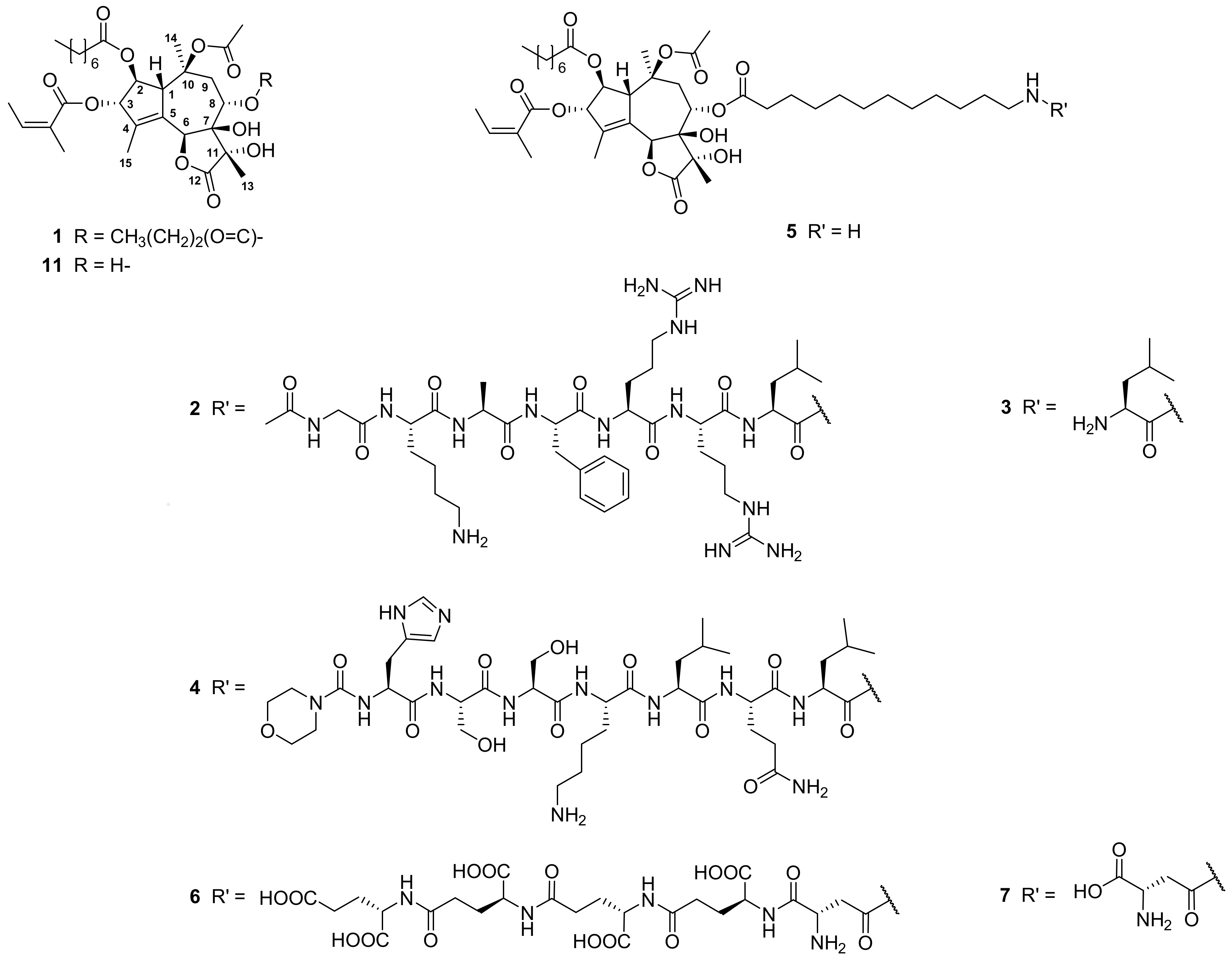
3.4. Synthesis and Purification of Compounds 2, 4, 5, 6, and 8
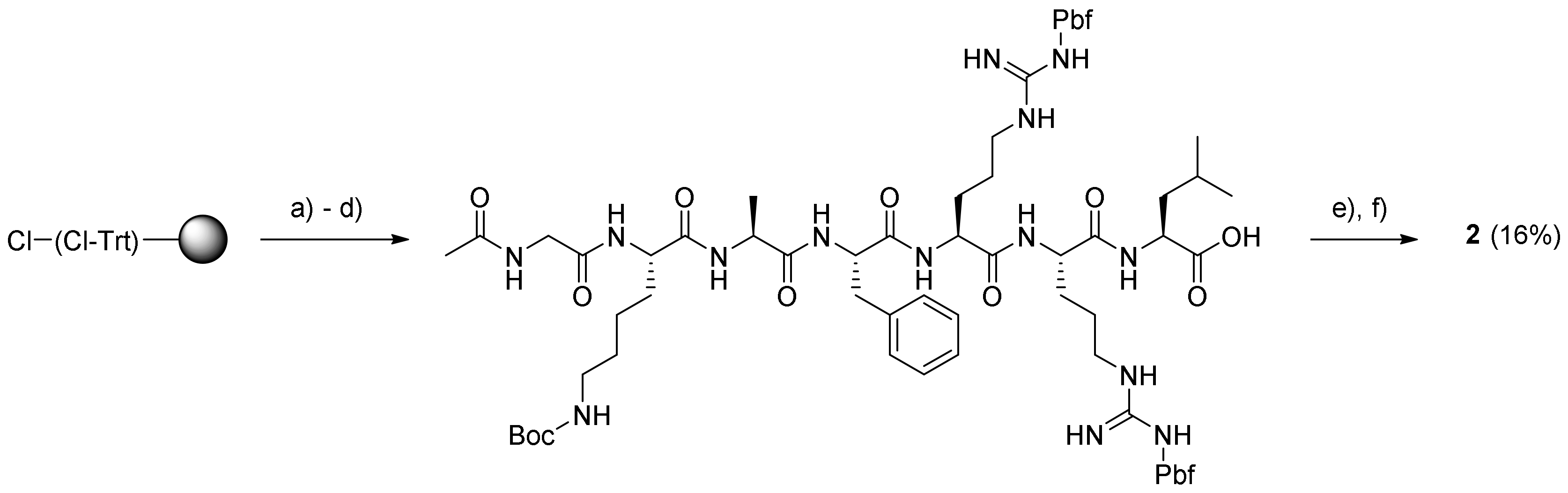
3.4.1. Ac-Gly-Lys-Ala-Phe-Arg-Arg-Leu-12-Aminododecanoate-(O-8)-Debutanoyl-Thapsigargin (2)
Loading of the C-terminal residue was performed by swelling 2-chlorotrityl chloride (CTC) resin (1.6 mmol/g; 1.0 g) in DCM (4 mL), followed by the addition of Fmoc-Leu-OH (564 mg; 1.6 mmol) dissolved in DCM (4 mL) and DIPEA (2.7 mL; 16 mmol). The mixture was shaken for 3.5 h at rt, and then the resin was washed with DCM, capped by treatment with DIPEA–MeOH–DCM 5:15:80 (8 mL; 2 × 5 min) and subsequently, washed with DMF, MeOH, and DCM (each with 3 × 10 mL for 3 min). Test cleavage with 20% HFIP–DCM of a small amount of dry loaded resin, followed by careful evaporation and weighing of the amount of cleaved Fmoc-Leu-OH showed a loading of ~0.9 mmol/g.
The peptide Ac-GKAFRRL-OH was synthesized on a Liberty BlueTM microwave peptide synthesizer starting from the above Fmoc-Leu-O-2-CTC resin (0.9 mmol/g; 110 mg; 0.1 mmol) swelled in DMF (5 mL). Calculated amounts of Nα-Fmoc protected amino acids in 0.2 M solution (added as volumes corresponding to 5 equivalents): Fmoc-Gly-OH (0.33 g; 11 mL DMF), Fmoc-Lys(Boc)-OH (0.52 g; 11 mL DMF), Fmoc-Ala-OH (0.35 g; 11 mL DMF), Fmoc-Phe-OH (0.43 g; 11 mL DMF), and Fmoc-Arg(Pbf)-OH (2.08 g; 32 mL DMF). The conditions for chain elongation were double coupling at 45 °C for 15 min, while triple coupling at 45 °C during 15 min was needed for Arg; Fmoc removal was performed with 20% piperidine in DMF at 45 °C for 30 s and 180 s.
