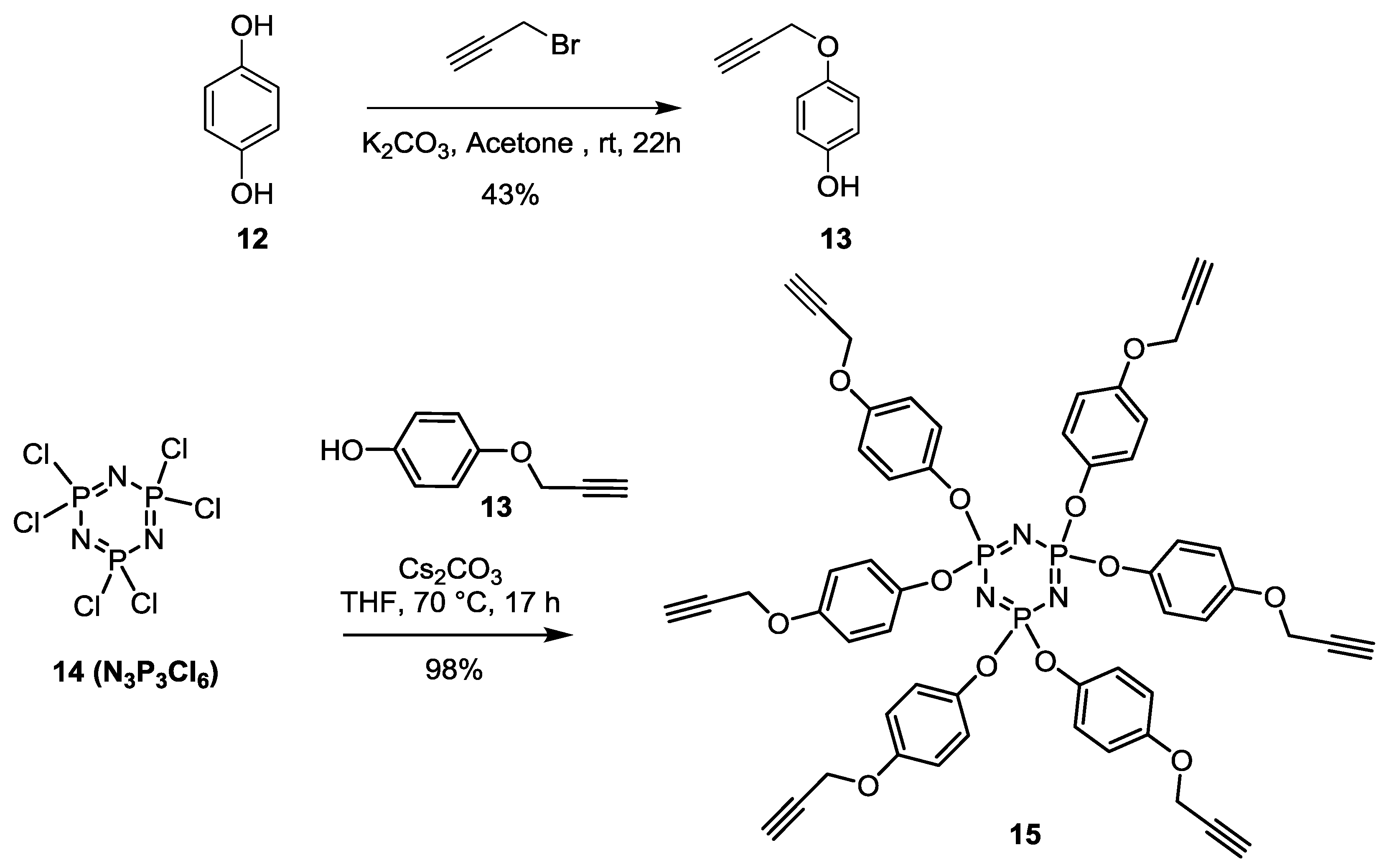
Monopropagyloxyphenol (
13) [
50]. Hydroquinone (10.28 g, 93.37 mmol) was dissolved in acetone (150 mL) followed by the addition of K
2CO
3 (15.42 g, 111.58 mmol). The mixture was reflux for 30 min, propargyl bromide (10.28 mL, 93.50 mmol, 0.98 equiv.) was added dropwise, then stirred over night at room temperature. Residue was dissolved in DCM (100 mL) and washed with water (3 × 50 mL) and brine (3 × 50 mL). The organic layer was dried over Na
2SO
4, filtered and concentered under vacuum. The crude product was purified by silica gel flash chromatography using 0–30% EtOAc in Hexane as eluents to afford the mono substituted propargyl hydroquinone
13 as a brownish powder (3.11 g, 20.96 mmol, 43%).
Rf = 0.35 (EtOAc/Hexane, 3:7).
1H-NMR (300 MHz, CDCl
3) δ 6.85 (d,
J = 9.1 Hz, 2H, aromatic), 6.78 (d,
J = 9.1 Hz, 2H, aromatic), 4.66 (d,
J = 1.4 Hz, 2H, C
H2), 2.53 (t,
J = 2.4 Hz, 1H,
≡C
H).
Synthesis of15 [
50,
51,
52,
53,
54,
55]. Cs
2CO
3 (3.4 g, 10.4 mmol) is added into a reaction mixture of 4-(prop-2-yn-1-yloxy)phenol (
13) (6.7 mmol) and N
3P
3Cl
6 (
14) (0.6 mmol) in THF (20 mL). The mixture was heated at 40 °C for 17 h, then filtered on Celite and concentrated. The pure
15 was isolated as a white crystal yielding 98% after crystallization in EtOH.
1H-NMR (300 MHz, CDCl
3) δ 6.87 (d,
J = 9.65 Hz, 6H, OC
6H4O), δ 6.80 (d,
J = 9.65 Hz, 6H, OC
6H4O), 4.65 (d,
J = 2.4 Hz, 6H, OC
H2), 2.52 (t,
J = 2.4 Hz, 3H, C
H).
31P-NMR (122 MHz, CDCl
3) δ 9.82 (s).
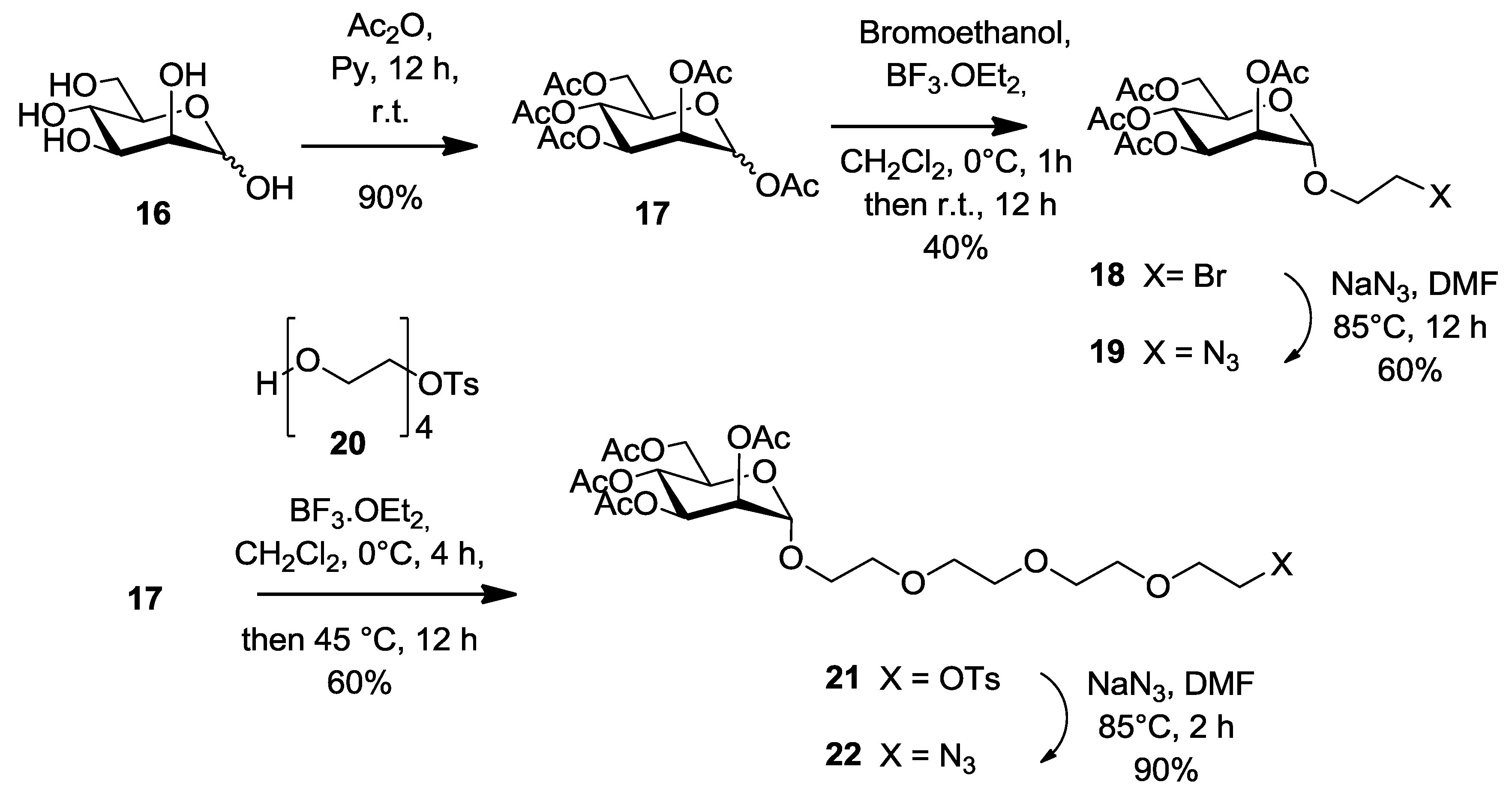
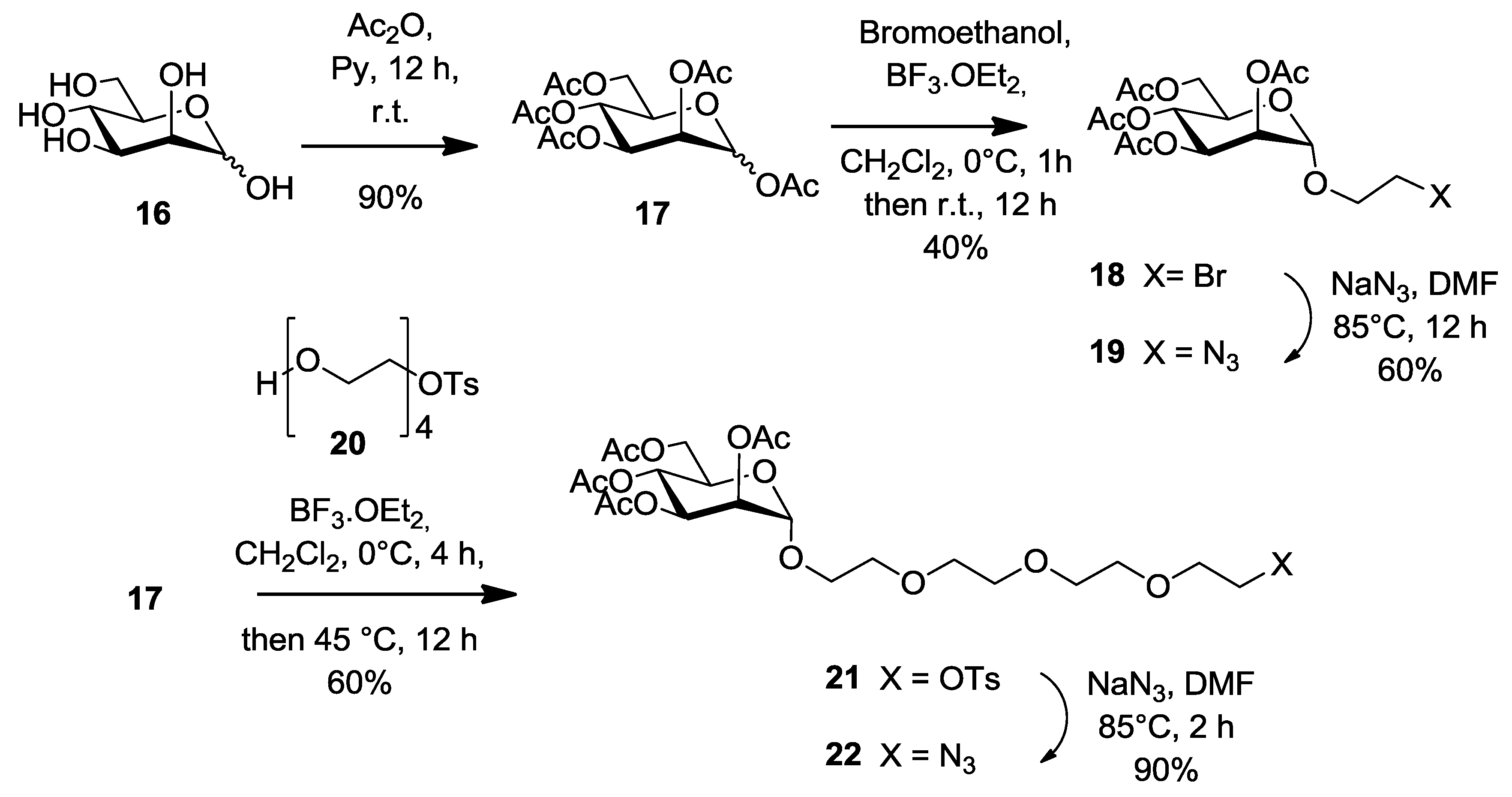
1,2,3,4,6-Penta-O-acetyl-α/β-d-mannopyranose (
17). 1,2,3,4,6-penta-
O-acetyl-α/β-
d-mannopyranose was prepared according to the procedure reported [
52] with a slight modification.
