New Chromane-Based Derivatives as Inhibitors of Mycobacterium tuberculosis Salicylate Synthase (MbtI): Preliminary Biological Evaluation and Molecular Modeling Studies

 ,
, 
 ,
,  , ,
, , 
 and
and
Abstract

1. Introduction
2. Results and Discussion
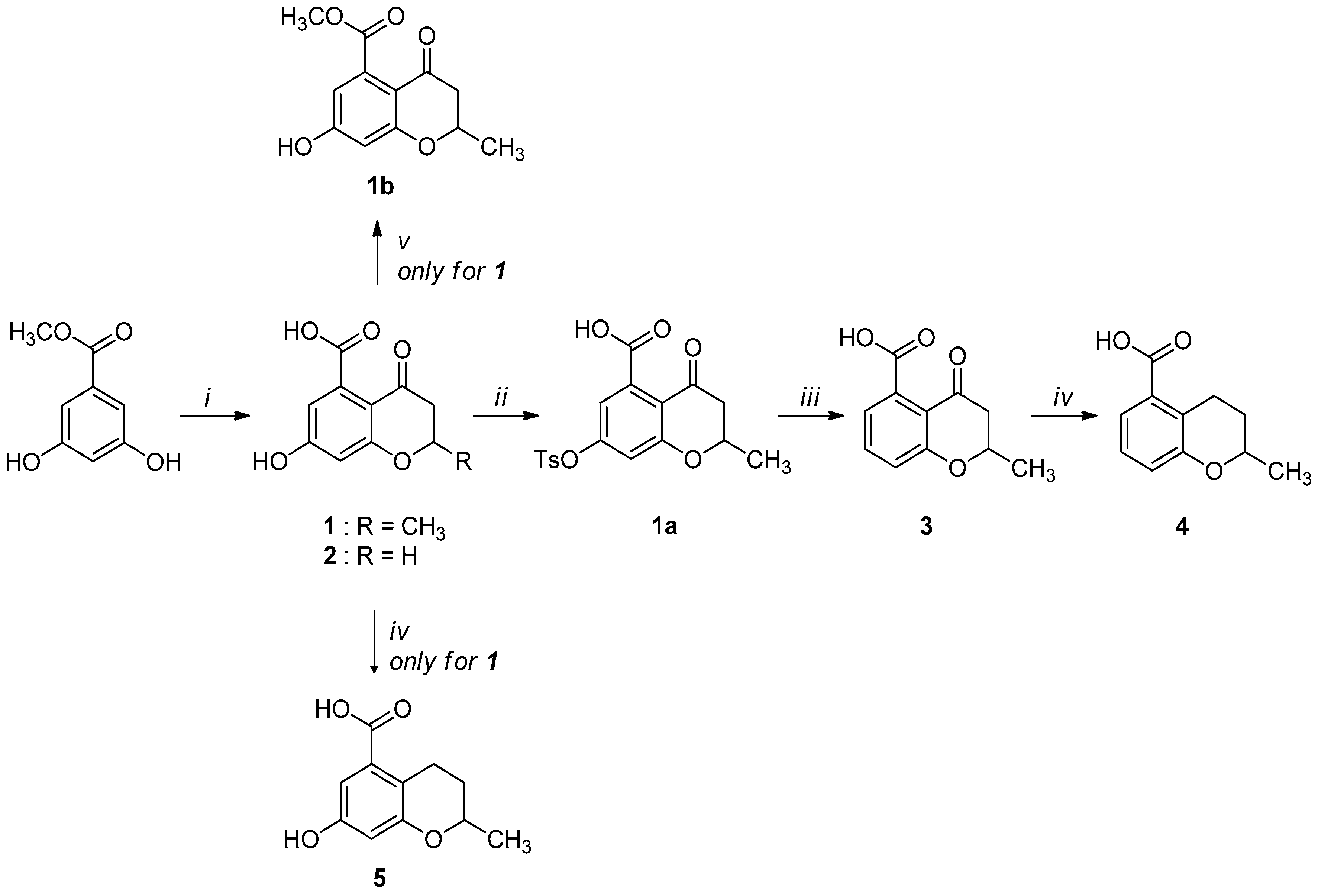
2.1. Synthesis
2.2. Biological Evaluation and Preliminary Structure-Activity Relationships (SAR)
2.3. Binding Mode Evaluation of 1–5 within the MbtI Catalytic Site
3. Materials and Methods
3.1. General Chemistry
3.1.1. Synthesis of 7-Hydroxy-2-methyl-4-oxo-3,4-dihydro-2H-benzopyran-5-carboxylic acid (1) and of 7-Hydroxy-4-oxo-3,4-dihydro-2H-benzopyran-5-carboxylic acid (2)
3.1.2. Synthesis of 2-Methyl-4-oxo-7-(toluene-p-sulfonyloxy)-3,4-dihydro-2H-benzopyran-5-carboxylic acid (1a)
3.1.3. Synthesis of 2-Methyl-4-oxo-3,4-dihydro-2H-benzopyran-5-carboxylic acid (3)
3.1.4. Synthesis of 2-Methyl-3,4-dihydro-2H-benzopyran-5-carboxylic acid (4)
3.1.5. Synthesis of 7-Hydroxy-2-methylchroman-5-carboxylic acid (5)
3.1.6. Synthesis of 7-Hydroxy-4-oxo-3,4-dihydro-2H-benzopyran-5-methyl ester (1b)
3.2. MbtI Inhibitory Activity
3.3. Docking Calculations
3.4. MD Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Global Tuberculosis Report 2017; World Health Organization: Geneva, Switzerland, 2017.
- Homolka, S.; Niemann, S.; Russell, D.G.; Rohde, K.H. Functional genetic diversity among Mycobacterium tuberculosis complex clinical isolates: Delineation of conserved core and lineage-specific transcriptomes during intracellular survival. PLoS Pathog. 2010, 6, e1000988. [Google Scholar] [CrossRef] [PubMed]
- Duncan, K. Progress in TB drug development and what is still needed. Tuberculosis 2003, 83, 201–207. [Google Scholar] [CrossRef]
- Gandhi, N.R.; Nunn, P.; Dheda, K.; Schaaf, H.S.; Zignol, M.; van Soolingen, D.; Bayona, J. Multidrug-resistant and extensively drug-resistant tuberculosis: A threat to global control of tuberculosis. Lancet 2010, 375, 1830–1843. [Google Scholar] [CrossRef]
- Zhang, Y.; Yew, W.W. Mechanisms of drug resistance in Mycobacterium tuberculosis. Int. J. Tuberc. Lung Dis. 2009, 13, 1320–1330. [Google Scholar] [PubMed]
- Falzon, D.; Gandhi, N.; Migliori, G.B.; Sotgiu, G.; Cox, H.; Holtz, T.H.; Hollm-Delgado, M.G.; Keshavjee, S.; DeRiemer, K.; Centis, R.; et al. Resistance to fluoroquinolones and second-line injectable drugs: Impact on MDR-TB outcomes. Eur. Respir. J. 2013, 42, 156–168. [Google Scholar] [CrossRef] [PubMed]