The resulting Gly-Lys-Ala-Phe-Arg-Arg-Leu-O-(Cl-Trt)-resin was washed twice with both DMF and DCM, and was then acetylated with Ac2O–DIPEA–NMP (1:2:3, 6 mL; 2 × 5 min). The resulting resin-bound peptide was washed with DMF, MeOH, and DCM (each 3 × 8 mL for 3 min) and cleaved from the resin with 20% HFIP–DCM. The drained cleavage solution was concentrated to give the protected peptide as a gel. To a solution of the residue (345 mg; 0.23 mmol) in DCM (1.5 mL) and PyBOP (48 mg; 0.092 mmol) in DMF (0.4 mL) and DIPEA (27 µL; 0.132 mmol) were added. The mixture was stirred for 20 min to pre-activate the carboxylic acid, and then a solution of compound 5 (60 mg; 0.077 mmol) in DCM (1.5 mL) was added. The mixture was stirred under argon for 14 h at rt, and the resulting reaction mixture was concentrated in vacuo. The obtained residue was deprotected with TFA (3 mL with four drops of H2O added) for 2 h at rt. The reaction mixture was evaporated, and the residue was subjected to purification by preparative HPLC using the gradient 20% → 80% B over 20 min. The purity of the product (retention time: 13.0–13.7 min) was estimated by analytical UHPLC to be >98% using the gradient 20% → 100% B for 10 min (retention time: 7.7 min). Upon concentration, the product was dissolved in dioxane–H2O and freeze-dried to provide prodrug 2 (20 mg; 15.7%) as a white solid. Characterization of prodrug 2: 1H NMR (400 MHz, methanol-d4) δ ppm = 7.32–7.28 (m, 2 H, HPhe-ε), 7.27–7.25 (m, 2 H, HPhe-δ), 7.22 (m, 1 H, HPhe-ζ), 6.18 (qq, J = 7.2, 1.5 Hz, 1 H, HAng-3), 5.71 (m, 1 H, H-6), 5.68 (m, 1 H, H-3), 5.59 (t, J = 3.7 Hz, 1 H, H-8), 5.52 (t, J = 2.9 Hz, 1 H, H-2), 4.47 (m, 1 H, HPhe-α), 4.36 (m, 1 H, H-1), 4.33 (m, 1 H, HLys-α), 4.30 (m, 1 H, HArg-α), 4.24 (m, 1 H, HArg-α), 4.20 (m, 1 H, HLeu-α), 4.17 (m, 1 H, HAla-α), 3.97–3.83 (m, 2 H, 2 HGly-α), 3.27–3.18 (m, 6 H, 2 HArg-δ and HPhe-β), 3.17–3.10 (m, 2 H, HLys-ε), 3.02 (m, 1 H, H-9a), 2.95 (m, 2 H, H12-AD-12), 2.37 (m, 2 H, Hoct-2), 2.32 (m, 1 H, H-9b), 2.29 (m, 2 H, H12-AD-2), 2.02–1.98 (m, 3 H, HAng-4), 