d-Mannose
16 (3 g, 16.6 mmol), pyridine (40 mL) and acetic anhydride (32 mL, 333 mmol) were stirred at room temperature. After stirring for 12 h, the reaction mixture was diluted with ice-water and extracted with DCM. The combined organic layer was washed with 1 M aqueous HCl, saturated sodium bicarbonate solution (NaHCO
3), H
2O and Brine. The organic layer was dried over Na
2SO
4 and the solvent was removed under reduced pressure. The product
17 was obtained as colorless syrup (6 g, 15.4 mmol, 90%) which was a mixture of α and β anomers with a ratio of 3:1. This product was used in the next synthetic step without any further purification.
1H-NMR (300 MHz, CDCl
3) δ 6.09 (d,
J = 1.8 Hz, 3H,
H-1a), 5.86 (d,
J = 1.1 Hz, 1H,
H-1b), 5.49 (dd,
J = 3.3, 1.1 Hz, 1H), 5.38–5.33 (m, 6H), 5.32 (d,
J = 2.8 Hz, 1H), 5.26 (t,
J = 2.0 Hz, 3H), 5.13 (dd,
J = 10.0, 3.3 Hz, 1H), 4.30 (ddd,
J = 12.4, 7.5, 5.1 Hz, 5H), 4.18–4.02 (m, 8H), 3.80 (ddd,
J = 9.8, 5.3, 2.4 Hz, 1H), 2.22 (s, 3H), 2.18 (s, 9H), 2.17 (s, 9H), 2.10 (s, 3H), 2.09 (s, 12H), 2.05 (s, 12H), 2.01 (s, 12H).
2-Bromoethyl 2,3,4,6-tetra-O-acetyl-α-d-manno-pyranoside (
18) [
27,
51]. Compound
17 (5.37 g, 13.8 mmol) and 2-bromoethanol (0.98 mL, 13.8 mmol) were dissolved in DCM (50 mL). Then, boron trifluoride etherate (5.8 mL, 47.2 mmol) was added to the solution and stirred under a nitrogen atmosphere for 3 h and monitored by TLC (EtOAc/Hexane, 1:1). After addition of DCM (100 mL), the reaction mixture was neutralized by adding saturated sodium bicarbonate solution (100 mL) and the resulting solution was washed with water (2 × 200 mL). The combined organic layers were dried over Na
2SO
4, filtered and concentrated to dryness under reduced pressure. The resulting oil was then purified using silica gel chromatography (EtOAc/Hexane, 1:1). The relevant fractions were collected, combined and concentrated to dryness under reduced pressure to yield
18 as a colorless powder (2.2 g, 40%).
1H-NMR (300 MHz, CDCl
3) δ 5.39–5.18 (m, 4H, H-2, 3 and 4), 4.85 (d,
J = 1.6 Hz, 1H,
H-1), 4.27 (dd,
J = 12.2, 5.3 Hz, 1H,
H-6), 4.16–3.98 (m, 2H,
H-6,
H-5), 3.85 (ddd,
J = 10.6, 6.6, 4.0 Hz, 1H, j), 3.72–3.60 (m, 1H, j′), 3.54–3.34 (m, 2H, C
H2Br), 2.12 (s, 3H, COC
H3), 2.08 (s, 3H, COC
H3), 2.03 (s, 3H, COC
H3), 1.97 (s, 3H, COC
H3).
2-Azidoethyl 2,3,4,6-tetra-O-acetyl-α-d-manno-pyranoside (
19) [
27,
51]. Compound
18 (1.40 g, 3.0 mmol) and sodium azide (1.00 g, 15.4 mmol) were dissolved in anhydrous DMF (30 mL) and stirred at 80 °C for 5 h. The reaction mixture was filtered and concentrated to dryness under reduced pressure and further processed as given in general procedure (
II) to afford compound
19 (0.75 g, 60%) as white powder.
1H-NMR (300 MHz, CDCl
3) δ 5.48–5.11 (m, 3H,
H-2, 3 and 4), 4.87 (d,
J = 1.6 Hz, 1H,
H-1), 4.29 (dd,
J = 12.3, 5.3 Hz, 1H,
H-6), 4.20–3.99 (m, 2H,
H-6′ and 5), 3.87 (m, 1H, j), 3.76–3.60 (m, 1H, j′), 3.56–3.36 (m, 2H, C
H2N
3), 2.16 (s, 3H, COC
H3), 2.11 (s, 3H, COC
H3), 2.06–2.03 (s, 3H, COC
H3), 2.00 (s, 3H, COC
H3).
Monotosylated tetraethylene glycol (
20) [
53]. To a solution of tetraethylene glycol (26.3 g, 135.4 mmol, 10 equiv.) in THF (60 mL) was added a solution of sodium hydroxide (1.79 g, 44.7 mmol, 3.3 equiv.) dissolved in water (5 mL). The mixture was cooled to 0 °C and toluensulfonyl chloride (2.57 g, 16.54 mmol, 1 equiv.) in THF (5 mL) was added dropwise. The reaction was stirred at 0 °C for 2 h. The solution was poured into water and the aqueous layer was extracted with dichloromethane. The organic layers were washed with water, dried over Na
2SO
4, filtered and concentrated under reduced pressure to yield
20 as a colorless oil (4.69 g, 97%) yield based on the
p-toluenesulfomyl chloride.
Rf = 0.25 (in 100% EtOAc).
1H-NMR (300 MHz, CDCl
3) δ 7.79 (d,
J = 8.0 Hz, 2H, b), 7.33 (d,
J = 8.0 Hz, 2H, d), 4.16 (t,
J = 4.7 Hz, 2H, a), 3.75–3.55 (m, 14H, e), 2.44 (s, 3H, c).
2-(2-{2-[2-(2-Tosyloxy-ethoxy)-ethoxy]-ethoxy}-ethyl) 2,3,4,6-tetra-O-acetyl-α-d-mannopyranoside (
21) [
53]. Into a solution of pentaacetate mannose
17 (1.68 g, 4.31 mmol, 1 equiv.) in anhydrous CH
2Cl
2 (20 mL) was added boron trifluoride etherate (1.23 mL, 9.93 mmol, 2.3 equiv.) at room temperature under nitrogen atmosphere. The solution was stirred for 4 h before compound
20 (3.76 g, 10.79 mmol, 2.5 equiv.) was added. Glycosylation was completed after stirring at 40 °C overnight. The crude product was washed with NaHCO
3 sat, dried over Na
2SO
4 and concentrated under reduced pressure. Further purification was processed on silica gel flash chromatography using 0–20% EtOAc in Hexane as eluents to afford compound
21 as a colorless oil (1.8 g, 60%).
Rf = 0.35 (EtOAc/Hexane, 1:4).
1H-NMR (300 MHz, CDCl
3) δ 7.80 (d,
J = 8.3 Hz, 2H, aromatic), 7.34 (d,
J = 8.0 Hz, 2H, aromatic), 5.38–5.25 (m, 3H,
H-3, 4 and 2), 4.87 (d,
J = 1.6 Hz, 1H,
H-1), 4.36–4.19 (m, 2H,
H-6), 4.18–4.01 (m, 3H, c and
H-5), 3.85–3.75 (m, 1H, j’), 3.73–3.56 (m, 13H, j, i, d, g, f, e), 2.45 (s, 3H, C
H3), 2.16 (d, 3H, COC
H3), 2.09 (s, 3H, COC
H3), 2.03 (s, 3H, COC
H3), 1.99 (s, 3H, COC
H3).
2-(2-{2-[2-(2-Azido-ethoxy)-ethoxy]-ethoxy}-ethyl) 2,3,4,6-tetra-O-acetyl-α-d-mannopyranoside (
22) [
53]. To a solution of
21 (520 mg, 0.766 mmol, 1 equiv.) in dry DMF (15 mL) under a nitrogen atmosphere were added sodium azide (996 mg, 15.3 mmol, 20 equiv.). After stirring at 80 °C for 2 h, the solution was diluted in EtOAc and further processed as given in general procedure (
II) to afford compound
22 as a colorless oil (380 mg, 90%),
Rf = 0.4 (EtOAc/Hexane, 1:4).
