Cyclic Octamer Peptoids: Simplified Isosters of Bioactive Fungal Cyclodepsipeptides


 ,
,  ,
, 
Abstract

1. Introduction
2. Results and Discussion
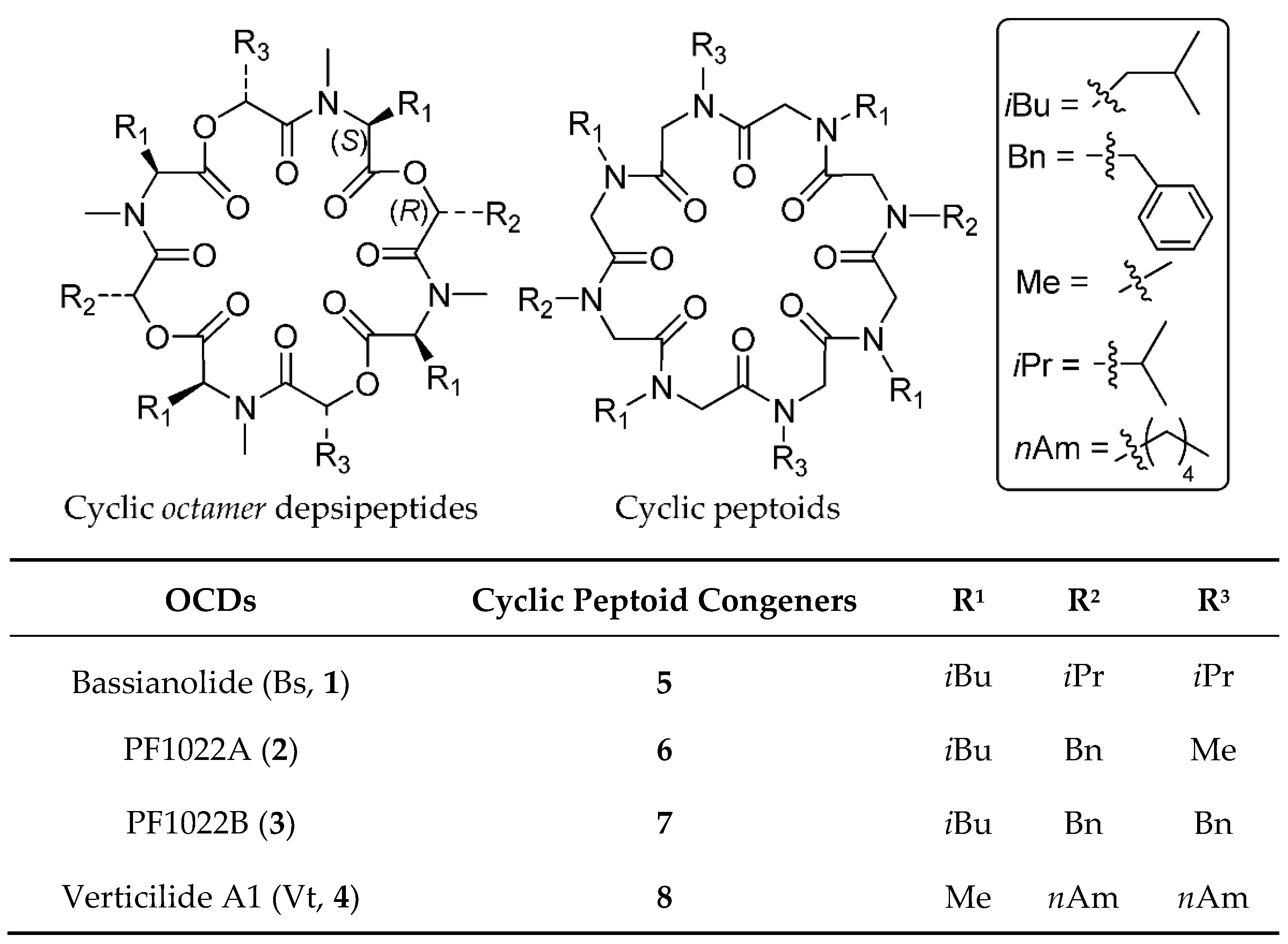
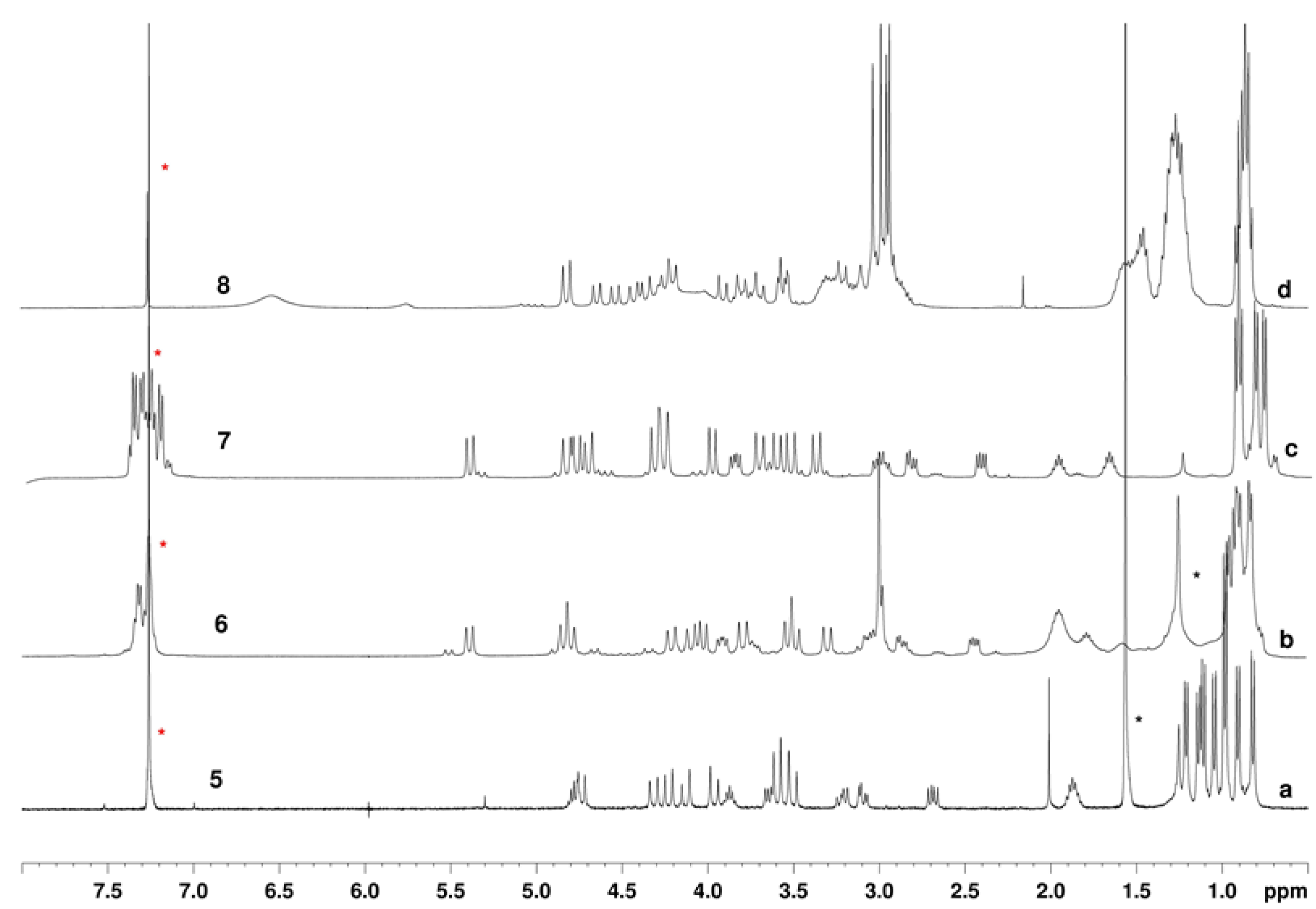
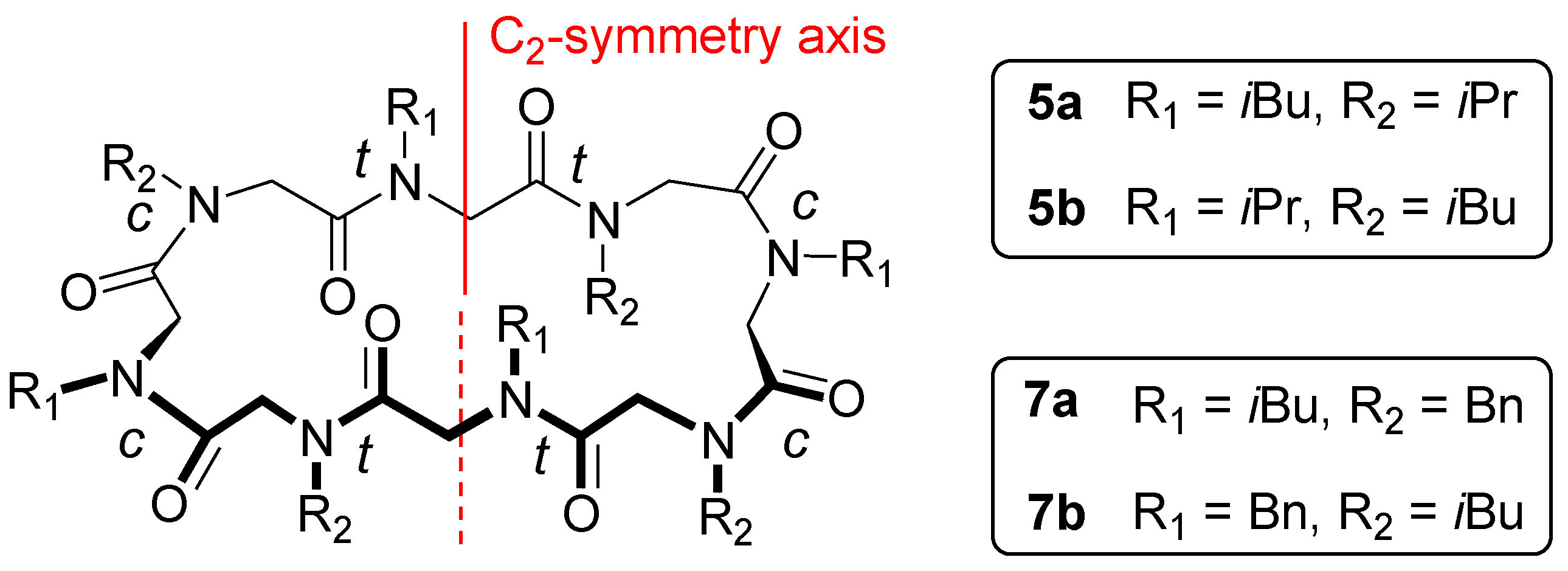
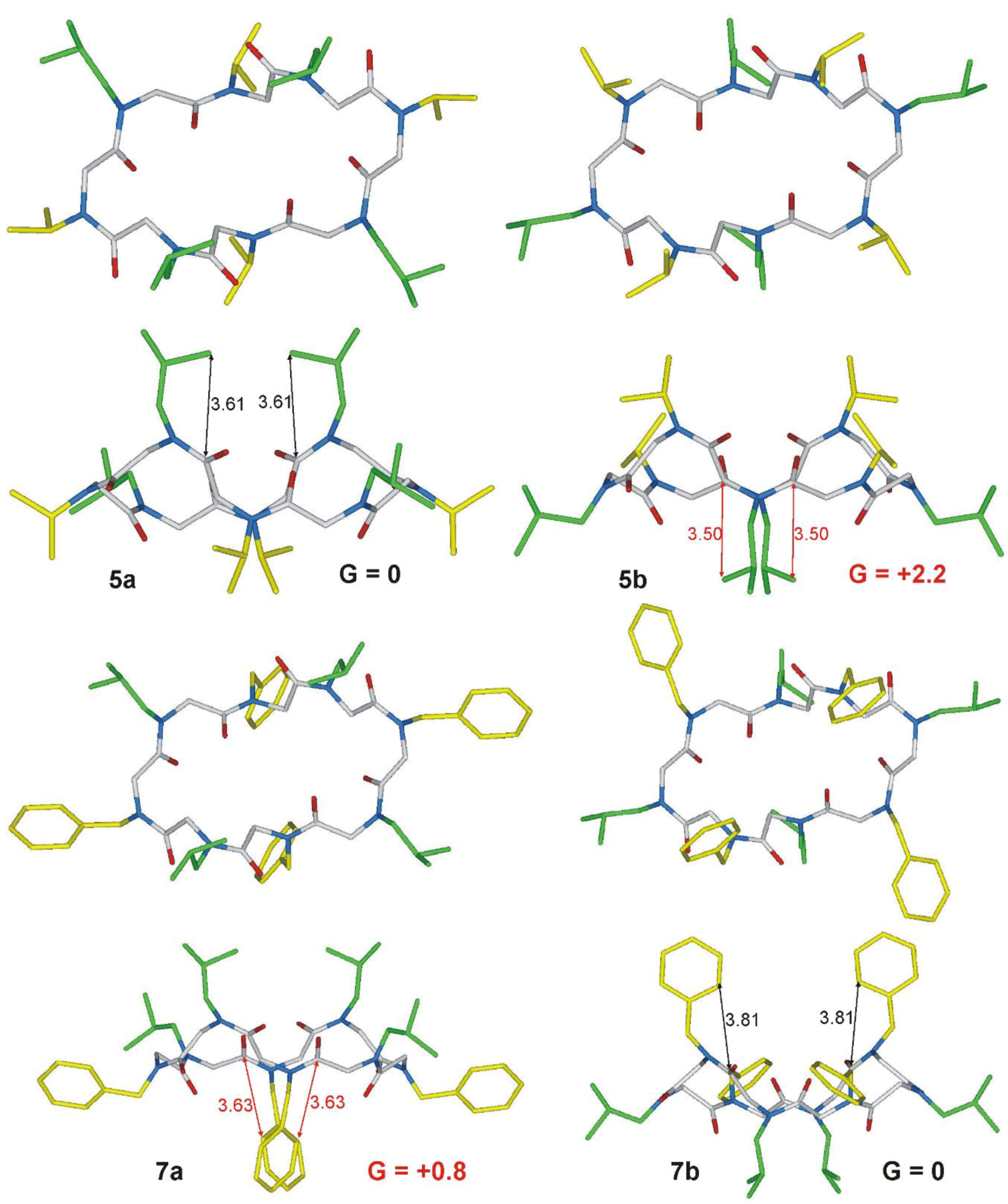
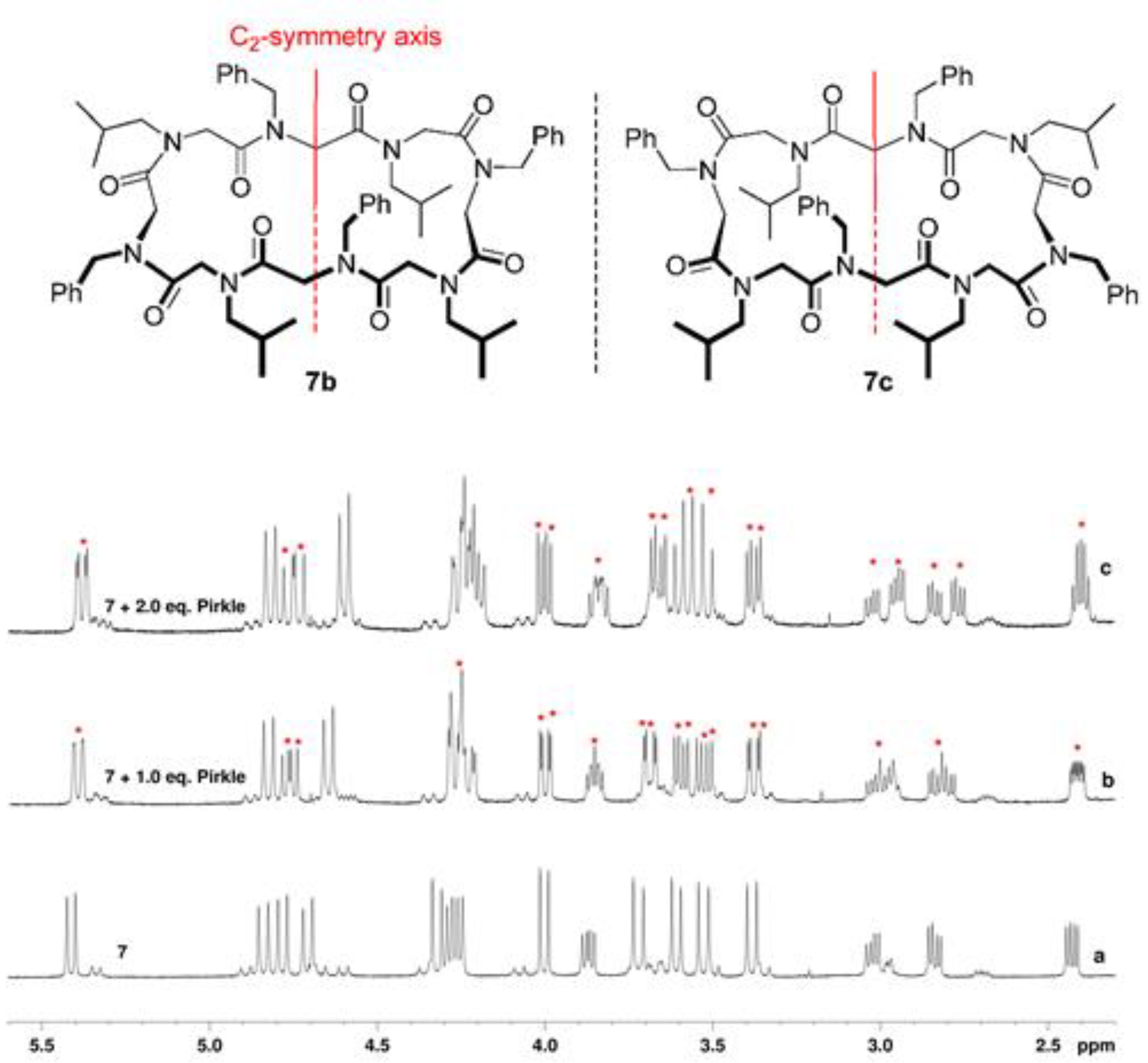
2.1. Chemistry and Conformational Studies
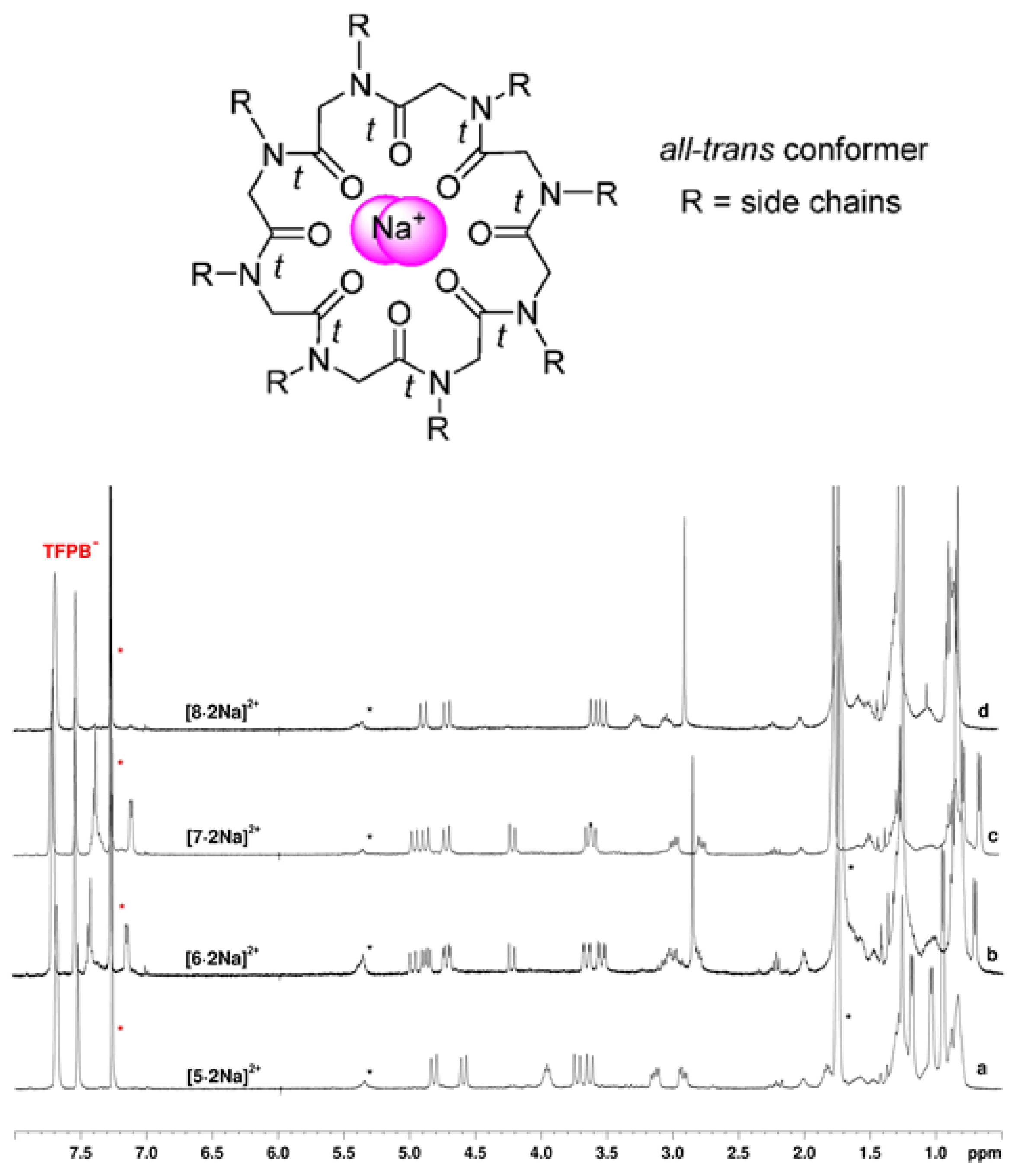
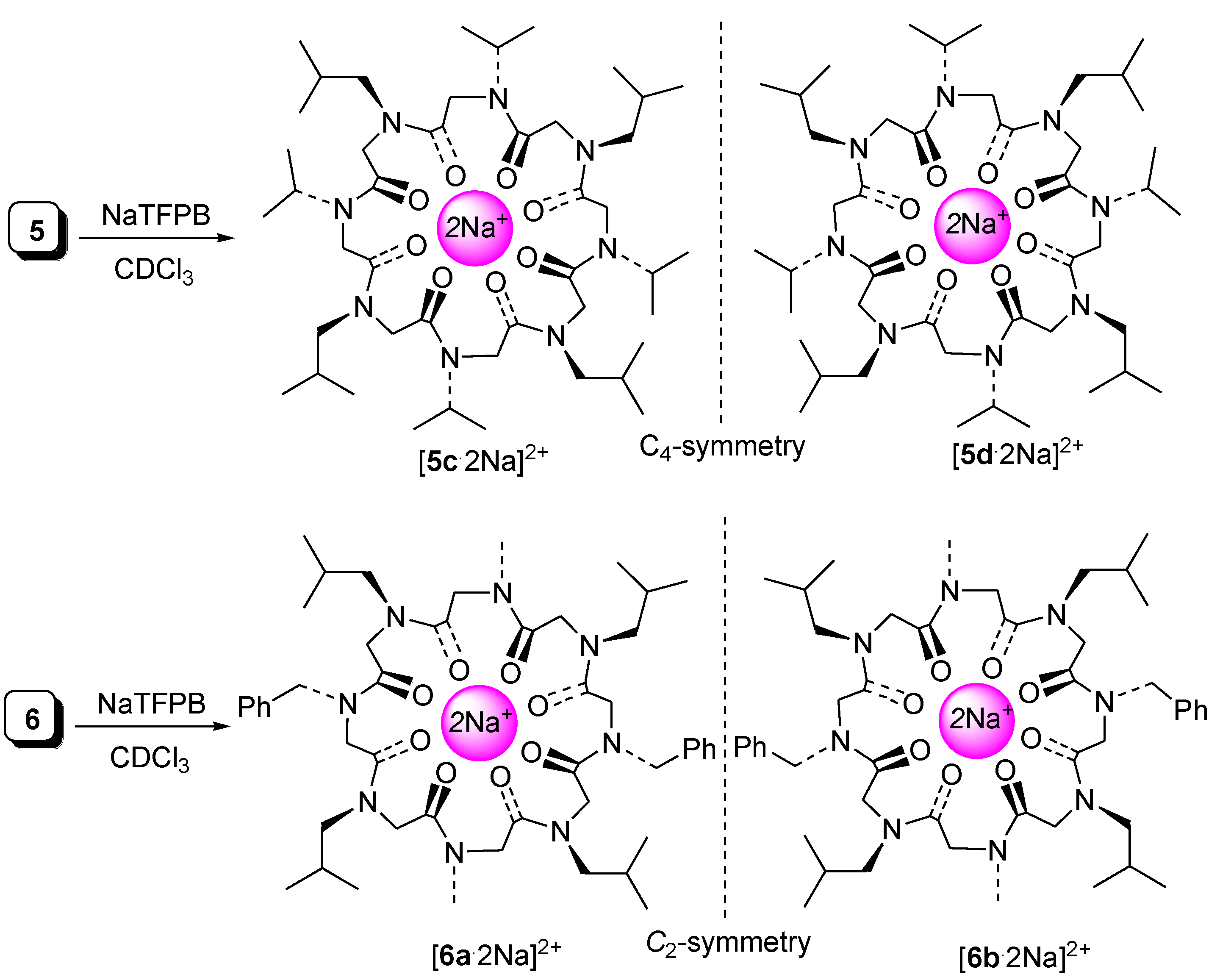
2.2. Complexation Studies
2.3. Insecticidal Activity against Silkworm Larvae
3. Experimental Section
3.1. Chemistry
3.1.1. General Methods
3.1.2. General Procedure for the Solid-Phase Synthesis of Linear Peptoids 9–12
3.1.3. General Procedure for the High Dilution Cyclization. Synthesis of Cyclic Peptoids 5–8
3.1.4. General Procedure for the Na+ Complexes Formation [5–8·2Na]2+·2[TFPB]2¯
3.1.5. 1H NMR Variable Temperature Experiments
3.1.6. General Procedure for the Pirkle’s Alcohol Addition to Racemic Mixture of Cyclopeptoid 7
3.2. Assay of Insecticidal Activity against Silkworm Larvae
3.3. Cell Lines and Viability Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Osbourn, A.E.; Lanzotti, V. (Eds.) Plant-Derived Natural Products: Synthesis, Function and Application; Springer: New York, NY, USA, 2009. [Google Scholar]