2.00 (s, 3 H, -(C=O)CH3), 1.93 (m, 3 H, CH3 from CAng-2), 1.89 (s, 3 H, AcTg), 1.86 (m, 3 H, H-15), 1.85–1.80 (m, 2 H, HArg-β), 1.80–1.74 (m, 2 H, HLys-β), 1.74–1.71 (m, 2 H, HArg-β), 1.71 (m, 1 H, HLeu-γ), 1.70–1.67 (m, 4 H, 2 HArg-γ), 1.66 (m, 2 H, HLeu-β), 1.64 (m, 2 H, HLys-δ), 1.62 (m, 2 H, Hoct-3), 1.61–1.56 (m, 2 H, H12-AD-3), 1.54–1.49 (m, 2 H, H12-AD-11), 1.49–1.44 (m, 2 H, HLys-γ), 1.42 (s, 3 H, H-14), 1.37 (s, 3 H, H-13), 1.36–1.28 (m, 25H, H12-AD-4 − 10, Hoct-4 − 7, HAla-β), 0.97–0.92 (m, 6 H, 2 HLeu-δ), 0.92–0.89 (m, 3 H, Hoct-8); 13C NMR (400 MHz, methanol-d4) δ ppm = 176.72 (1 C, C-12), 174.53 (1 C, CAla-1), 173.51 (1 C, CLeu-1), 172.98 (1 C, CPhe-1), 172.89 (2 C, CAc capped-1, C-1 Lys), 172.84 (1 C, Coct-1), 172.59 (1 C, CArg-1), 172.39 (1 C, CArg-1), 172.35 (1 C, C12-AD-1), 171.65 (1 C, CGly-1), 170.42 (1 C, CAc Tg-1), 167.18 (1 C, CAng-1), 157.25 (2 C, 2 CArg-6), 139.66 (1 C, C-4), 138.07 (1 C, CAng-3), 131.96 (1 C, C-5), 128.85 (1 C, CAng-2), 128.83 (2 C, CPhe-6 + 8), 128.19 (2 C, CPhe-5 + 9), 127.43 (1 C, CPhe-4), 126.53 (1 C, CPhe-7), 84.52 (1 C, C-3), 84.30 (1 C, C-10), 78.09 (1 C, C-11), 78.05 (1 C, C-7), 77.92 (1 C, C-2), 76.62 (1 C, C-6), 66.04 (1 C, C-8), 57.68 (1 C, C-1), 55.76 (1 C, CPhe-2), 54.60 (1 C, CLeu-2), 53.44 (2 C, 2 CArg-2), 52.14 (1 C, CLys-2), 50.47 (1 C, CLys-2), 42.92 (1 C, CGly-2), 40.55 (3 C, CLeu-3 Leu + 2 CArg-5), 39.07 (2 C, C12-AD-12 and CLys-6), 38.02 (1 C, C-9), 34.14 (1 C, C12-AD-2), 33.78 (1 C, Coct-2), 31.45 (1 C, Coct-7), 30.33 (1 C, CLys-3), 29.31–28.68 (9 C, C12-AD-4 − 9 and 11, Coct-4 − 5), 27.84 (2 C, 2 CArg-3), 26.73 (2 C, 2 CArg-4), 26.53 (1 C, C12-AD-11), 24.84 (1 C, CLys-5), 24.57 (2 C, Coct-3 and C12-AD-3), 24.18 (1 C, CLeu-4), 22.25 (1 C, Coct-6), 22.16 (2 C, CLys-3 and CLeu-5a), 21.95 (1 C, C-14), 21.29 (1 C, CAc Tg-2 Tg), 21.14 (1 C, CH3 in Ac-2), 20.42 (1 C, CLeu-5b), 19.35 (1 C, CH3 from CAng-2), 15.65 (1 C, CAla-3), 14.67 (1 C, CAng-4), 14.46 (1 C, C-13), 13.02 (1 C, Coct-8), 11.58 (1 C, C-15); HR-MALDI-TOF [C82H133N15O20 + H]+: m/z: 1648.9924. Found 1648.9929.
