
General Method for the Synthesis of (−)-Conduritol C and Analogs from Chiral Cyclohexadienediol Scaffolds

 and
and 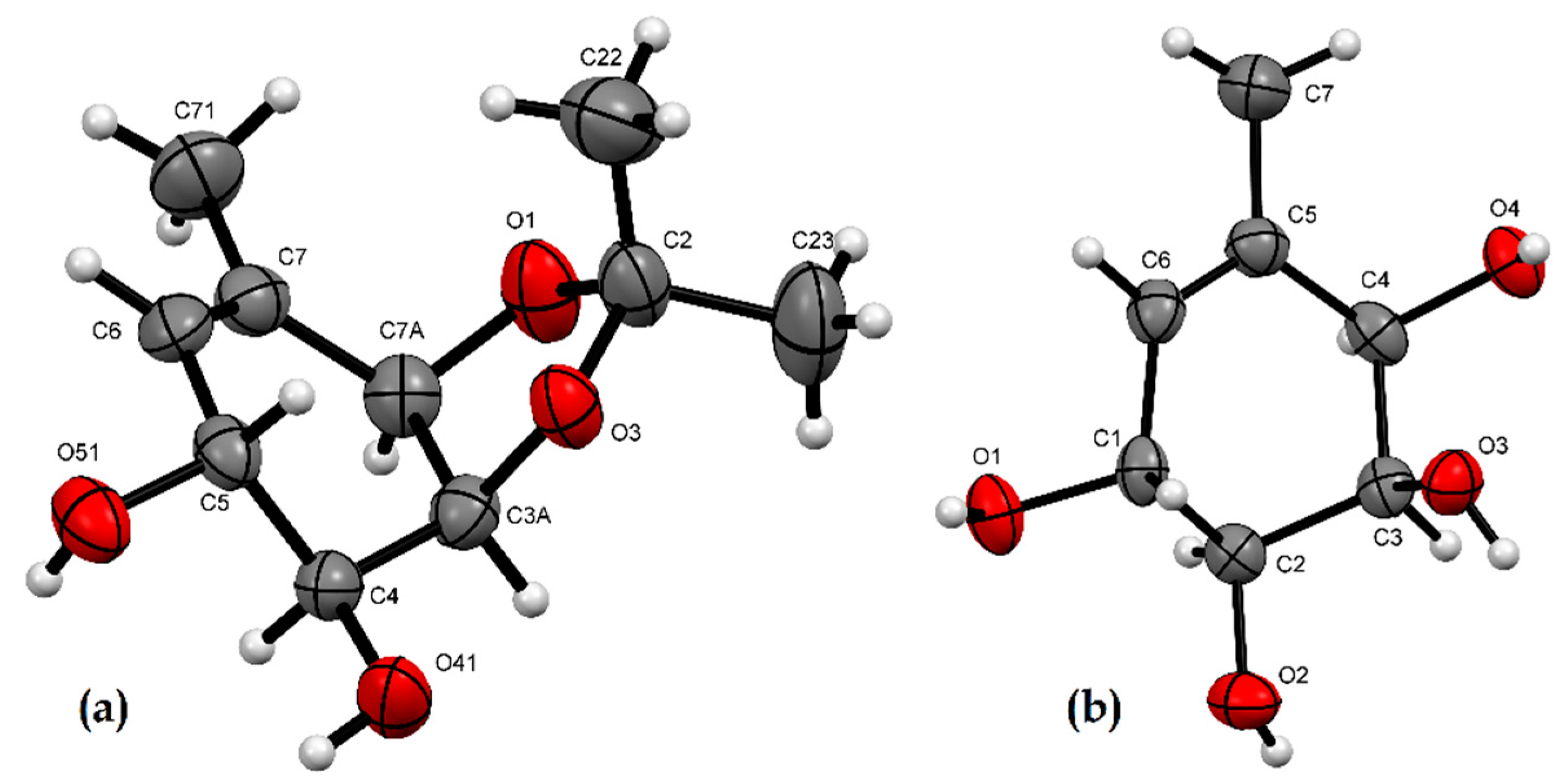{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
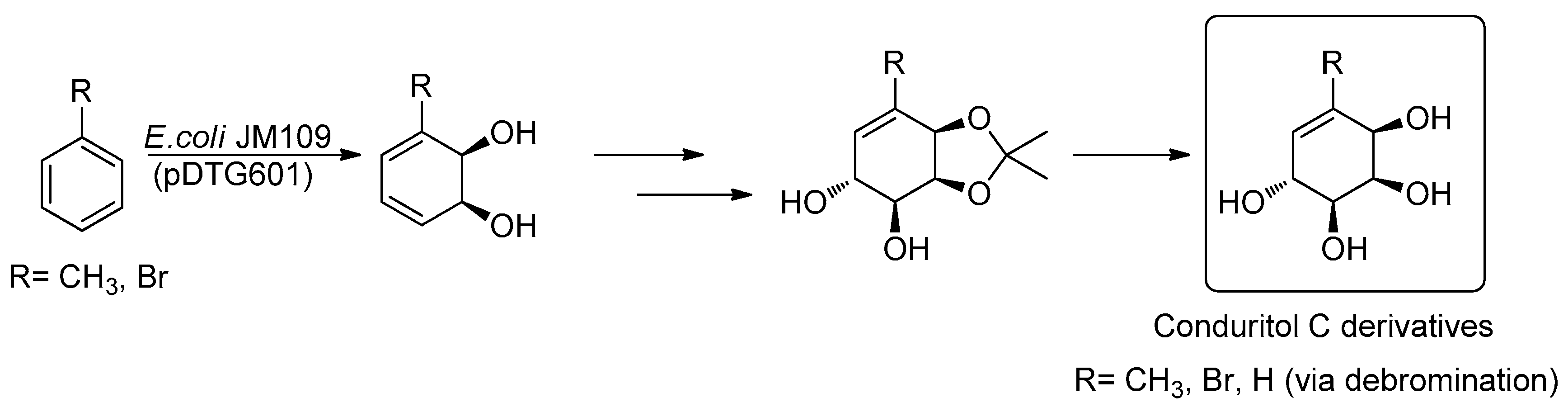
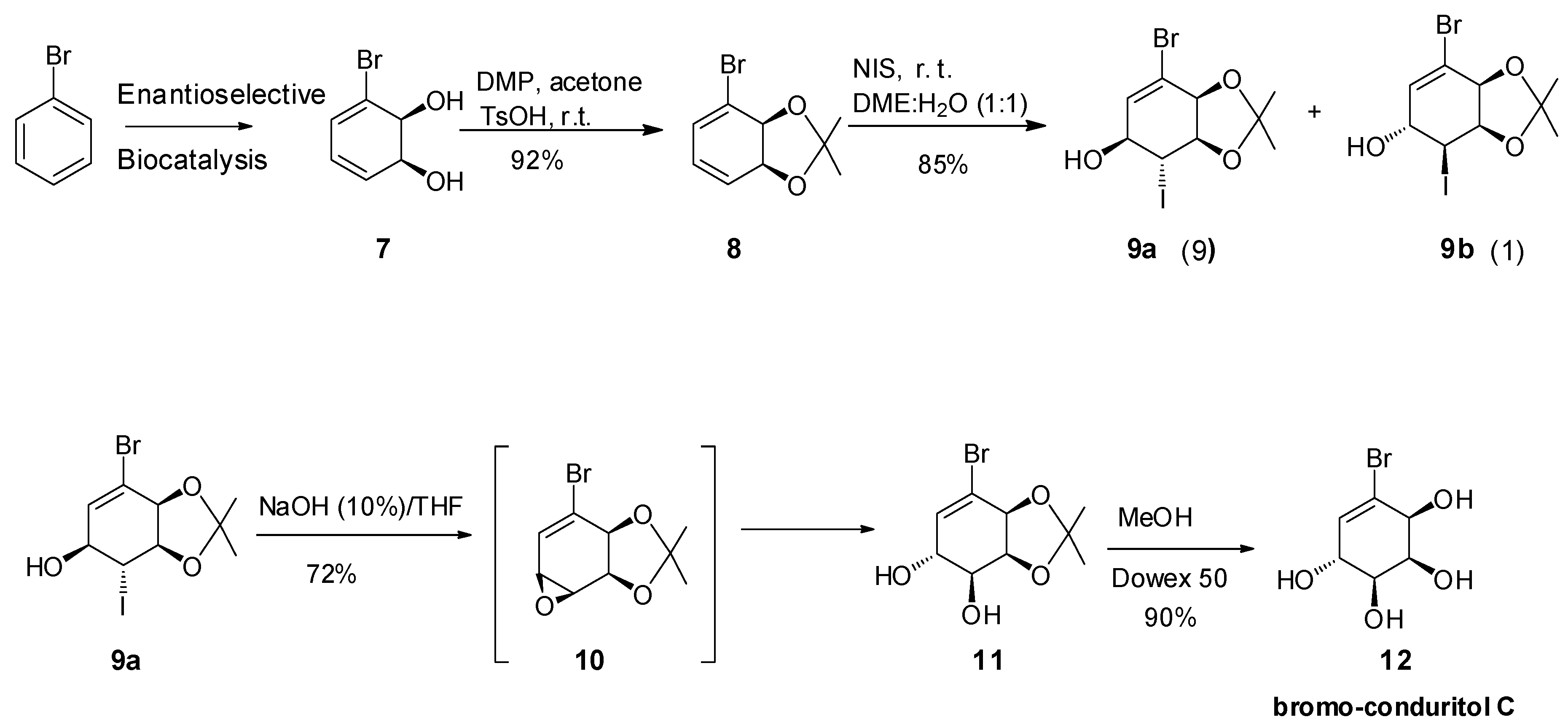
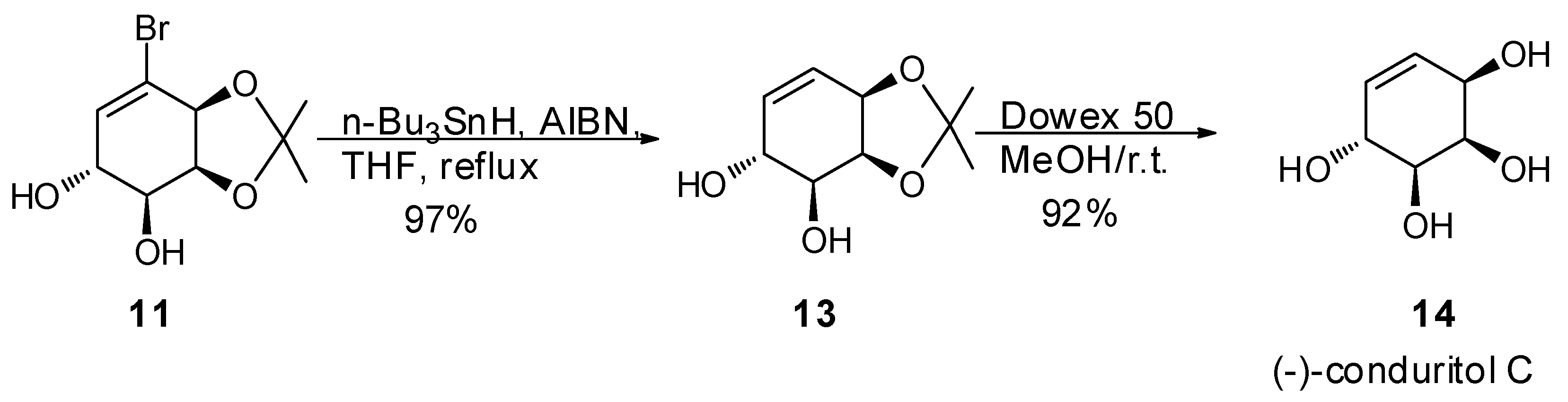
2. Results and Discussion
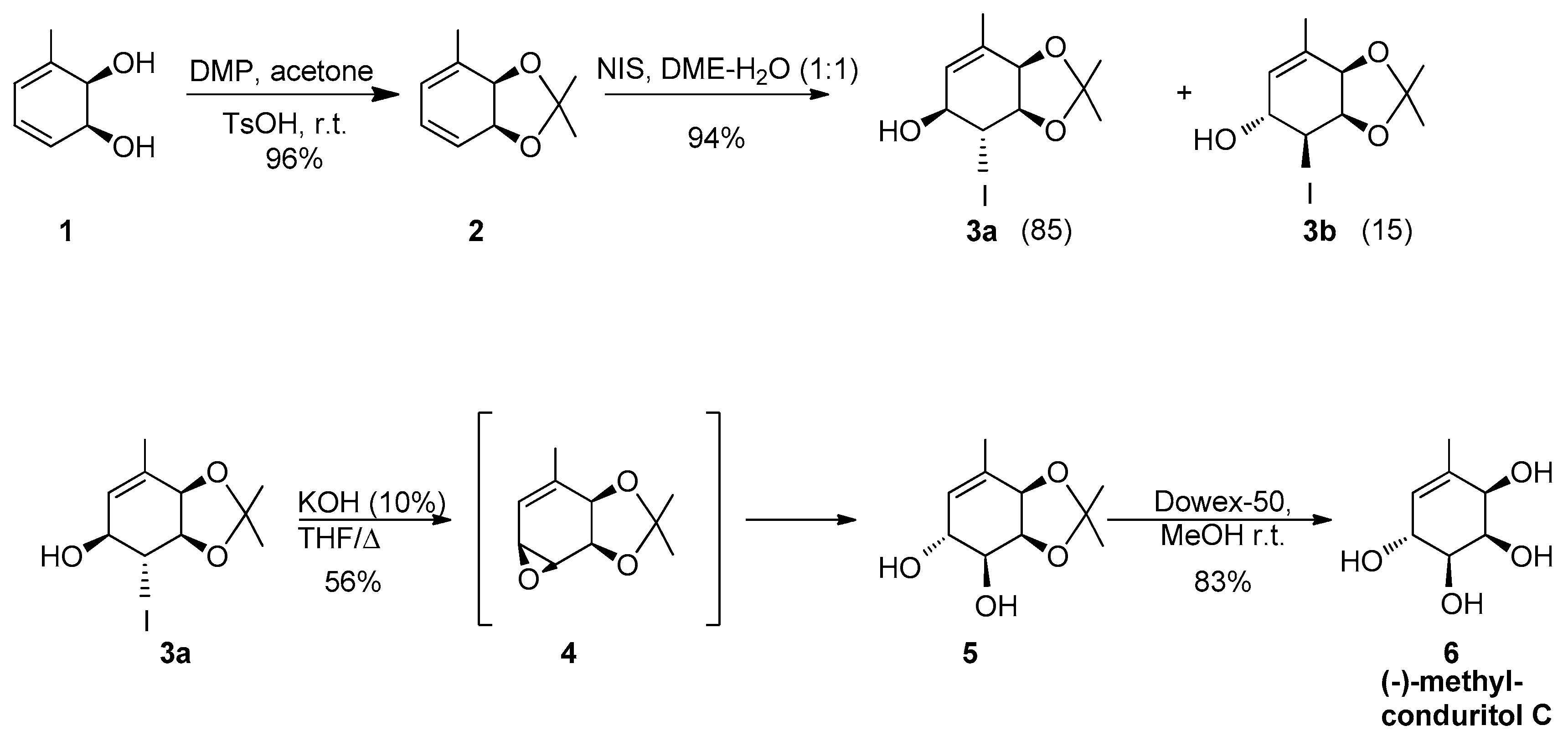
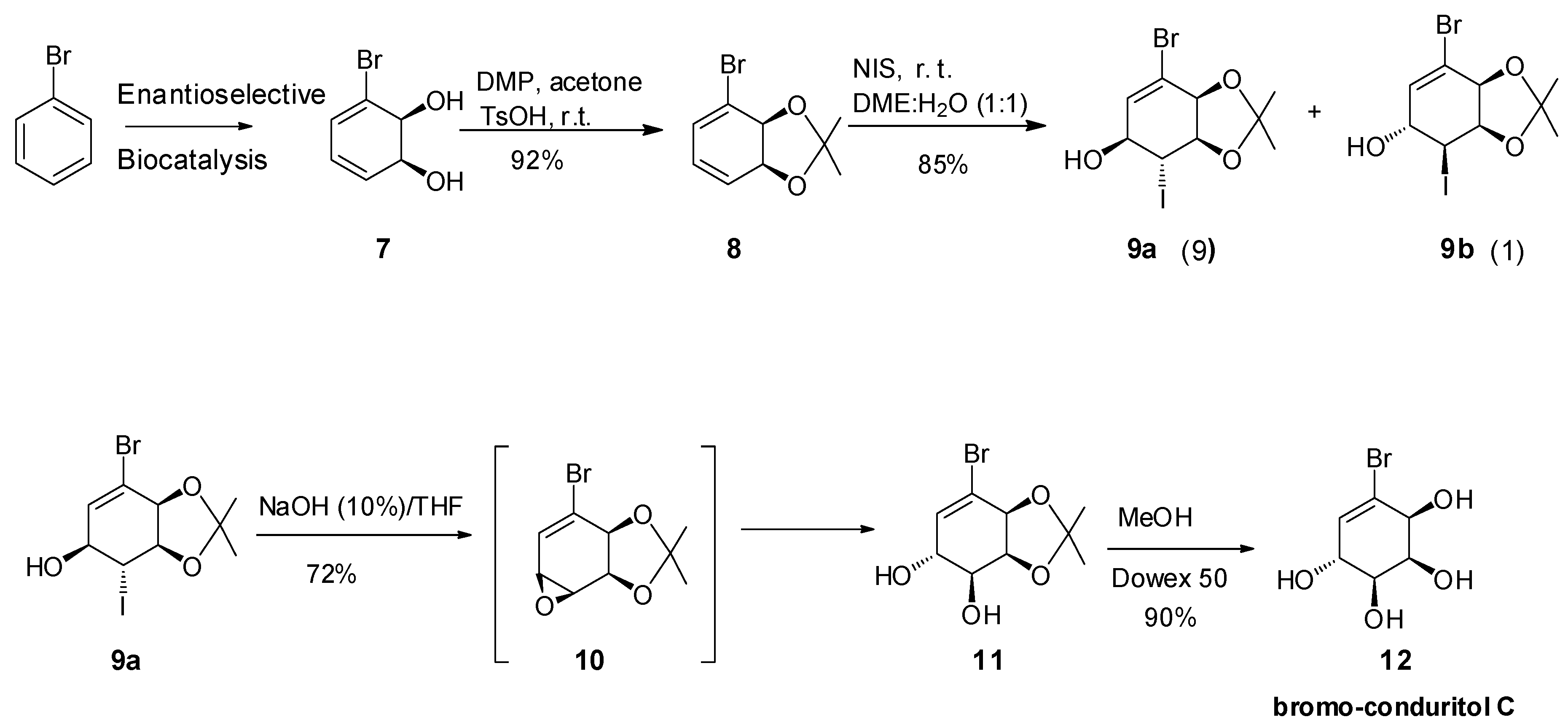
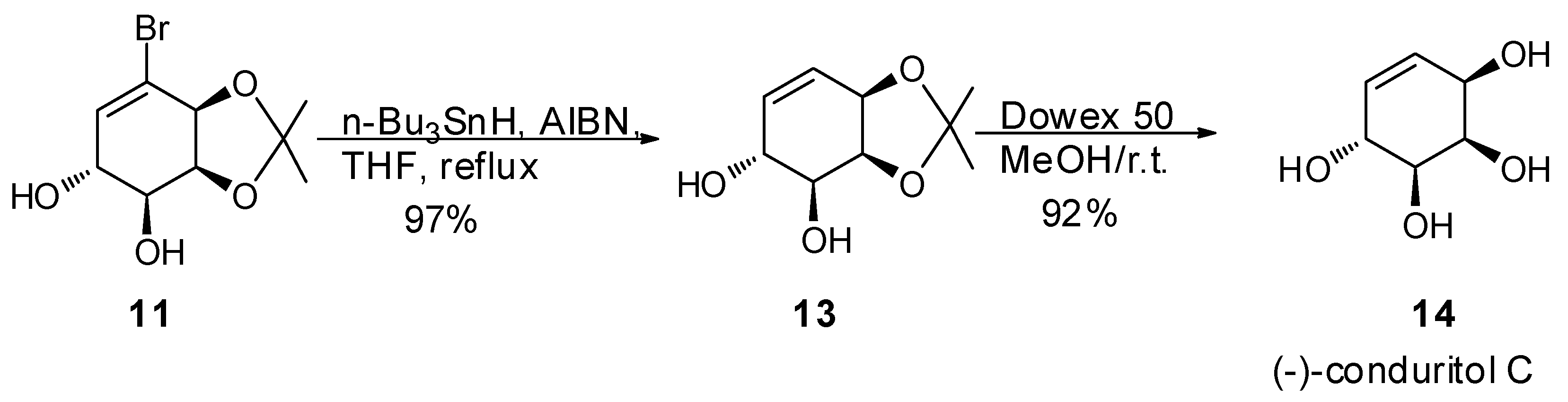
2.1. Synthetic Strategy
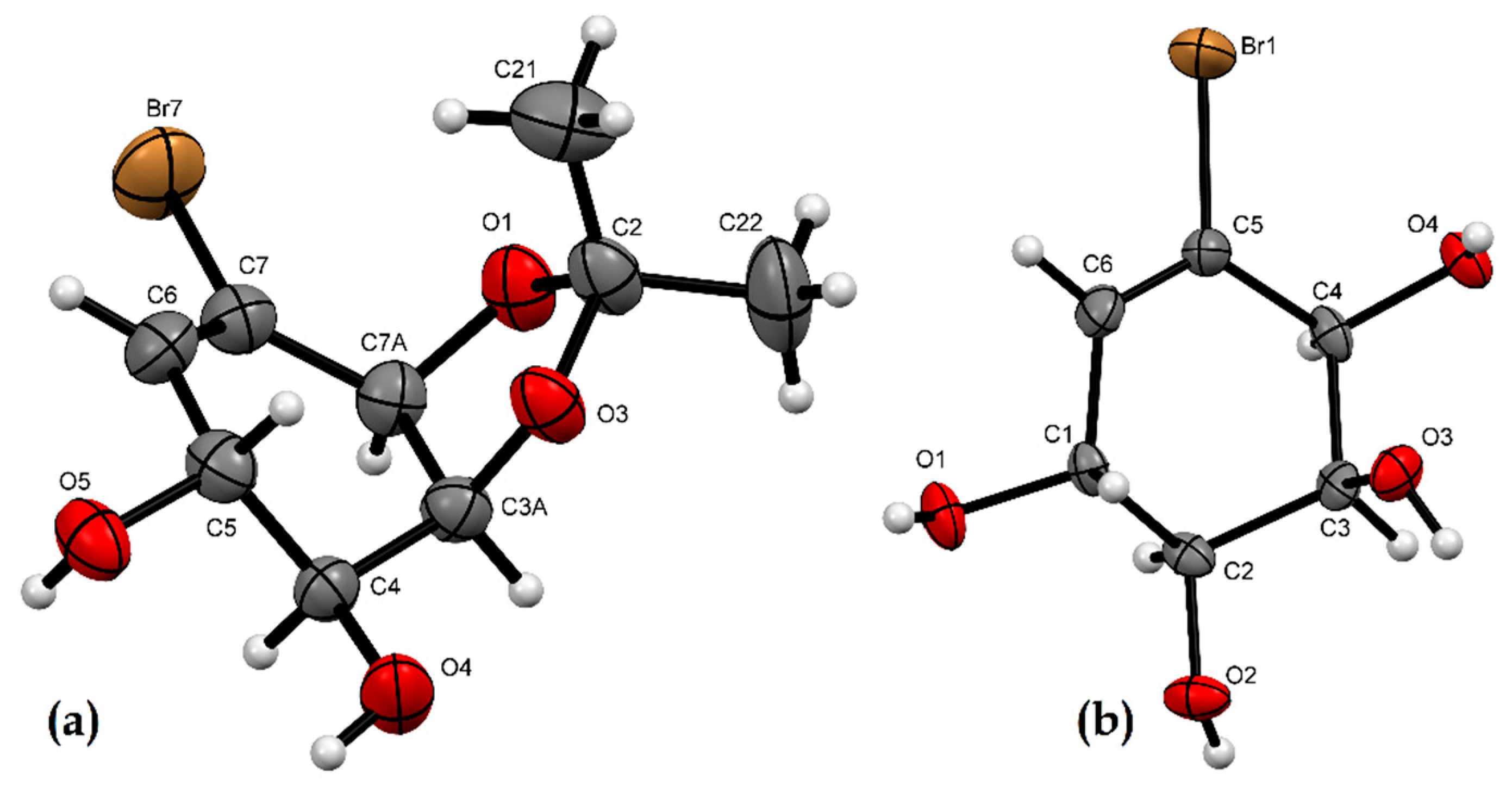
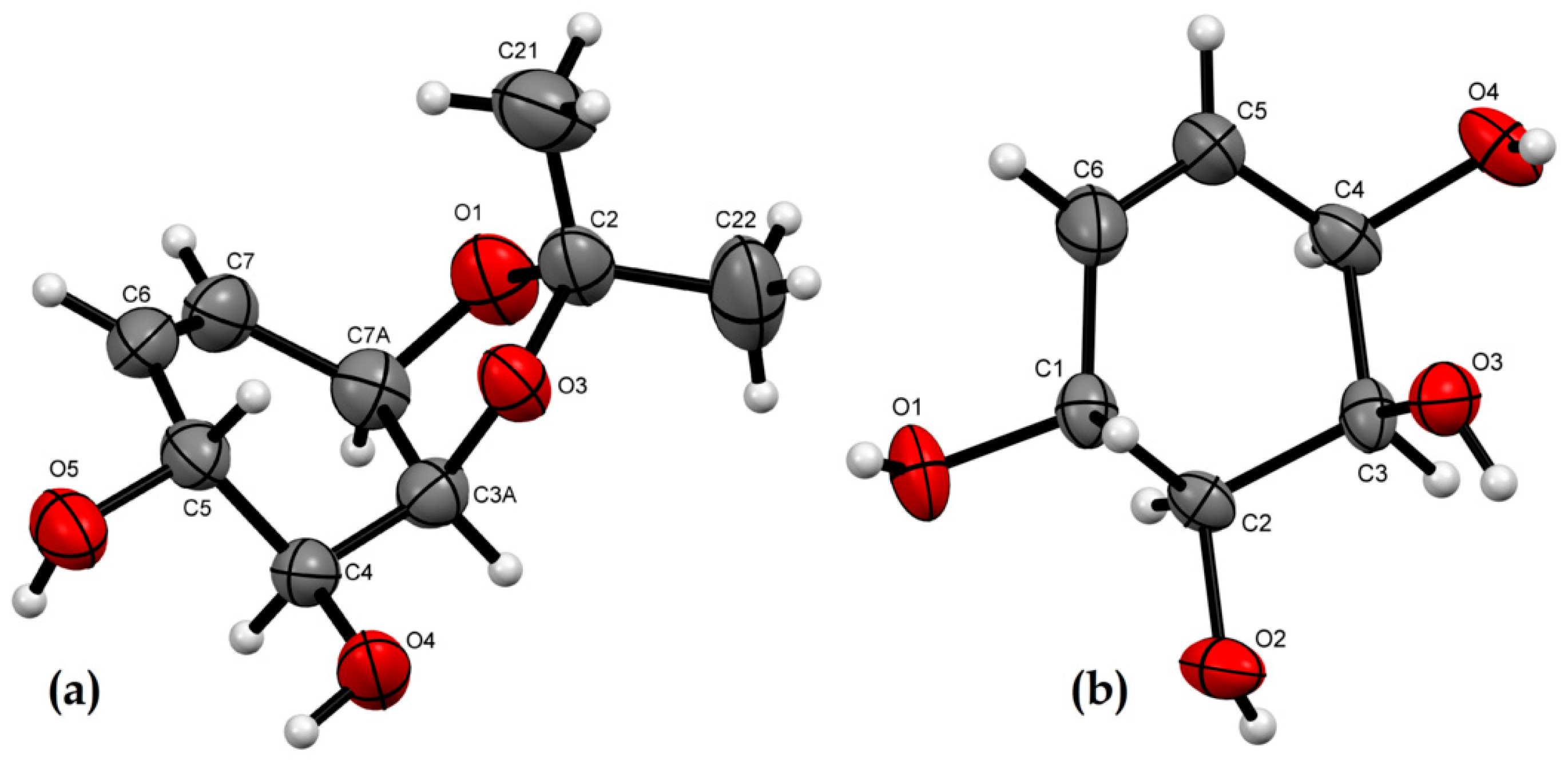
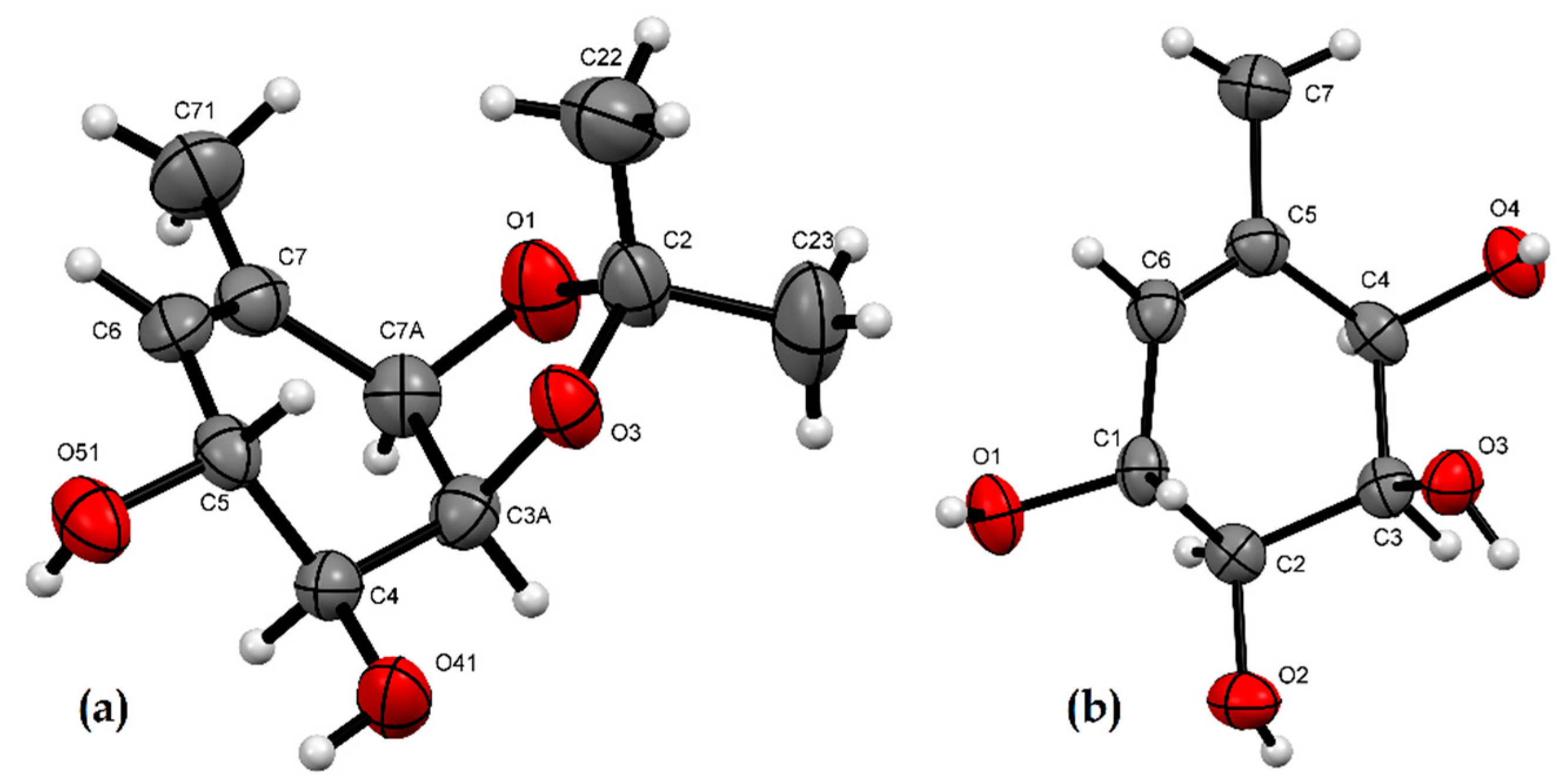
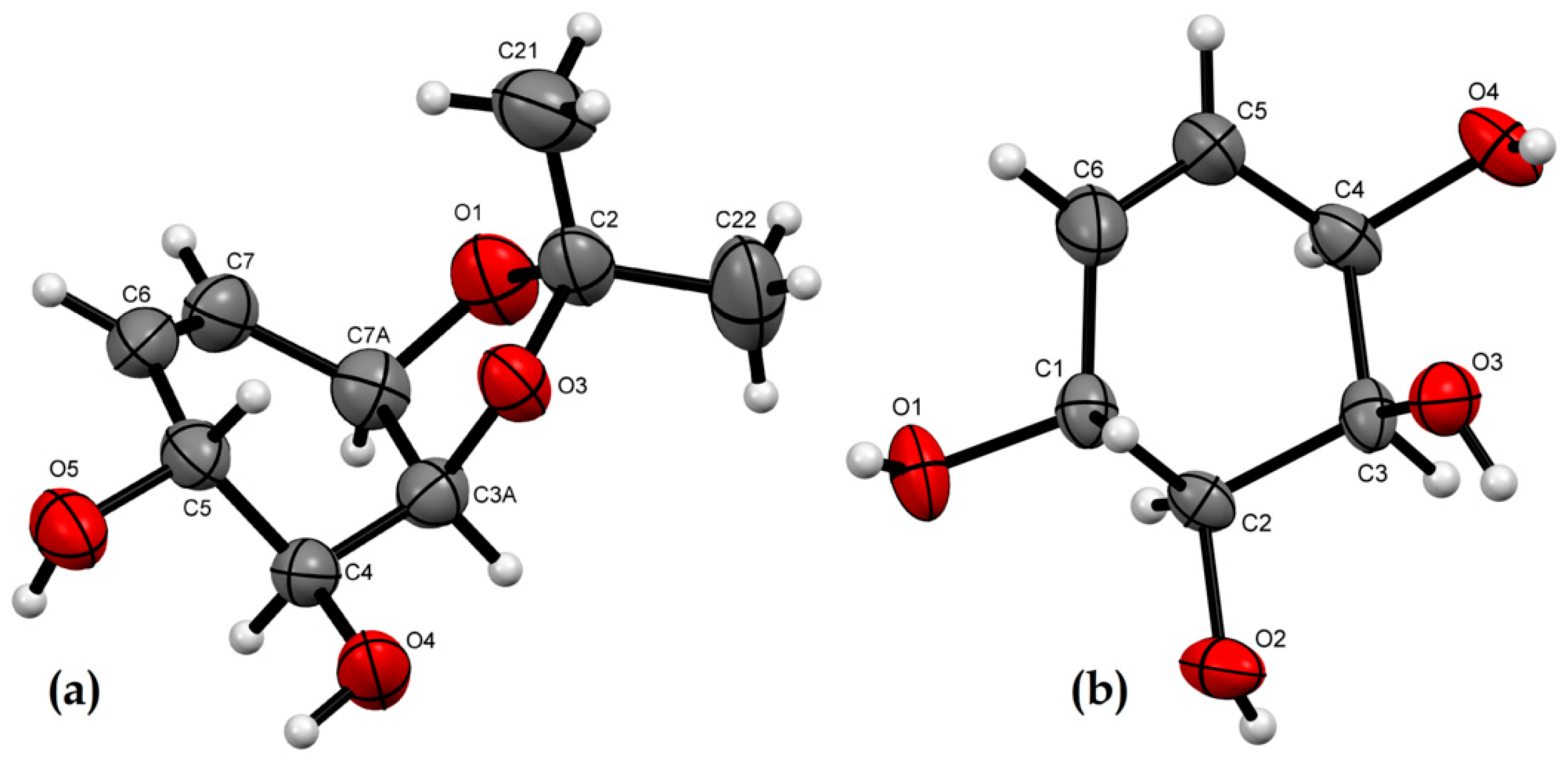
2.2. X-ray Crystallographic Analysis
3. Materials and Methods
3.1. General Procedure for the Synthesis of Diols 5 and 11
3.2. General Procedure for the Synthesis of Conduritol Derivatives