- Wenders, P.A.; Miller, B.L. Synthesis at the molecular frontier. Nature 2009, 460, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Yoo, B.; Kirshenbaum, K. Peptois architectures: Elaboration, actuation and application. Curr. Opin. Chem. Biol. 2008, 12, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Fowler, S.A.; Blackwell, H.E. Structure-function relationships in peptoids: Recent advances toward deciphering the structural requirements for biological function. Org. Biomol. Chem. 2009, 7, 1508–1524. [Google Scholar] [CrossRef] [PubMed]
- Zuckermann, R.N.; Kerr, J.M.; Kent, S.B.H.; Moos, W.H. Efficient method for the preparation of peptoids [oligo(N-substituted glycines)] by submonomer solid-phase synthesis. J. Am. Chem. Soc. 1992, 114, 10646–10647. [Google Scholar] [CrossRef]
- Simon, R.J.; Kania, R.S.; Zuckermann, R.N.; Huebner, V.D.; Jewell, D.A.; Banville, S.; Ng, S.; Wang, L.; Rosenberg, S.; Merlowe, C.K. Peptoids: A modular approach to drug discovery. Proc. Natl. Acad. Sci. USA 1992, 89, 9367–9371. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zuckermann, R.N. Peptoid polymers: A highly designable bioinspired material. ACS Nano 2013, 7, 4715–4732. [Google Scholar] [CrossRef] [PubMed]
- Zuckermann, R.N.; Kodadek, T. Peptoid as potential therapeutics. Curr. Opin. Mol. Ther. 2009, 11, 299–307. [Google Scholar] [PubMed]
- Webster, A.M.; Cobb, S.L. Recent advances in the synthesis of peptoid macrocycles. Chem. Eur. J. 2018, 24, 7560–7573. [Google Scholar] [CrossRef] [PubMed]
- D’Amato, A.; Schettini, R.; Della Sala, G.; Costabile, C.; Tedesco Izzo, I.; De Riccardis, F. Conformational isomerism in cyclic peptoids and its specification. Org. Biomol. Chem. 2017, 15, 9932–9942. [Google Scholar] [CrossRef] [PubMed]