- Borisov, S.E.; Dheda, K.; Enwerem, M.; Romero Leyet, R.; D’Ambrosio, L.; Centis, R.; Sotgiu, G.; Tiberi, S.; Alffenaar, J.W.; Maryandyshev, A.; et al. Effectiveness and safety of bedaquiline-containing regimens in the treatment of MDR- and XDR-TB: A multicentre study. Eur. Respir. J. 2017, 49, 1700387. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, L.; Centis, R.; Tiberi, S.; Tadolini, M.; Dalcolmo, M.; Rendon, A.; Esposito, S.; Migliori, G.B. Delamanid and bedaquiline to treat multidrug-resistant and extensively drug-resistant tuberculosis in children: A systematic review. J. Thorac. Dis. 2017, 9, 2093–2101. [Google Scholar] [CrossRef] [PubMed]
- Meneghetti, F.; Villa, S.; Gelain, A.; Barlocco, D.; Chiarelli, L.R.; Pasca, M.R.; Costantino, L. Iron acquisition pathways as targets for antitubercular drugs. Curr. Med. Chem. 2016, 23, 4009–4026. [Google Scholar] [CrossRef] [PubMed]
- Fanzani, L.; Porta, F.; Meneghetti, F.; Villa, S.; Gelain, A.; Lucarelli, A.P.; Parisini, E. Mycobacterium tuberculosis low molecular weight phosphatases (MPtpA and MPtpB): From biological insight to inhibitors. Curr. Med. Chem. 2015, 22, 3110–3132. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Mizrahi, V. Identification and validation of novel drug targets in Mycobacterium tuberculosis. Drug Discov. Today 2017, 22, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Koul, A.; Arnoult, E.; Lounis, N.; Guillemont, J.; Andries, K. The challenge of new drug discovery for tuberculosis. Nature 2011, 469, 483–490. [Google Scholar] [CrossRef]
- Harrison, A.J.; Yu, M.; Gårdenborg, T.; Middleditch, M.; Ramsay, R.J.; Baker, E.N.; Lott, J.S. The structure of MbtI from Mycobacterium tuberculosis, the first enzyme in the biosynthesis of the siderophore mycobactin, reveals it to be a salicylate synthase. J. Bacteriol. 2006, 188, 6081–6091. [Google Scholar] [CrossRef] [PubMed]
- De Voss, J.J.; Rutter, K.; Schroeder, B.G.; Su, H.; Zhu, Y.; Barry, C.E. The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages. Proc. Natl. Acad. Sci. USA 2000, 97, 1252–1257. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.V.; Puri, R.V.; Chauhan, P.; Kar, R.; Rohilla, A.; Khera, A.; Tyagi, A.K. Disruption of mycobactin biosynthesis leads to attenuation of Mycobacterium tuberculosis for growth and virulence. J. Infect. Dis. 2013, 208, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Raymond, K.N.; Dertz, E.A.; Kim, S.S. Enterobactin: An archetype for microbial iron transport. Proc. Natl. Acad. Sci. USA 2003, 100, 3584–3588. [Google Scholar] [CrossRef] [PubMed]