1H-NMR (300 MHz, CDCl
3) δ 5.43–5.24 (m, 3H,
H-3, 4 and 2), 4.87 (d,
J = 1.6 Hz, 1H,
H-1), 4.27 (td,
J = 12.6, 5.0 Hz, 1H,
H-6), 4.14–4.03 (m, 2H,
H-6′ and 5), 3.86–3.76 (m, 1H, j′), 3.71–3.64 (m, 13H, j, i, d, g, e, h, f), 3.39 (t,
J = 5.1 Hz, 2H,
CH2-N3), 2.15 (s, 3H,
CO
CH
3), 2.10 (s, 3H,
CO
CH
3), 2.04 (s, 3H,
CO
CH
3), 1.99 (s, 3H,
CO
CH
3).
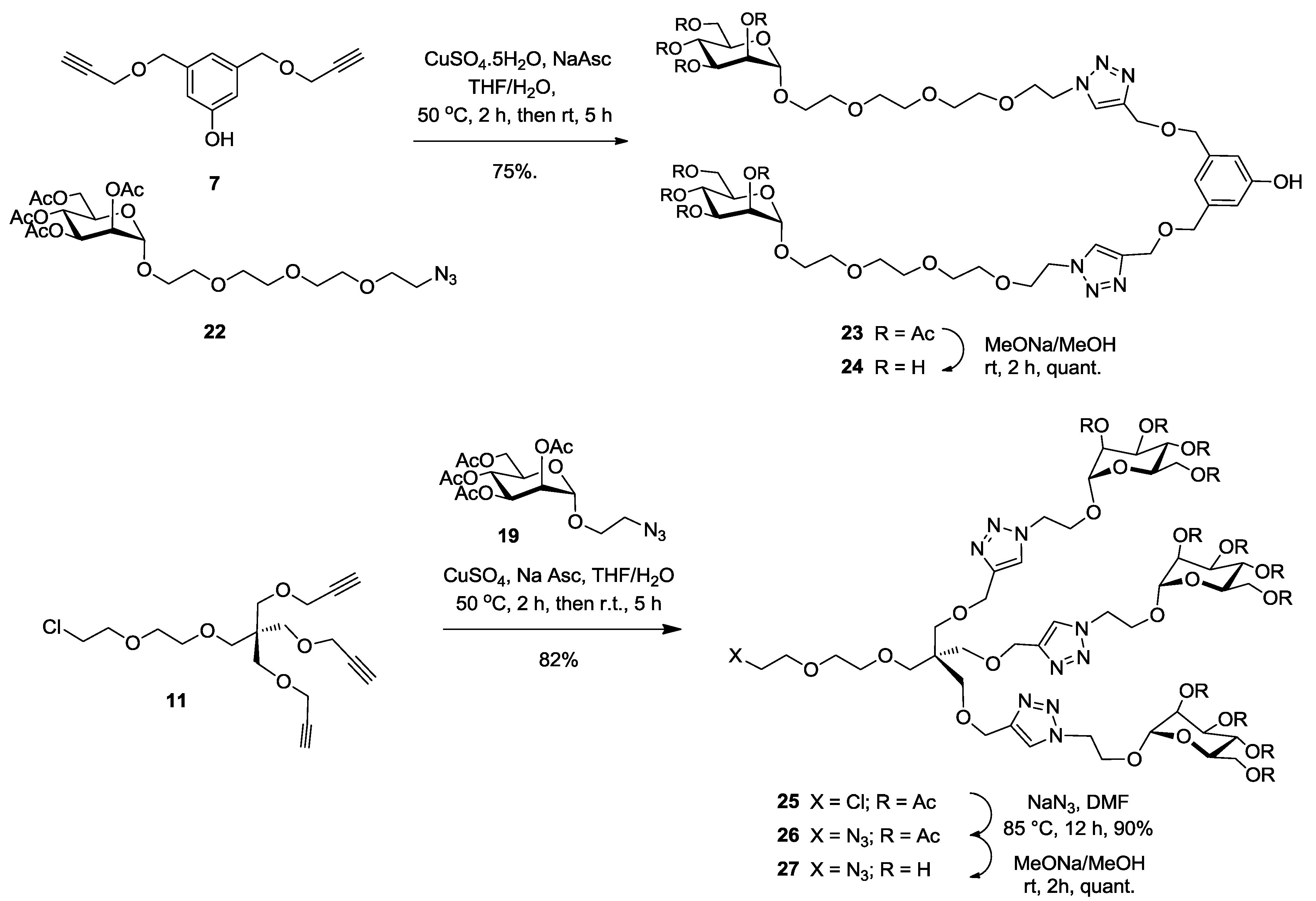
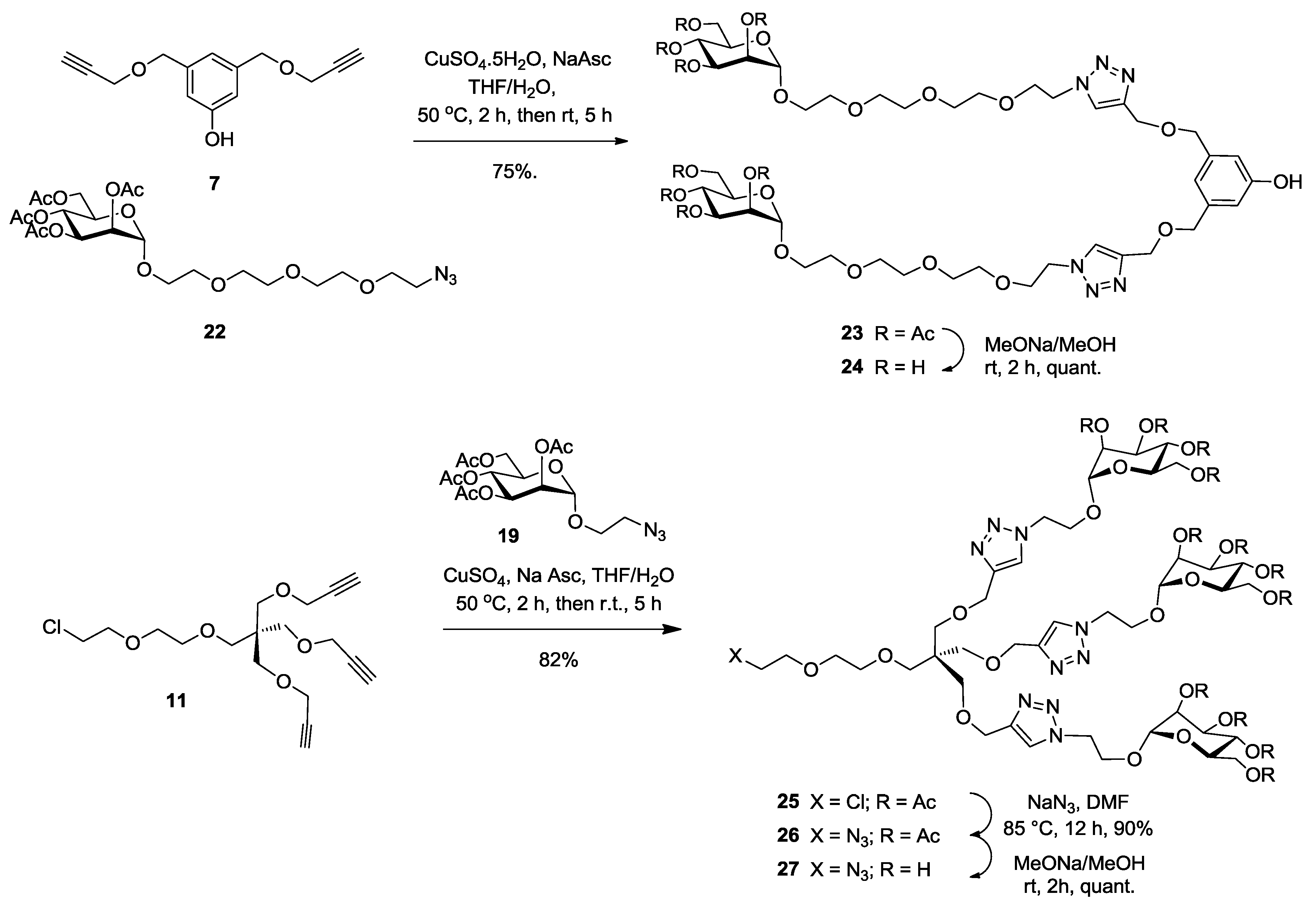
Synthesis of23. Into a solution of bispropargylated core (7) (30 mg, 0.13 mmol, 1.0 equiv.) in a mixture of THF/H2O (4 mL, 3:1, v/v), was added 2-(2-{2-[2-(2-Azido-ethoxy)-ethoxy]-ethoxy}-ethyl) 2,3,4,6-tetra-O-acetyl-α-d-mannopyranoside (22) (220 mg, 0.39 mmol, 3 equiv.), Na-ascorbate (18 mg, 0.09 mmol, 0.7 equiv.) and CuSO4·5H2O (19 mg, 0.08 mmol, 0.6 equiv.). The mixture was stirred at 50 °C for 2 h, then at room temperature for 5 h. Upon completion of the reaction, EtOAc was added to the reaction mixture and further processed as given in general procedure (I) to afford compound 23 (136 mg, 75%) as a white powder; Rf = 0.35 (with 5% MeOH in CH2Cl2 as eluents). 1H-NMR (600 MHz, CDCl3) δ 7.74 (s, 2H, H-triazole), 6.77 (s, 1H, HP-aromatic), 6.74 (s, 2H, HO-aromatic), 5.30 (dd, J = 10.0, 3.4 Hz, 2H, H-3), 5.25 (d, J = 9.9 Hz, 2H, H-4), 5.23–5.20 (m, 2H, H-2), 4.82 (s, 2H, H-1), 4.61 (s, 4H, b), 4.52–4.49 (m, 4H, c), 4.47 (s, 4H, m), 4.24 (dd, J = 12.2, 4.8 Hz, 2H, H-6), 4.05 (dd, J = 12.3, 4.1 Hz, 2H, H-6′), 4.03–3.99 (m, 2H, H-5), 3.83 (t, J = 4.9 Hz, 4H, d), 3.77–3.73 (m, 2H, j), 3.61 (m, 2H, j′), 3.60 (s, 4H, i), 3.58 (s, 16H, g, f, e, h), 2.11 (s, 6H, COCH3), 2.05 (s, 6H, COCH3, 1.99 (s, 6H, COCH3), 1.94 (s, 6H, COCH3). 13C-NMR (150 MHz, CDCl3) δ 170.6 (C=O), 169.9 (C=O), 169.8 (C=O), 169.6 (C=O), 156.8 (COH), 144.7 (C-triazole), 139.5 (Cm-aromatic), 123.8 (CH-triazole), 118.4 (Cp-aromatic), 113.9 (CO-aromatic), 97.5 (C-1), 71.9 (Cm), 70.5 (Cd), 70.4 (Cg), 70.3 (Cf), 69.8 (Ce), 69.4 (Ch), 69.4 (Cj), 69.3 (C-2), 68.9 (C-3), 68.2 (C-4), 67.2 (C-5), 65.9 (Ci), 63.4 (Cb), 62.2 (C-6), 50.1 (Cc), 20.8 (COCH3), 20.6 (COCH3), 20.6 (COCH3). ESI+-HRMS: [M + Na]+ calcd for C56H86N6NaO29, 1329.5331; found: 1329.5395.