- Yoo, B.; Shin, S.B.Y.; Huang, M.L.; Kirshenbaum, K. Peptoid macrocycles: Making the rounds with peptidomimetic oligomers. Chem. Eur. J. 2010, 16, 5528–5537. [Google Scholar] [CrossRef] [PubMed]
- D’Amato, A.; Volpe, R.; Vaccaro, M.C.; Terracciano, S.; Bruno, I.; Tosolini, M.; Tedesco, C.; Pierri, G.; Tecilla, P.; Costabile, C.; et al. Cyclic peptoids as mycotoxins mimics: An exploration of their structural and biological properties. J. Org. Chem. 2017, 82, 8848–8863. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Kanaoka, M.; Isogai, A.; Murakoshi, S.; Ichinoe, M.; Tamura, S. Bassianolide, a new insecticidal cyclodepsipeptide from Beauveria bassiana and Verticillium lecanii. Tetrahedron Lett. 1977, 25, 2167–2170. [Google Scholar] [CrossRef]
- Kanaoka, M.; Isogai, A.; Murakoshi, S.; Ichinoe, M.; Suzuki, A.; Tamura, S. Bassianolide, a new insecticidal cyclodepsipeptide from Beauveria bassiana and Verticillium lecanii. Agric. Biol. Chem. 1978, 43, 629–635. [Google Scholar] [CrossRef]
- Shiomi, K.; Matsui, R.; Kakei, A.; Yamaguchi, Y.; Masuma, R.; Hatano, H.; Arai, N.; Isozaki, M.; Tanaka, H.; Kobayashi, S.; et al. Verticilide, a new ryanodine-binding inhibitor, produced by Verticillium sp. FKI-1033. J. Antibiot. 2010, 63, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Monma, S.; Sunazuka, T.; Nagai, K.; Arai, T.; Shiomi, K.; Matsui, R.; Ōmura, S. Verticilide: Elucidation of absolute configuration and total synthesis. Org. Lett. 2006, 8, 5601–5604. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Takagi, M.; Yaguchi, T.; Miyadoh, S.; Okada, T.; Koyama, M. A new anthelmintic cyclodepsipeptide, PF1022A. J. Antibiot. 1992, 45, 692–697. [Google Scholar] [CrossRef] [PubMed]