- Chiarelli, L.R.; Mori, M.; Barlocco, D.; Beretta, G.; Gelain, A.; Pini, E.; Porcino, M.; Mori, G.; Stelitano, G.; Costantino, L.; et al. Discovery and development of novel salicylate synthase (MbtI) furanic inhibitors as antitubercular agents. Eur. J. Med. Chem. 2018. [Google Scholar] [CrossRef]
- Manos-Turvey, A.; Cergol, K.M.; Salam, N.K.; Bulloch, E.M.; Chi, G.; Pang, A.; Britton, W.J.; West, N.P.; Baker, E.N.; Lott, J.S.; et al. Synthesis and evaluation of M. tuberculosis salicylate synthase (MbtI) inhibitors designed to probe plasticity in the active site. Org. Biomol. Chem. 2012, 10, 9223–9236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-K.; Liu, F.; Fiers, W.D.; Sun, W.-M.; Guo, J.; Zheng, L.; Aldrich, C. Synthesis of transition-state inhibitors of chorismate utilizing enzymes from bromobenzene cis-1,2-dihydrodiol. J. Org. Chem. 2017, 82, 3432–3440. [Google Scholar] [CrossRef] [PubMed]
- Horton, D.A.; Bourne, G.T.; Smythe, M.L. The combinatorial synthesis of bicyclic privileged structures or privileged substructures. Chem. Rev. 2003, 103, 893–930. [Google Scholar] [CrossRef] [PubMed]
- Mujahid, M.; Gonnade, R.G.; Yogeeswari, P.; Sriram, D.; Muthukrishnan, M. Synthesis and antitubercular activity of amino alcohol fused spirochromone conjugates. Bioorg. Med. Chem. Lett. 2013, 23, 1416–1419. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Maddox, M.M.; Alam, M.Z.; Tsutsumi, L.S.; Narula, G.; Bruhn, D.F.; Wu, X.; Sandhaus, S.; Lee, R.B.; Simmons, C.J.; et al. Synthesis, structure-activity relationship studies, and antibacterial evaluation of 4-chromanones and chalcones, as well as olympicin A and derivatives. J. Med. Chem. 2014, 57, 8398–8420. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.C.; Peng, C.F.; Chen, I.H.; Tsai, I.L. Antitubercular chromones and flavonoids from Pisonia aculeata. J. Nat. Prod. 2011, 74, 976–982. [Google Scholar] [CrossRef] [PubMed]
- Duewell, H.; Haig, T.J. A neighbouring group effect of carbonyl as shown in the mass fragmentation patterns of 4-oxochroman-5-acetic acids and of some related compounds. Aust. J. Chem. 1988, 41, 535–548. [Google Scholar] [CrossRef]
- Ramanathan, V.; Venkataraman, K. A new method for the preparation of γ-resorcylic acid. Curr. Sci. 1952, 21, 283. [Google Scholar]