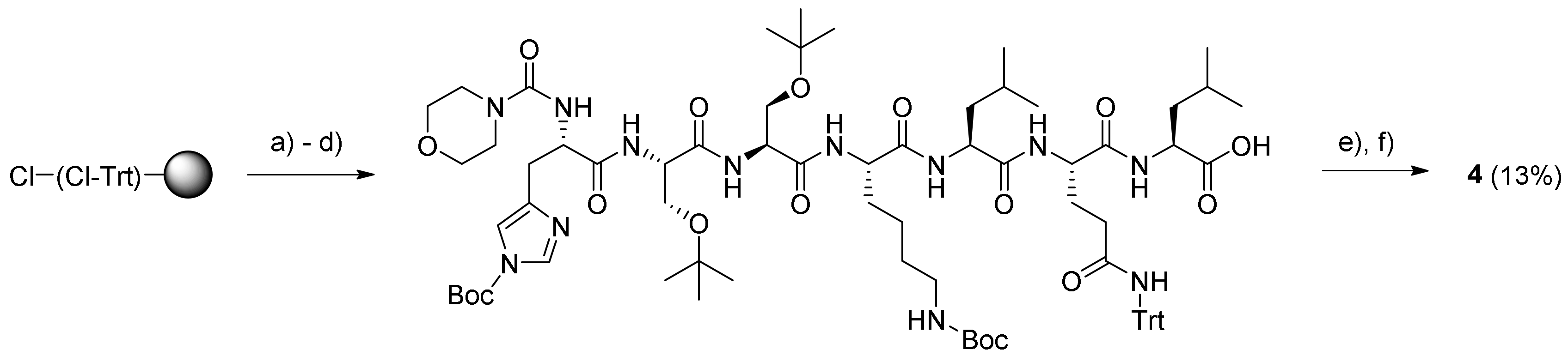
3.4.2. Morpholine-4-Carbonyl-His-Ser-Ser-Lys-Leu-Gln-Leu-12-Aminododecanoate-(O-8)- Debutanoyl-Thapsigargin (4)
Synthesis of the HSSKLQL peptide was performed on a Liberty BlueTM microwave peptide synthesizer starting from the above Fmoc-Leu-O-2-CTC resin (0.9 mmol/g; 110 mg; 0.1 mmol) swelled in DMF (5 mL). The calculated amounts of Nα-Fmoc protected amino acids in 0.2 M solution (added as volumes corresponding to 5 equivalents) were: Fmoc-His(Trt)-OH (0.38 g; 6 mL DMF), Fmoc-Ser(tBu)-OH (0.43 g; 11 mL DMF), Fmoc-Lys(Boc)-OH (0.29 g; 6 mL DMF), Fmoc-Leu-OH (0.22 g; 6 mL DMF), and Fmoc-Gln(Trt)-OH (0.37 g; 6 mL DMF). The conditions for chain elongation were single coupling at 45 °C for 15 min; Fmoc removal was performed with 20% piperidine in DMF at 45 °C for 30 s and 180 s.
The resulting protected His-Ser-Ser-Lys-Leu-Gln-Leu-O-(Cl-Trt) resin was placed in two reaction vessels, and then, the resin was washed twice with both DMF and DCM. Solutions of morpholine-4-carbonyl chloride–Et3N–NMP (1:4:20; 8 mL) were added to the two resin batches, which then were heated to 40 °C for 2 h. The resin was washed with DCM, and the procedure was repeated for 1 h with fresh reagents, followed by washing with DMF, MeOH, and DCM (each 3 × 8 mL for 3 min). The product was cleaved from the resin with 20% HFIP–DCM, and the concentration of the drained cleavage mixture gave the protected peptide as a gel (220 mg). A portion of this residue (165 mg) was dissolved in DCM (1.5 mL), and then, PyBOP (41 mg; 0.078 mmol) and DIPEA (23 µL; 0.178 mmol) were added. The mixture was stirred for 20 min to pre-activate the carboxylic acid, followed by the addition of compound 5 (52 mg; 0.067 mmol) dissolved in DCM (1.5 mL). The mixture was stirred under argon for 14 h at rt. The resulting reaction mixture was concentrated in vacuo, and the residue was deprotected with TFA (3 mL; with four drops of H2O added) for 2 h at rt. The drained cleavage mixture was evaporated, and the residue was subjected to purification by preparative HPLC using the gradient 20% → 80% B over 20 min. The purity of the product (retention time: 16.7–17.2 min) was determined by analytical UHPLC (>99%) using the