- Sivanathan, S.; Scherkenbeck, J. Cyclodepsipeptides: A rich source of biologically active compounds for drug research. Molecules 2014, 19, 12368–12420. [Google Scholar] [CrossRef] [PubMed]
- Süssmuth, R.; Müller, J.; Von Döhren, H.; Molnár, I. Fungal cyclooligomers depsipeptides: From classical biochemistry to combinatorial biosynthesis. Nat. Prod. Rep. 2011, 28, 99–124. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gong, X.; Li, P.; Lai, D.; Zhou, L. Structural diversity and biological activities of cyclic depsipeptides from fungi. Molecules 2018, 23, 169. [Google Scholar] [CrossRef] [PubMed]
- Krücken, J.; Harder, A.; Jeschke, P.; Holden-Dye, L.; O’Connor, V.; Welz, C.; von Samson-Himmelstjerna, G. Anthelmintic cyclooctadepsipeptides: Complex in structure and mode of action. Trends Parasitol. 2012, 28, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Harder, A.; Schmitt-Wrede, H.P.; Krücken, J.; Marinovski, P.; Wunderlich, F.; Willson, J.; Amliwala, K.; Holden-Dye, L.; Walker, R. Cyclooctadepsipeptides-an anthelmintically active class of compounds exhibiting a novel mode of action. Int. J. Antimicrob. Agents 2003, 22, 318–331. [Google Scholar] [CrossRef]