- Bavkar, S.N.; Salunke, D.B.; Hazra, B.G.; Pore, B.S.; Thierry, J.; Dodd, R.H. Pd-catalized one-pot chemoselective hydrogenation protocol for the preparation of carboxamides directly from azides. Tetrahedron Lett. 2010, 51, 3815–3819. [Google Scholar] [CrossRef]
- Porta, F.; Facchetti, G.; Ferri, N.; Gelain, A.; Meneghetti, F.; Villa, S.; Barlocco, D.; Masciocchi, D.; Asai, A.; Miyoshi, N.; et al. An in vivo active 1,2,5-oxadiazole Pt(II) complex: A promising anticancer agent endowed with STAT3 inhibitory properties. Eur. J. Med. Chem. 2017, 131, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.; Li, B. Enantioselective aldol reaction of α-ketoester and cyclopentanone catalyzed by L-proline. Chin. J. Chem. 2010, 28, 617–621. [Google Scholar] [CrossRef]
- Chowdhury, P.K.; Borah, P. A mild and chemoselective deoxygenation of ketones with zinc and hydrogen chloride generated in situ using a zinc-aluminium chloride hexahydrate-tetrahydrofuran-water system. J. Chem. Res. 1994, 230–231. [Google Scholar]
- Emami, S.; Ghanbarimasir, Z. Recent advances of chroman-4-one derivatives: Synthetic approaches and bioactivities. Eur. J. Med. Chem. 2015, 93, 539–563. [Google Scholar] [CrossRef] [PubMed]
- Tuccinardi, T.; Poli, G.; Romboli, V.; Giordano, A.; Martinelli, A. Extensive consensus docking evaluation for ligand pose prediction and virtual screening studies. J. Chem. Inf. Model. 2014, 54, 2980–2986. [Google Scholar] [CrossRef] [PubMed]
- Vasan, M.; Neres, J.; Williams, J.; Wilson, D.J.; Teitelbaum, A.M.; Remmel, R.P.; Aldrich, C.C. Inhibitors of the salicylate synthase (MbtI) from Mycobacterium tuberculosis discovered by high-throughput screening. ChemMedChem 2010, 5, 2079–2087. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Kerbarh, O.; Chirgadze, D.Y.; Blundell, T.L.; Abell, C. Crystal structures of Yersinia enterocolitica salicylate synthase and its complex with the reaction products salicylate and pyruvate. J. Mol. Biol. 2006, 357, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Berryman, J.T.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; et al. AMBER 2015; University of California: San Francisco, CA, USA, 2015. [Google Scholar]
- Maestro, version 9.0; Schrödinger Inc.: Portland, OR, USA, 2009; Available online: https://www.schrodinger.com/ (accessed on 31 May 2018).
- Macromodel, version 9.7; Schrödinger Inc.: Portland, OR, USA, 2009; Available online: https://www.schrodinger.com/ (accessed on 31 May 2018).