Synthesis of24. Compound 23 (50 mg, 0.036 mmol) and sodium methoxide (16 μL from 1 M solution in MeOH) in 3 mL of methanol were stirred for 4 h and the mixture was treated following the general procedure (III) described above. Deprotected compound 24 (38 mg, quant.) was obtained as a colorless solid. 1H-NMR (600 MHz, D2O) δ 8.04 (s, 2H), 6.89 (s, 1H), 6.82 (s, 2H), 4.84 (s, 2H), 4.70 (s, 4H), 4.66–4.59 (m, 4H), 4.57 (s, 4H), 3.98–3.94 (m, 4H), 3.93–3.80 (m, 4H), 3.80–3.72 (m, 4H), 3.67–3.60 (m, 12H), 3.60–3.55 (m, 8H), 3.55–3.52 (m, 4H). 13C-NMR (150 MHz, D2O) δ 215.4, 155.9, 143.9, 139.3, 125.5, 120.1, 114.7, 99.8, 72.7, 71.7, 70.5, 69.9, 69.7, 69.6, 69.5, 69.4, 69.4, 68.7, 66.7, 66.3, 62.4, 60.9, 49.9, 30.2. ESI+-HRMS: [M + Na] + calcd for C40H70N6NaO21, 993.4486; found: 993.4545.
Synthesis of25. Into a solution of tripropargyl pentaerythritol core (11) (125 mg, 0.35 mmol, 1.0 equiv.) in a mixture of THF/H2O (4 mL, 3:1, v/v), was added 2-azidoethyl 2,3,4,6-tetra-O-acetyl-α-d-mannopyranoside (19) (745 mg, 1.42 mmol, 5.1 equiv.), Na-ascorbate (70 mg, 0.29 mmol, 1.1 equiv.) and CuSO4·5H2O (78 mg, 0.31 mmol, 0.9 equiv.). The mixture was stirred at 50 °C for 2 h, then at room temperature for 5 h. Upon completion of the reaction, EtOAc was added to the reaction mixture and further processed as given in general procedure (I) to afford compound 25 (480 mg, 82%) as a white powder; Rf = 0.3 (with 5% MeOH in CH2Cl2 as eluents). 1H-NMR (300 MHz, CDCl3) δ 7.70 (s, 3H, HB-triazole), 5.31–5.19 (m, 9H, H-3, 4 and 2), 4.81 (d, J = 1.3 Hz, 3H, H-1), 4.61 (m, 6 H, i), 4.59 (s, 6H, h), 4.21 (dd, J = 12.3, 5.1 Hz, 3H, H-6), 4.13 (dt, J = 10.4, 4.9 Hz, 3H, j′), 4.04 (dd, J = 12.3, 2.3 Hz, 3H, H-6′), 3.91 (dt, J = 10.6, 5.2 Hz, 3H, j), 3.74 (m, 2H, d), 3.61 (m, 7 H, H-5, f and CH2-Cl), 3.57–3.53 (m, 2H, e), 3.49 (s, 6H, g′), 3.44 (s, 2H, g), 2.13 (s, 9H, COCH3), 2.09 (s, 9H, COCH3), 2.04 (s, 9H, COCH3), 1.99 (s, 9H, COCH3).
Synthesis of26. To a solution of compound 25 (480 mg, 0.29 mmol, 1.0 equiv.) in dry DMF (2 mL) under a nitrogen atmosphere were added sodium azide (186 mg, 2.9 mmol, 10 equiv.). After stirring at 80 °C for 2 h, the solution was diluted in EtOAc and further processed as given in general procedure (II) to afford compound 26 as a white powder (430 mg, 90%). 1H-NMR (300 MHz, CDCl3) δ 7.65 (s, 3H, HB-triazole), 5.28–5.09 (m, 9H, H-3, 4 and 2), 4.76 (d, J = 1.1 Hz, 3H, H-1), 4.6–4.57 (m, 6H, i), 4.53 (s, 6H, h), 4.15 (dd, J = 12.4, 5.1 Hz, 3H, H-6), 4.07 (dt, J = 10.5, 5.7 Hz, 3H, j’), 3.98 (dd, J = 12.3, 2.3 Hz, 3H, H-6′), 3.86 (dt, J = 10.4, 5.1 Hz, 3H, j), 3.63–3.59 (m, 2H, d), 3.56–3.53 (m, 5H, H-5, f), 3.49 (m, 2H, e), 3.44 (s, 6H, g′), 3.39 (s, 2H, g), 3.33–3.27 (m, 2H, c, CH2-N3), 2.07 (s, 9H, COCH3), 2.03 (s, 9H, COCH3), 1.98 (s, 9H, COCH3), 1.94 (s, 9H, COCH3). 13C-NMR (75 MHz, CDCl3) δ 170.6 (C=O), 170.0 (C=O), 169.6 (C=O), 169.1 (C=O), 145.6 (C-triazole), 123.7 (CHB-triazole), 97.6 (C-1), 77.2 (Cd), 71.0 (Cf), 70.3 (Ce), 70.0 (Cg), 69.2 (C-2), 69.1 (C-3), 68.9 (C-4), 66.3 (C-5), 65.7 (Cj), 64.8 (Ch), 62.2 (C-6), 51.8 (Ci), 50.8 (Cc), 49.5 (Cq), 20.8 (COCH3), 20.7 (COCH3).
Synthesis of27. Compound 26 (200 mg, 0.02 mmol) and sodium methoxide (16 μL from 1 M solution in MeOH) in 3 mL of methanol were stirred for 4 h and the mixture was treated following the general procedure (III) described above. Deprotected compound 27 (150 mg, quant.) was obtained as a white powder. 1H-NMR (300 MHz, MeOD) δ 8.00 (s, 3H), 4.69–4.60 (m, 9H), 4.55 (s, 6H), 4.11 (dt, J = 10.3, 4.9 Hz, 3H), 3.88 (dt, J = 5.8, 4.6 Hz, 3H), 3.81–3.71 (m, 6H), 3.65 (ddd, J = 18.4, 7.4, 5.7 Hz, 18H), 3.58–3.49 (m, 4H), 3.46 (s, 6H), 3.40 (dd, J = 9.5, 4.3 Hz, 6H), 3.21 (dd, J = 7.6, 5.1 Hz, 4H). 13C-NMR (75 MHz, MeOD) δ 146.08, 126.31, 101.33, 79.41, 74.58, 72.26, 72.06, 71.69, 71.35, 70.99, 70.1, 68.1, 66.9, 65.2, 62.4, 51.8, 51.4, 50.1, 49.9, 49.6, 49.3, 49.0, 49.0, 48.7, 48.4, 46.5, 31.3, 24.6. ESI+-HRMS: [M + 2H]2+ calcd for C42H72N12O23, 556.2418; found: 556.2470.
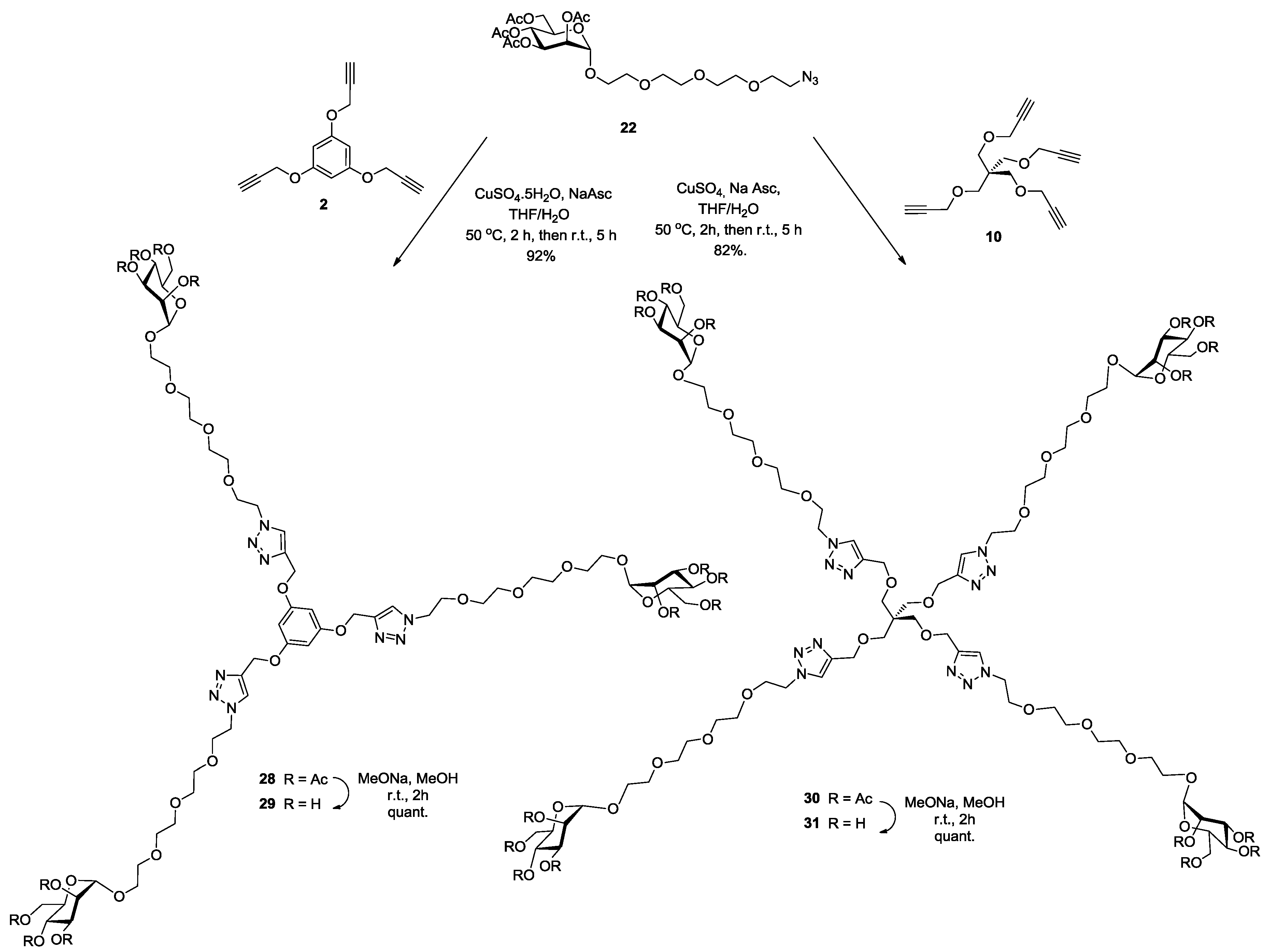
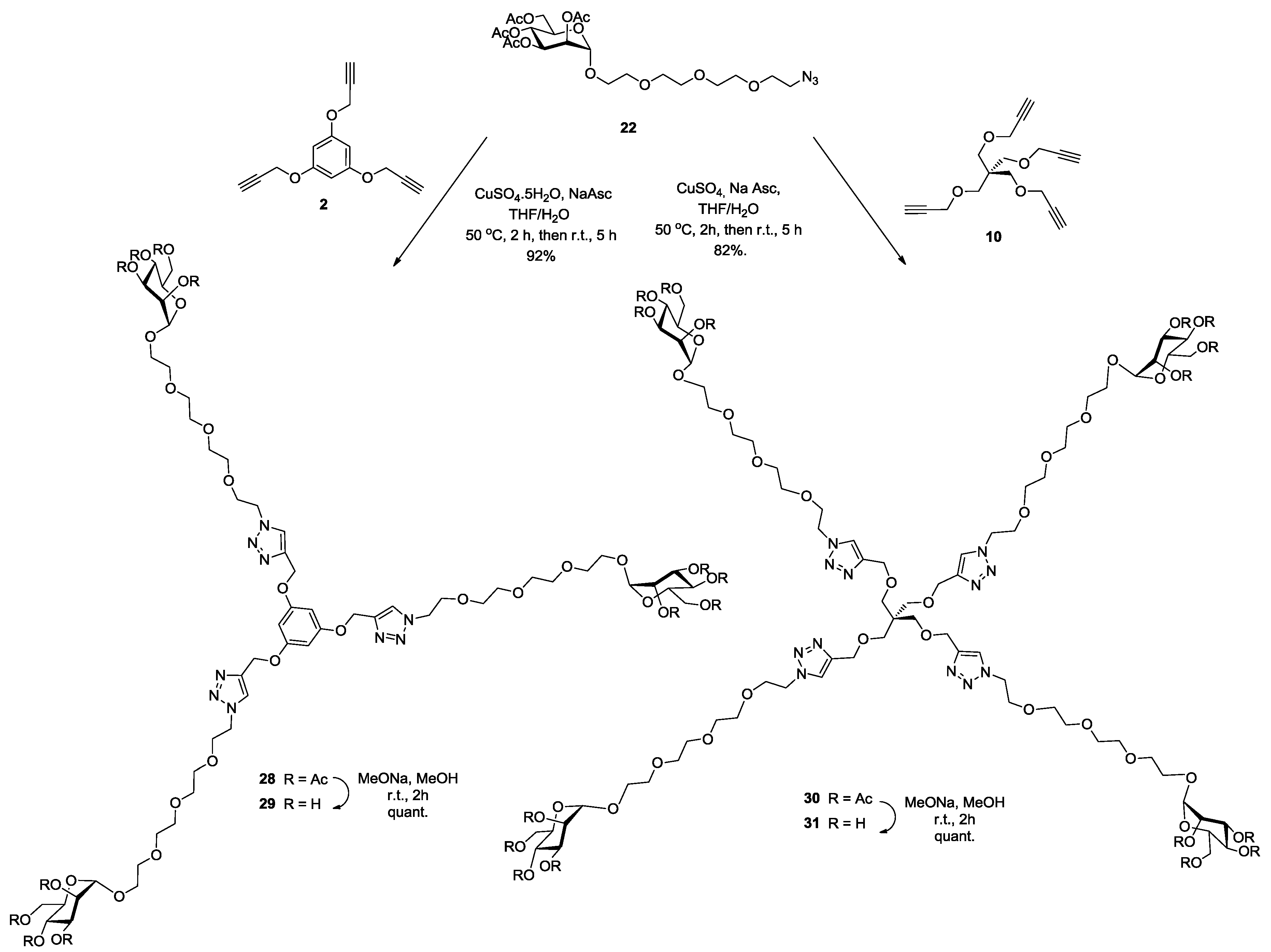