- Greenberg, R.M. Ion channels and drug transporters as targets for anthelmintics. Curr. Clin. Microbiol. Rep. 2014, 1, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Masuda, Y.; Tanaka, R.; Kai, K.; Ganesan, A.; Doi, T. Total synthesis and biological evaluation of PF1171A, C, F and G, cyclic hexapeptides with insecticidal activity. J. Org. Chem. 2014, 79, 7844–7853. [Google Scholar] [CrossRef] [PubMed]
- Masuda, Y.; Tanaka, R.; Ganesan, A.; Doi, T. Systematic analysis of relationship among 3D structure, bioactivity and membrane permeability of PF1171F, a cyclic hexapeptide with paralyzing effects on silkworms. J. Org. Chem. 2017, 82, 11447–11463. [Google Scholar] [CrossRef] [PubMed]
- De Cola, C.; Fiorillo, G.; Meli, A.; Aime, S.; Gianolio, E.; Izzo, I.; De Riccardis, F. Gadolinium-binding cyclic hexapeptoids: Synthesis and relaxometric properties. Org. Biomol. Chem. 2014, 12, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Scherkenbeck, J.; Plant, A.; Harder, A.; Mencke, N. A highly efficient synthesis of the anthelmintic cyclooctadepsipeptide FP1022A. Tetrahedron 1995, 51, 8459–8470. [Google Scholar] [CrossRef]
- Dutton, F.E.; Lee, B.H.; Johnson, S.S.; Coscarelli, E.M.; Lee, P.H. Restricted Conformation Analogues of an Anthelmintic Cyclodepsipeptide. J. Med. Chem. 2003, 46, 2057–2073. [Google Scholar] [CrossRef] [PubMed]
- Dale, J.; Titlestad, K. Cyclic oligopeptides of sarcosine (N-methylglycine). Chem. Commun. 1969, 656–659. [Google Scholar] [CrossRef]