- Poli, G.; Giuntini, N.; Martinelli, A.; Tuccinardi, T. Application of a flap-consensus docking mixed strategy for the identification of new fatty acid amide hydrolase inhibitors. J. Chem. Inf. Model. 2015, 55, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Martinelli, A.; Tuccinardi, T. Reliability analysis and optimization of the consensus docking approach for the development of virtual screening studies. J. Enzym. Inhib. Med. Chem. 2016, 31, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C.; Caligiuri, I.; Bertelli, E.; Poli, G.; Rizzolio, F.; Macchia, M.; Martinelli, A.; Minutolo, F.; Tuccinardi, T. Development of terphenyl-2-methyloxazol-5(4H)-one derivatives as selective reversible MAGL inhibitors. J. Enzym. Inhib. Med. Chem. 2017, 32, 1240–1252. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Monecke, P.; Hessler, G.; Stutzle, T.; Exner, T.E. pharmACOphore: Multiple flexible ligand alignment based on ant colony optimization. J. Chem. Inf. Model. 2010, 50, 1669–1681. [Google Scholar] [CrossRef] [PubMed]
- Tuccinardi, T.; Poli, G.; Dell’Agnello, M.; Granchi, C.; Minutolo, F.; Martinelli, A. Receptor-based virtual screening evaluation for the identification of estrogen receptor beta ligands. J. Enzym. Inhib. Med. Chem. 2015, 30, 662–670. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of compounds 1, 1b, 3, 4 and 5 are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Structure | MbtI Residual Activity at 100 μM | MbtI IC50 (μM) |
|---|---|---|---|
| I |  | 11.0 ± 3.9 | 11.6 ± 2.4 |
| 1 |  | 23.3 ± 3.4 | 55.8 ± 4.2 |
| 1b |  | >75 | ND |
| 2 |  | 28.1 ± 2.9 | 61.3 ± 5.3 |
| 3 |  | >75 | ND |
| 4 |  | >75 | ND |
| 5 |  | 50.1 ± 7.1 | ND |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pini, E.; Poli, G.; Tuccinardi, T.; Chiarelli, L.R.; Mori, M.; Gelain, A.; Costantino, L.; Villa, S.; Meneghetti, F.; Barlocco, D. New Chromane-Based Derivatives as Inhibitors of Mycobacterium tuberculosis Salicylate Synthase (MbtI): Preliminary Biological Evaluation and Molecular Modeling Studies. Molecules 2018, 23, 1506. https://doi.org/10.3390/molecules23071506
Pini E, Poli G, Tuccinardi T, Chiarelli LR, Mori M, Gelain A, Costantino L, Villa S, Meneghetti F, Barlocco D. New Chromane-Based Derivatives as Inhibitors of Mycobacterium tuberculosis Salicylate Synthase (MbtI): Preliminary Biological Evaluation and Molecular Modeling Studies. Molecules. 2018; 23(7):1506. https://doi.org/10.3390/molecules23071506
Chicago/Turabian StylePini, Elena, Giulio Poli, Tiziano Tuccinardi, Laurent Roberto Chiarelli, Matteo Mori, Arianna Gelain, Luca Costantino, Stefania Villa, Fiorella Meneghetti, and Daniela Barlocco. 2018. "New Chromane-Based Derivatives as Inhibitors of Mycobacterium tuberculosis Salicylate Synthase (MbtI): Preliminary Biological Evaluation and Molecular Modeling Studies" Molecules 23, no. 7: 1506. https://doi.org/10.3390/molecules23071506
APA StylePini, E., Poli, G., Tuccinardi, T., Chiarelli, L. R., Mori, M., Gelain, A., Costantino, L., Villa, S., Meneghetti, F., & Barlocco, D. (2018). New Chromane-Based Derivatives as Inhibitors of Mycobacterium tuberculosis Salicylate Synthase (MbtI): Preliminary Biological Evaluation and Molecular Modeling Studies. Molecules, 23(7), 1506. https://doi.org/10.3390/molecules23071506