Synthesis of28. Into a solution of trispropargylated core (2) (25 mg, 0.104 mmol, 1.0 equiv.) in a mixture of THF/H2O (4 mL, 3:1, v/v), was added 2-(2-{2-[2-(2-Azido-ethoxy)-ethoxy]-ethoxy}-ethyl) 2,3,4,6-tetra-O-acetyl-α-d-mannopyranoside (22) (281 mg, 0.5 mmol, 5 equiv.), Na-ascorbate (22 mg, 0.11 mmol, 0.5 equiv.) and CuSO4·5H2O (23 mg, 0.09 mmol, 0.9 equiv.). The mixture was stirred at 50 °C for 2 h, then at room temperature for 5 h. Upon completion of the reaction, EtOAc was added to the reaction mixture and further processed as given in general procedure (I) to afford compound 28 (188 mg, 92%) as a white powder; Rf = 0.32 (with 5% MeOH in CH2Cl2 as eluents). 1H-NMR (600 MHz, CDCl3) δ 7.83 (s, 3H, H-triazole), 6.28 (s, 3H, k) 5.32 (dd, J = 10.0, 2.8 Hz, 3H, H-3), 5.27 (t, J = 10.0 Hz, 3H, H-4), 5.25 (dd, J = 3.0, 1.7 Hz, 3H, H-2), 5.12 (s, 6H, b), 4.85 (d, J = 1.4 Hz, 3H, H-1), 4.55 (t, J = 4.9 Hz, 6H, c), 4.27 (dd, J = 12.4, 5.1 Hz, 3H, H-6), 4.07 (d, J = 12.8 Hz, 3H, H-6′), 4.03 (dd, J = 5.7, 3.5 Hz, 3H, H-5), 3.89 (t, J = 4.9 Hz, 6H, d), 3.82–3.73 (m, 3H, j), 3.65 (m, 3H, j′), 3.63 (s, 6H, i), 3.61 (s, 24H, g, e, f, h), 2.13 (s, 9H, COCH3), 2.07 (s, 9H, COCH3), 2.01 (s, 9H, COCH3), 1.96 (s, 9H, COCH3). 13C-NMR (150 MHz, CDCl3) δ 170.8 (C=O), 170.1 (C=O), 170.0 (C=O), 169.8 (C=O), 160.3 (COCb-aromatic), 143.7 (C-triazole), 124.2 (CH-triazole), 97.8 (C-1), 95.1 (Ck-aromatic), 70.8 (Cd), 70.7 (Cg), 70.6 (Cf), 70.1 (Ce), 69.7 (Ch), 69.6 (Cj), 69.5 (C-2), 69.2 (C-3), 68.5 (C-4), 67.5 (C-5), 66.2 (Ci), 62.5 (Cb), 62.1 (C-6), 50.4 (Cc), 21.0 (COCH3), 20.9 (COCH3), 20.8 (COCH3), 20.8 (COCH3). MALDI-TOF: [M + H]+ calcd for C81H118N9O42: 1888.737; found, 1888.789.
Synthesis of29. Compound 28 (100 mg, 0.05 mmol) and sodium methoxide (16 μL from 1 M solution in MeOH) in 3 mL of methanol were stirred for 4 h and the mixture was treated following the general procedure (III) described above. Deprotected compound 29 (74 mg, quant.) was obtained as a white solid. 1H-NMR (600 MHz, MeOD) δ 8.16 (s, 3H), 6.37 (s, 3H), 5.19 (s, 6H), 4.65–4.63 (m, 8H), 3.95–3.92 (m, 4H), 3.88–3.87 (m, 2H), 3.82 (m, 10.0, 3.1 Hz, 4H), 3.77–3.71 (m, 4H), 3.68–3.54 (m, 23H). 13C-NMR (150 MHz, MeOD) δ 159.9, 143.3, 125.4, 100.1, 95.5, 72.9, 70.9, 70.3, 69.9, 69.8, 69.8, 68.9, 66.9, 66.4, 61.1, 50.2, 48.5, 47.9, 47.9, 47.8, 47.8, 47.8. MALDI-TOF: [M]+ calcd for C57H92N9O30: 1382.592; found, 1382.801.
Synthesis of30. Into a solution of tetrapropargyl pentaerythritol core (10) (67 mg, 0.23 mmol, 1.0 equiv.) in a mixture of THF/H2O (4 mL, 3:1, v/v), was added 2-(2-{2-[2-(2-Azido-ethoxy)-ethoxy]-ethoxy}-ethyl) 2,3,4,6-tetra-O-acetyl-α-d-mannopyranoside (22) (790 mg, 1.4 mmol, 6 equiv.), Na-ascorbate (65 mg, 0.32 mmol, 1.4 equiv.) and CuSO4·5H2O (70 mg, 0.28 mmol, 1.2 equiv.). The mixture was stirred at 50 °C for 2 h, then at room temperature for 5 h. Upon completion of the reaction, EtOAc was added to the reaction mixture and further processed as given in general procedure (I) to afford compound 30 as a white powder (484 mg, 82%). Rf = 0.27 (with 5% MeOH in CH2Cl2 as eluents). 1H-NMR (600 MHz, CDCl3) δ 7.68 (s, 4H, H-triazole), 5.32 (dd, J = 10.0, 3.4 Hz, 4H, H-3), 5.26 (t, J = 10.0 Hz, 4H, H-2), 5.24 (dd, J = 3.2, 1.6 Hz, 4H, H-4), 4.85 (s, 4H, H-1), 4.52 (s, 8H, b), 4.51 (t, J = 5.5 Hz, 8H, c), 4.27 (dd, J = 12.2, 4.9 Hz, 4H, H-6), 4.07 (dd, J = 12.2, 2.2 Hz, 4H, H-6′), 4.04 (ddd, J = 9.8, 4.8, 2.3 Hz, 4H, H-5), 3.87 (t, J = 5.3 Hz, 8H, d), 3.81–3.75 (m, 4H, j), 3.66 (dd, J = 5.8, 3.7 Hz, 4H, j′), 3.64–3.63 (m, 8H, i), 3.62–3.58 (m, 32H, g, f, e, h), 3.45 (s, 8H, k), 2.13 (s, 12H, COCH3), 2.08 (s, 12H, COCH3), 2.01 (s, 12H, COCH3), 1.96 (s, 12H, COCH3). 13C-NMR (150 MHz, CDCl3) δ 170.7 (C=O), 170.0 (C=O), 169.9 (C=O), 169.7 (C=O), 145.1 (C-triazole), 123.6 (CH-triazole), 97.7 (C-1), 70.7 (Cd), 70.6 (Cg), 70.5 (Cf), 69.9 (Ce), 69.6 (Ch), 69.5 (Cj), 69.5 (C-2), 69.2 (Cq), 69.1 (C-3), 68.4 (C-4), 67.4 (C-5), 66.1 (Ci), 64.9 (Cb), 62.4 (C-6), 50.1 (Cc), 45.3 (Ck), 20.9 (COCH3), 20.8 (COCH3), 20.7 (COCH3), 20.7 (COCH3). ESI+-HRMS: [M + 2H]2+ calcd for C105H162N12O56, 1244.5021; found: 1244.0172.