- Titlestad, K. Cyclic peptides of sarcosine. syntheses and conformation. Acta Chem. Scand. B 1975, 29, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Lepage, M.L.; Meli, A.; Bodlenner, A.; Tarnus, C.; De Riccardis, F.; Izzo, I.; Compain, P. Synthesis of the first examples of iminosugar clusters based on cyclopeptoid cores. Beilstein J. Org. Chem. 2014, 10, 1406–1412. [Google Scholar] [CrossRef] [PubMed]
- Vollrath, S.B.L.; Bräse, S.; Kirshenbaum, K. Twice tied tight: Enforcing conformational order in bicyclic peptoid oligomers. Chem. Sci. 2012, 3, 2726–2731. [Google Scholar] [CrossRef]
- Vollrath, S.B.L.; Hu, C.; Bräse, S.; Kirshenbaum, K. Peptoid nanotubes: An oligomer macrocycle that reversibly sequesters water via single-crystal-to-single-crystal transformations. Chem. Commun. 2013, 49, 2317–2319. [Google Scholar] [CrossRef] [PubMed]
- Tedesco, C.; Meli, A.; Macedi, E.; Iuliano, V.; Ricciardulli, A.G.; De Riccardis, F.; Vaughan, G.B.M.; Smith, V.J.; Barbour, L.J.; Izzo, I. Ring size effect on the solid state assembly of propargyl substituted hexa- and octacyclic peptoids. Cryst. Eng. Comm. 2016, 18, 8838–8848. [Google Scholar] [CrossRef]
- Pirkle, W.H.; Beare, S.D. Optically active solvents in nuclear magnetic resonance spectroscopy. IX. Direct determinations of optical purities and correlations of absolute configurations of alpha.-amino acids. J. Am. Chem. Soc. 1969, 91, 5150–5155. [Google Scholar] [CrossRef]
- De Cola, C.; Licen, S.; Comegna, D.; Cafaro, E.; Bifulco, G.; Izzo, I.; Tecilla, P.; De Riccardis, F. Size-dependent cation transport by cyclic α-peptoid ion carriers. Org. Biomol. Chem. 2009, 7, 2851–2854. [Google Scholar] [CrossRef] [PubMed]