3.3. X-ray Crystallographic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DMP | dimetoxypropane |
| NIS | N-Iodosuccinimide |
| DME | Dimetoxyethane |
| THF | Tetrahydrofurane |
References
- Balci, M.; Sütbeyaz, Y.; Seceb, H. Conduritols and related compounds. Tetrahedron 1990, 46, 3715–3742. [Google Scholar] [CrossRef]
- McCasland, G.E.; Reeves, J.M. Ciclitols VIII. Bromination of epi-Inositol. Synthesis of Conduritol-C. J. Am. Chem. Soc. 1955, 77, 1812–1814. [Google Scholar] [CrossRef]
- Nakajima, M.; Tomida, I.; Takei, S. Zur Chemie des Benzolglykols, III. Über die synthese von vier stereoisomeren 3.4.5.6tetrahydroxy-cyclohexenen (Konduriten). Eur. J. Inorg. Chem. 1957, 90, 246–250. [Google Scholar]
- Yurev, Y.K.; Zefmv, K. Furans. 13. New stereospecific method of synthesis of hexahydroxy cyclohexane. Zhurnal Obshchei Khimii 1961, 31, 685–686. [Google Scholar]
- Le Drian, C.; Vieira, E.; Vogel, P. Synthesis of (−)-conduritol C (1L-cyclohex-5-ene-1,2,3,4-tetrol). Helv. Chem. Acta 1989, 72, 338–347. [Google Scholar] [CrossRef]
- Boyd, D.R.; Dorrity, M.R.J.; Hand, M.V.; Malone, J.F.; Sharma, N.D.; Dalton, H.; Gray, D.J.; Sheldrake, G.N. Enantiomeric excess and absolute configuration determination of -Dihydrodiols from bacterial metabolism of monocyclic arenes. J. Am. Chem. Soc. 1991, 113, 666–667. [Google Scholar] [CrossRef]
- Carless, H.A.J.; Oak, O.Z. Enantiospecific synthesis of (4S,5S,6S)-4,5,6-trihydroxycyclohex-2-enone and (+)-conduritol C from fluorobenzene via microbial oxidation. J. Chem. Soc. Chem. Commun. 1991, 2, 61–62. [Google Scholar] [CrossRef]