Synthesis of31. Compound 30 (100 mg, 0.038 mmol) and sodium methoxide (16 μL from 1 M solution in MeOH) in 3 mL of methanol were stirred for 4 h and the mixture was treated following the general procedure (III) described above. Deprotected compound 31 (70 mg, quant.) was obtained as a colorless solid. 1H-NMR (600 MHz, D2O) δ 8.01 (s, 4H), 4.87 (s, 4H), 4.60 (t, J = 4.9 Hz, 8H), 4.52 (s, 8H), 3.95 (d, J = 4.8 Hz, 12H), 3.90–3.86 (m, 4H), 3.85 (dd, J = 7.4, 3.7 Hz, 4H), 3.81 (dd, J = 9.1, 3.2 Hz, 4H), 3.75 (dd, J = 12.2, 5.4 Hz, 4H), 3.72–3.65 (m, 12H), 3.65–3.60 (m, 24H), 3.60–3.57 (m, 16H), 3.40 (s, 8H). 13C-NMR (150 MHz, D2O) δ 144.1, 125.3, 99.9, 72.7, 70.5, 69.9, 69.9, 69.7, 69.6, 69.5, 69.5, 68.7, 68.1, 66.7, 66.3, 63.5, 60.9, 49.9, 44.6. ESI+-HRMS: [M + 2H]2+ calcd for C73H130N12O40, 907.4248; found: 907.4258.
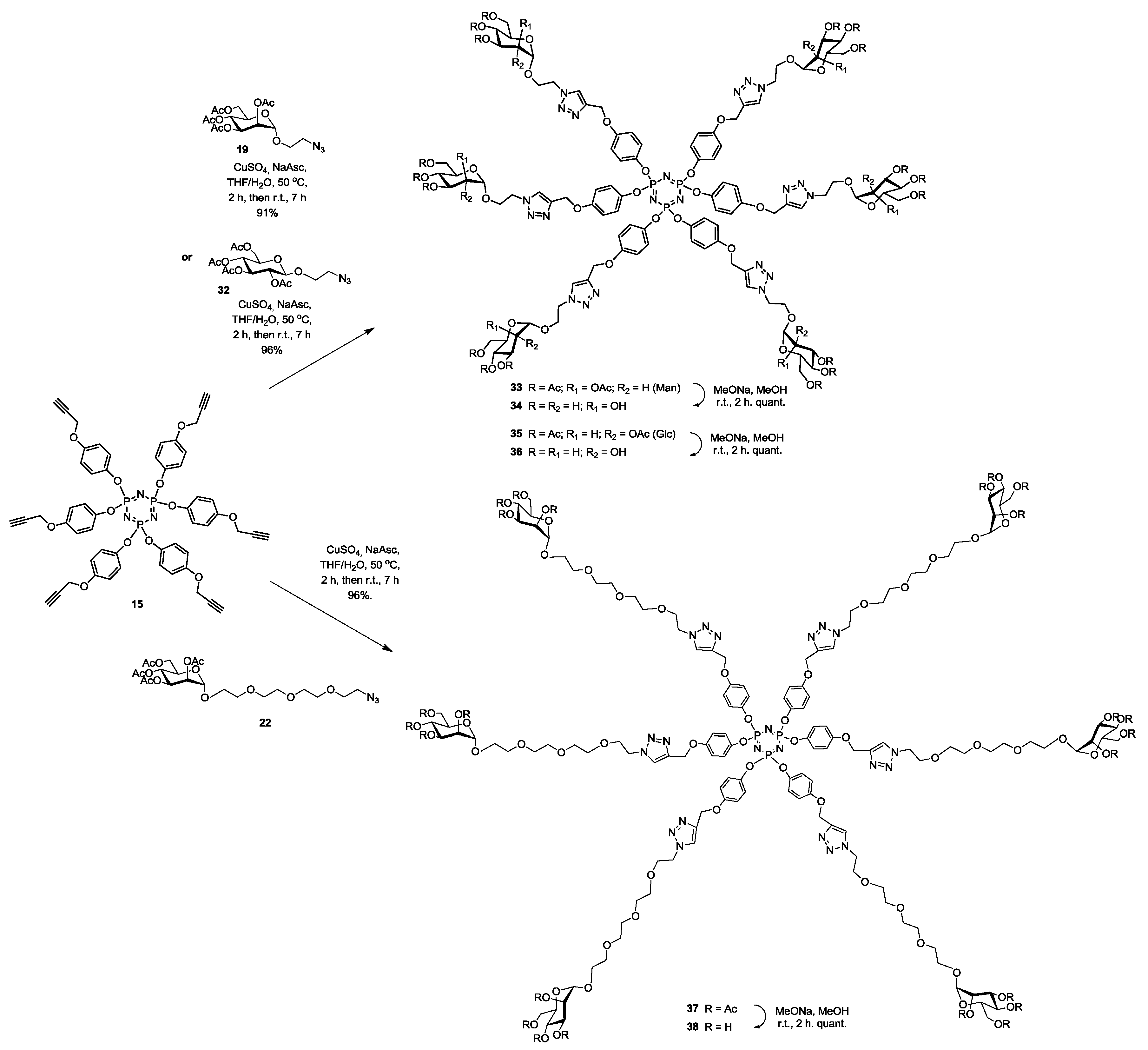
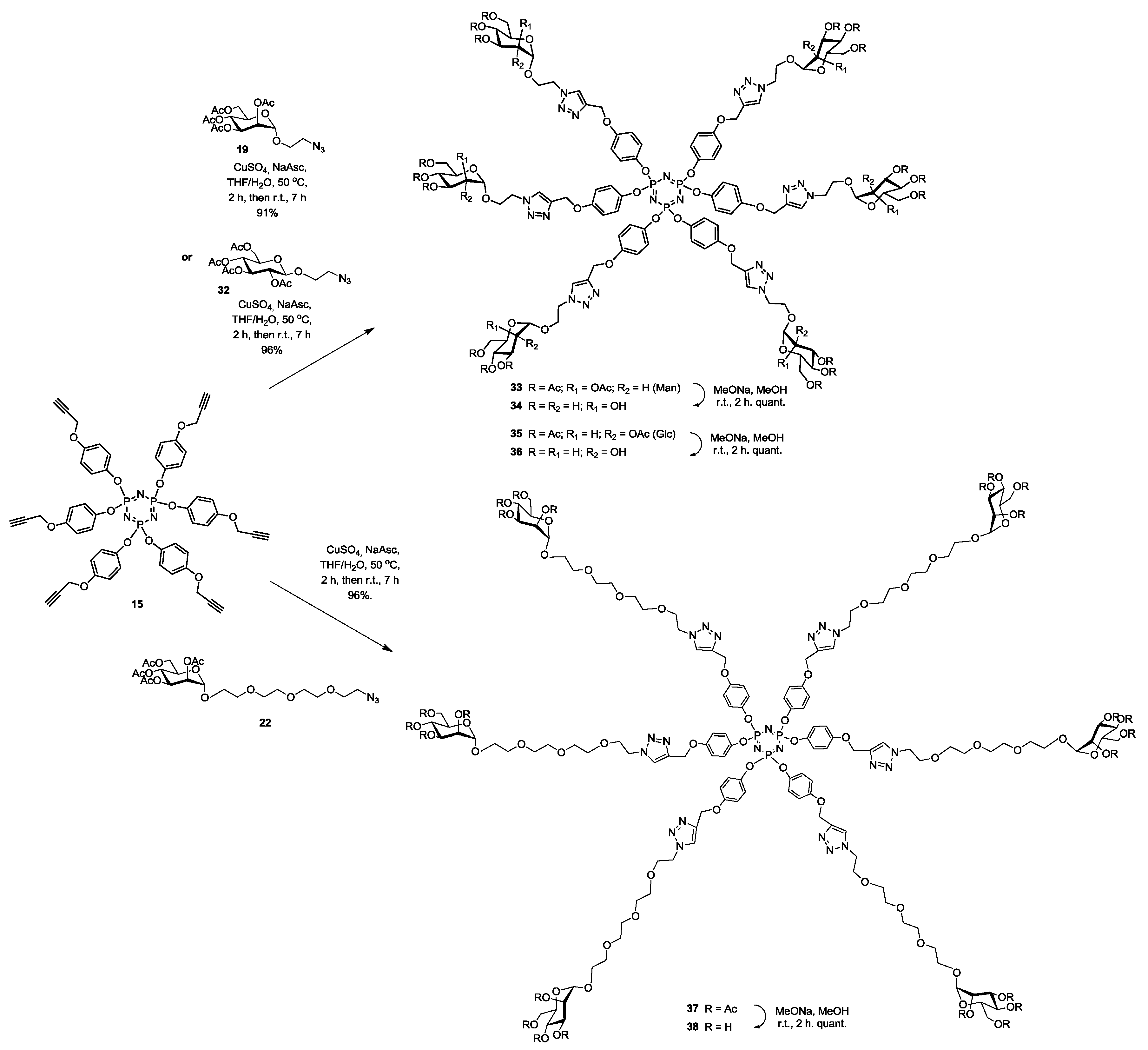
Synthesis of33. Into a solution of hexapropargylated core (15) (30 mg, 0.03 mmol, 1.0 equiv.) in a mixture of THF/H2O (4 mL, 3:1, v/v), was added 19 (152 mg, 0.29 mmol, 9.6 equiv.), Na-ascorbate (12 mg, 0.06 mmol, 1.98 equiv.) and CuSO4·5H2O (14 mg, 0.05 mmol, 1.8 equiv.). The mixture was stirred at 50 °C for 2 h, then at room temperature for 5 h. Upon completion of the reaction, EtOAc was added to the reaction mixture and further processed as given in general procedure (I) to afford compound 33 as a white powder (97 mg, 91%); Rf = 0.3 (with 5% MeOH in CH2Cl2 as eluents). 1H-NMR (600 MHz, CDCl3) δ 7.83 (s, 6H, H-triazole), 6.85 (d, J = 9.0 Hz, 12H, aromatic), 6.79 (d, J = 9.1 Hz, 12H, aromatic), 5.31–5.19 (m, 18H, H-2, 3 and 4), 5.16–5.08 (m, 12H, Ar-OCH2), 4.82 (s, 6H, H-1), 4.64–4.54 (m, 12H, Man-OCH2), 4.21 (dd, J = 12.3, 5.1 Hz, 6H, CHH′N), 4.16–4.09 (m, 6H, H-6), 4.04 (dd, J = 12.3, 2.2 Hz, 6H, H-6′), 3.90 (dt, J = 10.4, 5.1 Hz, 6H, CHH′N), 3.65–3.58 (m, 6H, H-5), 2.13 (s, 18H, COCH3), 2.09 (s, 18H, COCH3), 1.98 (s, 18H, COCH3), 1.97 (s, 18H, COCH3). 13C-NMR (151 MHz, CDCl3) δ 170.8 (C=O), 170.2 (C=O), 170.2 (C=O), 169.9 (C=O), 155.7 (CO-aromatic), 145.1 (C-triazole), 144.5 (Cp-aromatic), 124.5 (CH-triazole), 122.2 (Cm-aromatic), 115.7 (CO-aromatic), 97.9 (C-1), 77.2 (C-2), 69.6 (C-3), 69.5 (C-4), 69.3 (C-5), 66.6 (Cj), 66.2 (Cb), 62.7 (C-6), 50.0 (Ci), 21.1 (COCH3), 21.0 (COCH3), 20.9 (COCH3). 31P-NMR (243 MHz, CDCl3) δ 9.84. ESI+-HRMS: [M + 3H]3+ calcd for C150H183N21O72P3, 1174.34752; found: 1174.6753.