- Fielding, L. Determination of Association Constants (Ka) from Solution NMR Data. Tetrahedron 2000, 56, 6151–6170. [Google Scholar] [CrossRef]
- Meli, A.; Gambaro, S.; Costabile, C.; Talotta, C.; Della Sala, G.; Tecilla, P.; Milano, D.; Tosolini, M.; Izzo, I.; De Riccardis, F. Synthesis and complexing properties of cyclic benzylopeptoids–a new family of extended macrocyclic peptoids. Org. Biomol. Chem. 2016, 14, 9055–9062. [Google Scholar] [CrossRef] [PubMed]
- Juvvadi, P.; Vunnam, S.; Merrifield, R.B. Synthetic Melittin, Its Enantio, Retro and Retroenantio Isomers and Selected Chimeric Analogs: Their Antibacterial, Hemolytic and Lipid Bilayer Action. J. Am. Chem. Soc. 1996, 118, 8989–8997. [Google Scholar] [CrossRef]
- Shemyakin, M.M.; Ovchinnikov, Y.A.; Ivanov, V.T. Topochemical Investigations on Peptide Systems. Angew. Chem. Int. Ed. Engl. 1969, 8, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Ohshiro, T.; Matsudo, D.; Kazuhiro, T.; Uchida, R.; Nonaka, K.; Masuma, R.; Tomada, H. New verticilides, inhibitors of acyl-CoA: Cholesterol acyltransferase, produced by Verticillium sp. FKI-2679. J. Antibiot. 2012, 65, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Kurland, R.J.; Rubin, M.B.; Wise, M.B. Inversion Barrier in Singly Bridged Biphenyls. J. Chem. Phys. 1964, 40, 2426–2427. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 5, 7 and 8 are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence a (Oligomer) | ESI/MS | Yield | Purity |
|---|---|---|---|
| H-[NVal-NLeu]4-OH (9) | 867.5 [M + H]+ | 100% | 82% |