- Carless, H.A.J. Enantiospecific and Stereoselective synthesis of (−)-Conduritol C from chlorobenzene via microbial oxidation and epoxidation. J. Chem. Soc. Chem. Commun. 1992, 3, 234–235. [Google Scholar] [CrossRef]
- Hudlicky, T.; Fan, R.; Tsunoda, T.; Luna, H. Biocatalysis as a rational approach to enantiodivergent synthesis of highly oxygenated compounds: (+)- and (−)-pinitol and other cyclitols’. Isr. J. Chem. 1991, 31, 229–238. [Google Scholar] [CrossRef]
- Duchek, J.; Adams, D.R.; Hudlicky, T. Chemoenzymatic synthesis of inositols, conduritols, and cyclitol analogues. Chem. Rev. 2011, 111, 4223–4258. [Google Scholar] [CrossRef] [PubMed]
- Cantekin, S.; Baran, A.; Çalıskan, R.; Balci, M. Synthesis of bromo-conduritol-B and bromo-condutirol C as glycosidase inhibitors. Carbohydr. Res. 2009, 344, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Macías, M.A.; Suescun, L.; Pandolfi, E.; Schapiro, V.; Tibhe, G.D.; Mombru, A.W. Crystal structure and absolute configuration of (3aS,4S,5R,7aR)-2,2,7-trimethyl-3a,4,5,7a-tetrahydro-1,3-benzodioxole-4,5-diol. Acta Crystallogr. 2015, E71, 1013–1016. [Google Scholar] [CrossRef] [PubMed]
- Vila, M.A.; Brovetto, M.; Gamenara, D.; Bracco, P.; Zinola, G.; Seoane, G.; Rodríguez, S.; Carrera, I. Production of cis-1,2-dihydrocatechols of high synthetic value by whole-cell fermentation using Escherichia coli JM109 (pDTG601): A detailed study. J. Mol. Catal. B Enzym. 2013, 96, 14–20. [Google Scholar] [CrossRef]
- Pazos, M.; Martínez, S.; Vila, M.A.; Rodríguez, P.; Veiga, N.; Seoane, G.; Carrera, I. Aza and oxo Diels–Alder reactions using cis-cyclohexadienediols of microbial origin: Chemoenzymatic preparation of synthetically valuable heterocyclic scaffolds. Tetrahedron Asymmetry 2015, 26, 1436–1447. [Google Scholar] [CrossRef]