Synthesis of34. Compound 33 (97 mg, 0.03 mmol) and sodium methoxide (16 μL from 1 M solution in MeOH) in 3 mL of methanol were stirred for 4 h and the mixture was treated following the general procedure (III) described above. Deprotected compound 34 (69 mg, quant.) was obtained as a white solid. 1H-NMR (600 MHz, D2O) δ 7.80 (s, 6H), 6.56 (s, 24H), 4.84 (s, 18H), 4.65 (s, 6H), 4.36 (s, 12H), 3.87 (s, 6H), 3.74 (s, 6H), 3.71–3.50 (m, 24H), 3.05 (d, J = 3.2 Hz, 6H). 13C-NMR (151 MHz, D2O) δ 154.9, 143.8, 143.1, 125.1, 121.6, 115.5, 99.5, 72.8, 70.5, 69.9, 66.4, 65.2, 61.3, 60.6, 49.8. 31P-NMR (243 MHz, CDCl3) δ 10.16. ESI+-HRMS: [M + 3H]3+ calcd for C102H135N21O48P3, 838.26603; found: 838.5901.
Synthesis of35. Into a solution of hexapropargylated core (15) (50 mg, 0.049 mmol, 1.0 equiv.) in a mixture of THF/H2O (4 mL, 3:1, v/v), was added 32 (185 mg, 0.44 mmol, 6 equiv.), Na-ascorbate (20 mg, 0.1 mmol, 0.1 equiv.) and CuSO4·5H2O (22 mg, 0.28 mmol, 0.09 equiv.). The mixture was stirred at 50 °C for 2 h, then at room temperature for 5 h. Upon completion of the reaction, EtOAc was added to the reaction mixture and further processed as given in general procedure (I) to afford compound 35 as a white powder (171 mg, 96%); Rf = 0.2 (with 5% MeOH in CH2Cl2 as eluents). 1H-NMR (600 MHz, CDCl3) δ 7.74 (s, 6H, H-triazole), 6.92–6.74 (m, 24H, aromatic), 5.16 (t, J = 9.5 Hz, 6H, H-3), 5.11 (d, J = 6.3 Hz, 12H, b), 5.05 (t, J = 9.7 Hz, 6H, H-4), 4.98 (dd, J = 9.6, 8.0 Hz, 6H, H-2), 4.60 (dt, J = 14.4, 4.0 Hz, 6H, j), 4.53–4.47 (m, 12H, j′, H-1), 4.24 (dt, J = 8.4, 4.5 Hz, 12H, i, H-6), 4.13 (dd, J = 12.3, 2.2 Hz, 6H, H-6′), 3.93 (ddd, J = 10.6, 8.8, 3.5 Hz, 6H, i′), 3.70 (ddd, J = 10.0, 4.6, 2.4 Hz, 6H, H-5), 2.07 (s, 18H, COCH3), 2.01 (s, 18H, COCH3), 1.97 (s, 18H, COCH3), 1.92 (s, 18H, COCH3). 13C-NMR (150 MHz, CDCl3) δ 169.6 (C=O), 169.1 (C=O), 168.4 (C=O), 168.4 (C=O), 154.2 (CO-aromatic), 143.6 (C-triazole), 142.7 (Cp-aromatic), 123.3 (CH-triazole), 120.9 (Cm-aromatic), 114.3 (CO-aromatic), 99.5 (C-1), 71.5 (C-2), 70.9 (C-3), 69.9 (C-4), 67.2 (C-5), 66.7 (Cj), 61.2 (Cb), 60.7 (C-6), 48.9 (Ci), 19.7 (COCH3), 19.7 (COCH3), 19.6 (COCH3), 19.5 (COCH3). 31P-NMR (243 MHz, CDCl3) δ 9.72. MALDI-TOF: [M]+ calcd for C150H179N21O72P3: 3519.020; found, 3519.489.
Synthesis of36. Compound 35 (100 mg, 0.03 mmol) and sodium methoxide (16 μL from 1 M solution in MeOH) in 3 mL of methanol were stirred for 4 h and the mixture was treated following the general procedure (III) described above. Deprotected compound 36 (78 mg, quant.) was obtained as a white powder. 1H-NMR (600 MHz, MeOD) δ 8.05 (s, 6H), 6.70 (dd, J = 40.7, 9.1 Hz, 24H), 5.02 (s, 12H), 4.51 (t, J = 5.0 Hz, 12H), 4.26 (d, J = 7.9 Hz, 6H), 4.17–4.07 (m, 6H), 4.01–3.84 (m, 6H), 3.75 (dd, J = 12.1, 1.9 Hz, 6H), 3.55 (dd, J = 12.1, 5.8 Hz, 6H), 3.31 (t, J = 9.0 Hz, 6H), 3.27–3.22 (m, 6H), 3.18 (d, J = 9.6 Hz, 6H), 3.11 (dd, J = 9.2, 8.0 Hz, 6H). 13C-NMR (150 MHz, MeOD) δ 156.3, 145.2, 144.2, 126.5, 122.7, 116.6, 103.8, 77.3, 77.1, 74.3, 70.9, 68.9, 62.4, 62.1, 51.4, 49.4, 49.0, 24.2. 31P-NMR (243 MHz, MeOD) δ 10.25. MALDI-TOF: [M + H]+ calcd for C102H132N21O48P3: 2512.778; found, 2512.851.
Synthesis of37. Into a solution of hexapropargylated core (15) (25 mg, 0.025 mmol, 1.0 equiv.) in a mixture of THF/H2O (4 mL, 3:1, v/v), was added compound 22 (140 mg, 0.25 mmol, 10 equiv.), Na-ascorbate (10 mg, 0.05 mmol, 2.1 equiv.) and CuSO4·5H2O (10.8 mg, 0.04 mmol, 1.8 equiv.). The mixture was stirred at 50 °C for 2 h, then at room temperature for 5 h. Upon completion of the reaction, EtOAc was added to the reaction mixture and further processed as given in general procedure (I) to afford compound 37 as a white powder (101 mg, 96%); Rf = 0.22 (with 4% MeOH in CH2Cl2 as eluents). 1H-NMR (600 MHz, CDCl3) δ 7.85 (s, 6H, H-triazole), 6.81 (dd, J = 29.7, 9.0 Hz, 24H, aromatic), 5.33 (dd, J = 10.0, 3.4 Hz, 6H, H-3), 5.28 (t, J = 10.0 Hz, 6H, H-2), 5.25 (dd, J = 3.2, 1.6 Hz, 6H, H-4), 5.13 (d, J = 7.6 Hz, 12H, b), 4.86 (s, 6H, H-1), 4.54 (t, J = 5.1 Hz, 12H, c), 4.28 (dd, J = 12.2, 4.9 Hz, 6H, H-6), 4.08 (dd, J = 12.3, 2.2 Hz, 6H, H-6′), 4.05 (ddd, J = 9.7, 4.8, 2.3 Hz, 6H, H-5), 3.88 (t, J = 5.2 Hz, 12H, d), 3.81–3.76 (m, 6H, j), 3.67–3.65 (m, 6H, j′), 3.63 (d, J = 3.7 Hz, 12H, i), 3.61 (d, J = 7.6 Hz, 48H, g, f, e, h), 2.14 (s, 18H, COCH3), 2.09 (s, 18H, COCH3), 2.02 (s, 18H, COCH3), 1.98 (s, 18H, COCH3). 13C-NMR (150 MHz, CDCl3) δ 170.7 (C=O), 170.0 (C=O), 169.9 (C=O), 169.7 (C=O), 155.3 (CO-aromatic), 144.6 (C-triazole), 143.6 (Cp-aromatic), 124.2 (CH-triazole), 121.8 (Cm-aromatic), 115.3 (CO-aromatic), 97.7 (C-1), 70.7 (Cd), 70.6 (Cg), 70.5 (Cf), 69.9 (Ce), 69.6 (Ch), 69.5 (Cj), 69.4 (C-2), 69.1 (C-3), 68.4 (C-4), 67.4 (C-5), 66.1 (Ci), 62.4 (Cb), 62.3 (C-6), 50.2 (Cc), 20.9 (COCH3), 20.8 (COCH3), 20.7 (COCH3), 20.7 (COCH3). 31P-NMR (243 MHz, CDCl3) δ 9.68. ESI+-HRMS: [M + 2H]2+ calcd for C186H254N21O90P3, 2158.2604; found: 2158.2588.
Synthesis of38. Compound 37 (100 mg, 0.023 mmol) and sodium methoxide (16 μL from 1 M solution in MeOH) in 3 mL of methanol were stirred for 4 h and the mixture was treated following the general procedure (III) described above. Deprotected compound 38 (77 mg, quant.) was obtained as a white solid. 1H-NMR (300 MHz, D2O) δ 7.96 (s, 6H, H-triazole), 6.71 (s, 24H, aromatic), 4.99 (s, 12H, b), 4.82 (s, 6H, H-1), 4.49 (s, 12H, c), 3.93–3.75 (m, 36H, H-2,3,4,5,6 and 6′), 3.74–3.38 (m, 84H, d, e, f, g, h, i, j). 31P-NMR (122 MHz, D2O) δ 10.11. ESI+-HRMS: [M + 2H]2+ calcd for C138H206N21O66P3, 1653.6321; found: 1653.6342.