| H-[NPhe-NLeu-NAla-NLeu]2-OH (10) | 908.2 [M + H]+ | 49% | 92% |
| H-[NPhe-NLeu]4-OH (11) | 1059.6 [M + H]+ | 89% | 71% |
| H-[Nam-NAla]4-OH (12) | 811.5 [M + H]+ | 100% | 83% |
| Compound | Number of Dead Larvae (n = 10) | |||||
|---|---|---|---|---|---|---|
| Time after Injection (h) | ||||||
| 1 | 6 | 24 | 72 | 120 | 168 | |
| vehicle | 0 | 0 | 0 | 0 | 0 | 0 |
| 5 | 0 | 0 | 0 | 2 | 2 | 2 |
| 6 | 0 | 0 | 0 | 1 | 2 | 2 |
| 7 | 0 | 0 | 0 | 2 | 2 | 2 |
| 8 | 0 | 0 | 0 | 1 | 1 | 1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Amato, A.; Della Sala, G.; Izzo, I.; Costabile, C.; Masuda, Y.; De Riccardis, F. Cyclic Octamer Peptoids: Simplified Isosters of Bioactive Fungal Cyclodepsipeptides. Molecules 2018, 23, 1779. https://doi.org/10.3390/molecules23071779
D’Amato A, Della Sala G, Izzo I, Costabile C, Masuda Y, De Riccardis F. Cyclic Octamer Peptoids: Simplified Isosters of Bioactive Fungal Cyclodepsipeptides. Molecules. 2018; 23(7):1779. https://doi.org/10.3390/molecules23071779
Chicago/Turabian StyleD’Amato, Assunta, Giorgio Della Sala, Irene Izzo, Chiara Costabile, Yuichi Masuda, and Francesco De Riccardis. 2018. "Cyclic Octamer Peptoids: Simplified Isosters of Bioactive Fungal Cyclodepsipeptides" Molecules 23, no. 7: 1779. https://doi.org/10.3390/molecules23071779
APA StyleD’Amato, A., Della Sala, G., Izzo, I., Costabile, C., Masuda, Y., & De Riccardis, F. (2018). Cyclic Octamer Peptoids: Simplified Isosters of Bioactive Fungal Cyclodepsipeptides. Molecules, 23(7), 1779. https://doi.org/10.3390/molecules23071779