- Tibhe, G.D.; Macías, M.A.; Pandolfi, E.; Suescun, L.; Schapiro, V. Total synthesis of gabosine H and two non-natural gabosines. Synthesis 2017, 49, 565–570. [Google Scholar]
- Smietana, M.; Gouverneur, V.; Mioskowski, C. An improved synthesis of iodohydrins from alkenes. Tetrahedron Lett. 2000, 41, 193–195. [Google Scholar] [CrossRef]
- Carrera, I.; Brovetto, M.C.; Seoane, G. Selectivity in the halohydroxylation of cyclohexadienediols. Tetrahedron 2007, 63, 4095–4107. [Google Scholar] [CrossRef]
- Fonseca, G.; Seoane, G.A. Chemoenzymatic synthesis of enantiopure alfa-substituted cyclohexanones from aromatic compounds. Tetrahedron Asymmetry 2005, 16, 1393–1402. [Google Scholar] [CrossRef]
- Hudlicky, T.; Nora Restrepo-Sanchez, N.; Kary, P.D.; Jaramillo-Gomez, L.M. A short, stereoselective synthesis of neo-inositol. Carbohydr. Res. 2000, 324, 200–203. [Google Scholar] [CrossRef]
- Nguyen, B.V.; York, C.; Hudlicky, T. Chemoenzymatic Synthesis of deoxyfluoroinositols: 5-deoxy-5-fluoro-myo-inositol and 3-deoxy-3-fluoro-l-chiro-inositol. Tetrahedron 1997, 53, 8807–8814. [Google Scholar] [CrossRef]
- Hudlicky, T.; Gonzalez, D.; Gibson, D.T. Enzimatic dihydroxylation of aromatics in enantioselective synthesis: Expanding asymmetric methodology. Aldrichim. Acta 1999, 32, 35–62. [Google Scholar]
Sample Availability: Samples of all compounds are available from the authors. |


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tibhe, G.D.; Macías, M.A.; Schapiro, V.; Suescun, L.; Pandolfi, E. General Method for the Synthesis of (−)-Conduritol C and Analogs from Chiral Cyclohexadienediol Scaffolds. Molecules 2018, 23, 1653. https://doi.org/10.3390/molecules23071653
Tibhe GD, Macías MA, Schapiro V, Suescun L, Pandolfi E. General Method for the Synthesis of (−)-Conduritol C and Analogs from Chiral Cyclohexadienediol Scaffolds. Molecules. 2018; 23(7):1653. https://doi.org/10.3390/molecules23071653
Chicago/Turabian StyleTibhe, Gaurao D., Mario A. Macías, Valeria Schapiro, Leopoldo Suescun, and Enrique Pandolfi. 2018. "General Method for the Synthesis of (−)-Conduritol C and Analogs from Chiral Cyclohexadienediol Scaffolds" Molecules 23, no. 7: 1653. https://doi.org/10.3390/molecules23071653
APA StyleTibhe, G. D., Macías, M. A., Schapiro, V., Suescun, L., & Pandolfi, E. (2018). General Method for the Synthesis of (−)-Conduritol C and Analogs from Chiral Cyclohexadienediol Scaffolds. Molecules, 23(7), 1653. https://doi.org/10.3390/molecules23071653