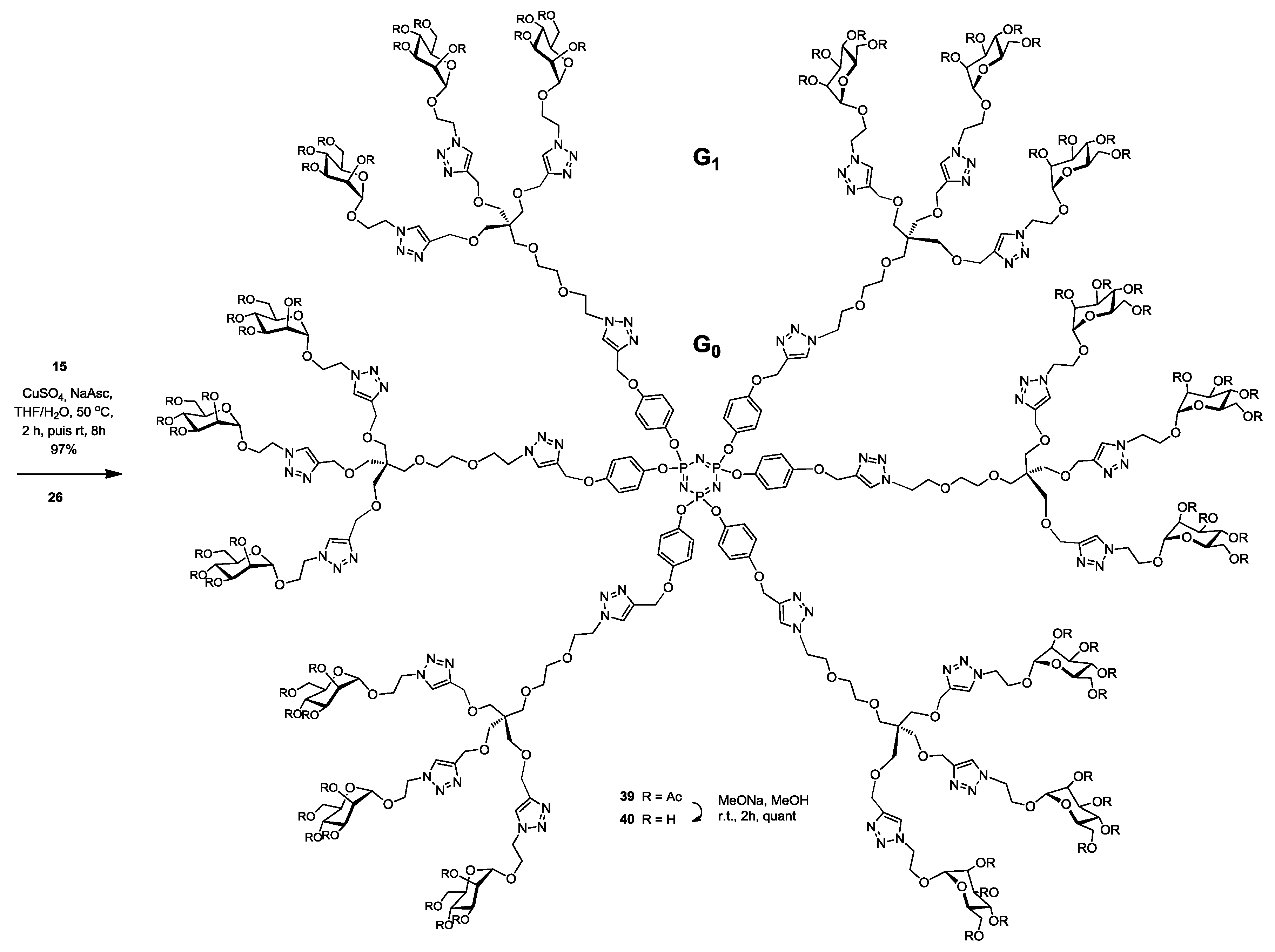
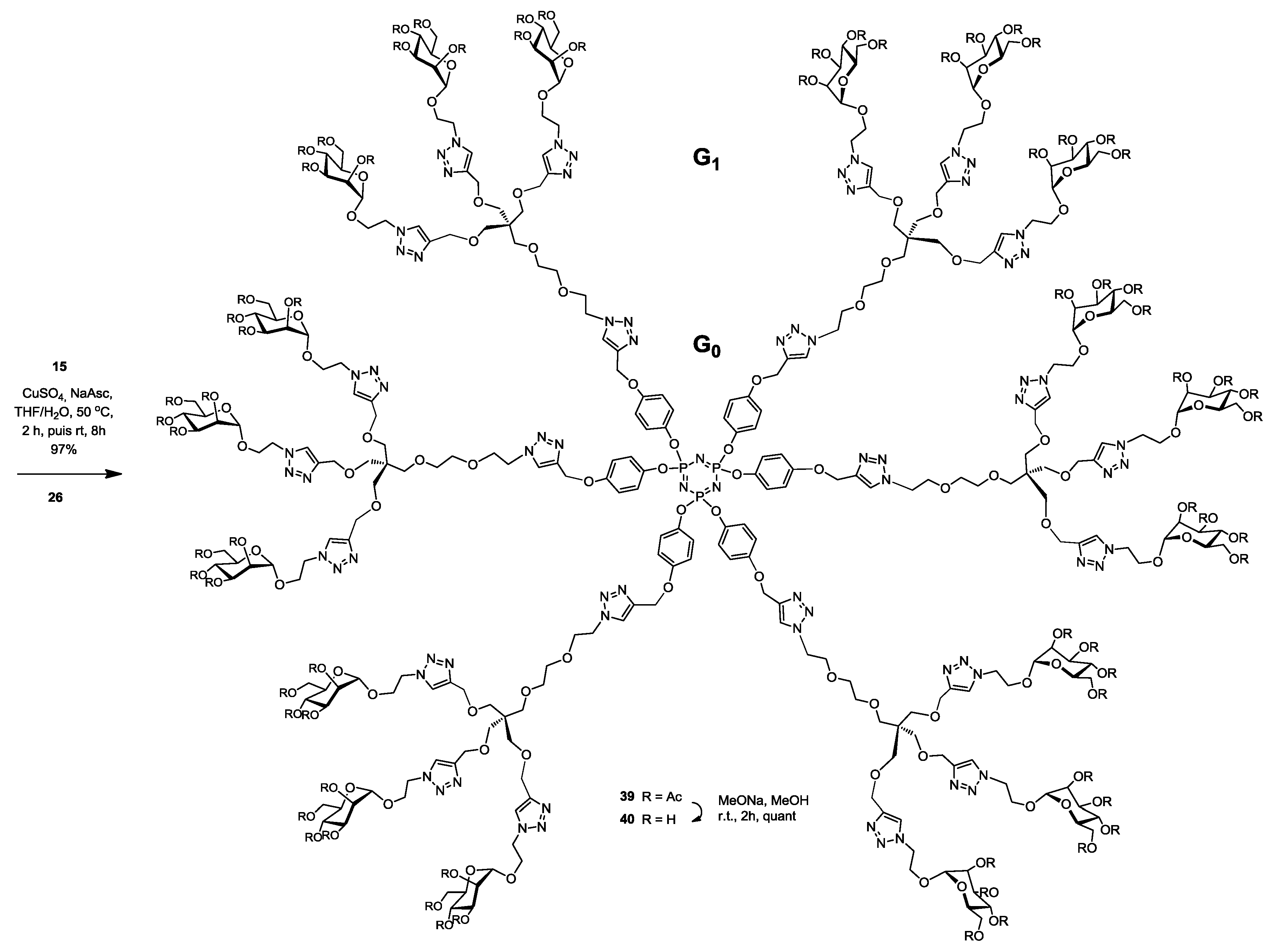
Synthesis of39. Into a solution of hexapropargylated core (15) (26 mg, 0.026 mmol, 1.0 equiv.) in a mixture of THF/H2O (4 mL, 3:1, v/v), was added (26) (400 mg, 0.24 mmol, 9.6 equiv.), Na-ascorbate (11 mg, 0.06 mmol, 2.1 equiv.) and CuSO4·5H2O (12 mg, 0.05 mmol, 1.8 equiv.). The mixture was stirred at 50 °C for 2 h, then at room temperature for 5 h. Upon completion of the reaction, EtOAc was added to the reaction mixture and further processed as given in general procedure (I) to afford compound 39 as a white powder (272 mg, 97%); Rf = 0.2 (with 5% MeOH in CH2Cl2 as eluents). 1H-NMR (600 MHz, CDCl3) δ 7.82 (s, 6H, HA-triazole), 7.69 (s, 18H, HB-triazole), 6.82 (dd, J = 32.8, 8.8 Hz, 24H, aromatic), 5.22 (t, J = 10.0 Hz, 18H, H-3), 5.19 (d, J = 3.2 Hz, 12H, H-4), 5.17 (s, 12H, H-2), 5.09 (s, 12H, b), 4.79 (s, 18H, H-1), 4.59 (dd, J = 10.5, 5.7 Hz, 36H, i), 4.54 (s, 36H, h), 4.49 (t, J = 5.0 Hz, 12H, c), 4.19 (dd, J = 12.3, 5.0 Hz, 18H, H-6), 4.14–4.07 (m, 18H, j′), 4.02 (dd, J = 12.3, 1.8 Hz, 18H, H-6′), 3.89 (m, 18H, j), 3.86 (t, J = 5.2 Hz, 12H, d), 3.62–3.59 (m, 18H, H-5), 3.55–3.48 (m, 24H, e, f), 3.46 (s, 36H, g′), 3.40 (s, 36H, g), 2.11 (s, 54H, COCH3), 2.06 (s, 54H, COCH3), 2.01 (s, 54H, COCH3), 1.96 (s, 54H, COCH3). 13C-NMR (150 MHz, CDCl3) δ 170.6 (C=O), 169.9 (C=O), 169.6 (C=O), 169.1 (C=O), 155.4 (CO-aromatic), 145.5 (CB-triazole), 144.6 (CA-triazole), 143.5 (Cp-aromatic), 124.2 (CAH-triazole), 123.7 (CBH-triazole), 121.8 (Cm-aromatic), 115.3 (CO-aromatic), 97.5 (C-1), 71.0 (Cf), 70.3 (Ce), 69.7 (Cg), 69.4 (Cd), 69.2 (C-2), 69.1 (C-3), 68.9 (C-4), 68.8 (C-5), 66.3 (Cj), 65.7 (Cb), 64.8 (Ch), 62.2 (C-6), 50.3 (Ci), 49.5 (Cc), 45.3 (Cq), 20.8 (COCH3), 20.7 (COCH3), 20.7 (COCH3), 20.7 (COCH3). 31P-NMR (243 MHz, CDCl3) δ 9.54. MALDI-TOF: [M + H]+ calcd for C450H607N75O222P3: 10705.772; found, 10706.833.
Synthesis of40. Compound 39 (200 mg, 0.02 mmol) and sodium methoxide (16 μL from 1 M solution in MeOH) in 3 mL of methanol were stirred for 4 h and the mixture was treated following the general procedure (III) described above. Deprotected compound 40 (150 mg, quant.) was obtained as a white solid. 1H-NMR (600 MHz, D2O) δ 8.05 (s, 1H), 7.92 (s, 3H), 6.74 (dd, J = 42.4, 7.8 Hz, 4H), 5.09 (s, 2H), 4.75 (s, 3H), 4.61–4.51 (m, 6H), 4.45 (s, 6H), 4.02 (s, 3H), 3.83 (s, 6H), 3.71 (d, J = 10.8 Hz, 3H), 3.63 (ddd, J = 25.4, 15.8, 7.5 Hz, 8H), 3.36 (dd, J = 75.3, 44.3 Hz, 12H), 3.11–3.02 (m, 3H). 13C-NMR (150 MHz, D2O) δ 215.7, 154.9, 144.3, 143.8, 142.9, 125.3, 125.1, 121.7, 115.8, 99.5, 72.8, 70.5, 69.9, 69.7, 68.9, 68.7, 68.3, 66.3, 65.4, 63.6, 61.4, 60.6, 50.1, 49.9, 44.8, 30.2, 29.7, 29.6, 29.4, 29.3, 29.2. 31P-NMR (243 MHz, D2O) δ 10.06. MALDI-TOF: [M + H]+ calcd for C306H463N75O150P3: 7681.011; found, 7681.383.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}