gradient 20% → 100% B for 10 min (retention time: 8.2 min). Upon concentration, the resulting residue was dissolved in dioxane–H2O and freeze-dried to provide prodrug 4 (15 mg; 13.4%) as a white solid. Characterization of prodrug 4: 1H NMR (400 MHz, methanol-d4) δ ppm = 8.82 (s, 1 H, HHis-ε1), 7.39 (s, 1 H, HHis-δ2), 6.19 (qq, J = 7.2, 1.5 Hz, 1 H, HAng-3), 5.71 (m, 1 H, H-6), 5.68 (m, 1 H, H-3), 5.60 (t, J = 3.7 Hz, 1 H, H-8), 5.53 (t, J = 2.9 Hz, 1 H, H-2), 4.61 (m, 1 H, HHis-α), 4.52–4.46 (m, 2 H, 2 HSer-α), 4.36 (m, 1 H, H-1), 4.32 (m, 1 H, HLys-α), 4.3 (m, 1 H, HGln-α), 4.29–4.24 (m, 2 H, 2 HLeu-α), 4.02–3.91 (m, 2 H, 2 HSer a-β), 3.87–3.80 (m, 2 H, 2 HSer b-β), 3.66 (t, J = 4.4 Hz, 4 H, HMorph-2 and 6), 3.41 (t, J = 5.1 Hz, 4 H, HMorph-3 and 5), 3.30 (m, 1 H, HHis a-β), 3.20 (m, 2 H, H12-AD-12), 3.15 (m, 1 H, HHis b-β), 3.01 (m, 1 H, H-9a), 2.96 (m, 2 H, HLys-ε), 2.37 (m, 2 H, Hoct-2), 2.35 (m, 2 H, HGln-γ), 2.33 (m, 1 H, H-9b), 2.30 (m, 2 H, H12-AD-2), 2.07–2.15 (m, 2 H, HGln-β), 2.00 (dq, J = 5.9. 1.0 Hz, 2 H, HAng-4), 1.94 (m, 2 H, CH3 from CAng-2), 1.91 (m, 1 H, HLys a-β), 1.90 (s, 3 H, Ac), 1.87 (s, 3 H, H-15), 1.80 (m, 1 H, HLys b-β), 1.75–1.71 (m, 2 H, 2 HLeu-γ), 1.70 (m, 2 H, HLys-δ), 1.69–1.66 (m, 4 H, 2 HLeu-β), 1.65 (m, 2 H, Hoct-3), 1.63 (m, 2 H, H12-AD-3), 1.54 (m, 2 H, H12-AD-11), 1.50 (m, 2 H, HLys-γ), 1.42 (s, 3 H, H-14), 1.37 (s, 3 H, H-13), 1.36–1.29 (m, 22 H, H12-AD-4 − 10, Hoct-4 − 7), 0.97 (m, 6 H, 2 HLeu-δ) 0.93 (m, 6 H, 2 HLeu-δ), 0.91 (m, 3 H, Hoct-8); 13C NMR (400 MHz, methanol-d4) δ ppm = 176.72 (1 C, C-12), 173.96 (1 C, CLeu-1), 173.61 (1 C, CGln-1), 173.51 (1 C, CLeu-1), 173.15 (1 C, CGln-5), 172.89 (1 C, C12-AD-1), 172.45 (1 C, CLys-1), 172.35 (1 C, Coct-1), 172.22 (1 C, CHis-1), 171.70 (2 C, 2 CSer-1), 170.43 (1 C, CAc-1), 167.18 (1 C, CAng-1), 157.99 (1 C, Morph. carbonyl), 139.66 (1 C, C-5), 138.07 (1 C, CAng-3), 133.60 (1 C, CHis-6) 131.98 (1 C,C-4), 129.94 (1 C, CAng-2), 127.43 (1 C, CHis-4), 117.12 (1 C, CHis-5), 84.52 (1 C, C-3), 84.30 (1 C, C-10), 78.09 (1 C, C-11), 78.04 (1 C, C-7), 77.93 (1 C, C-2), 76.62 (1 C, C-6), 66.12 (2 C, CMorph-3 + 5), 66.04 (1 C, C-8), 61.45 (1 C, CSer-3), 61.30 (1 C, CSer-3), 57.67 (1 C, C-1), 55.98 (1 C, CSer-2), 55.67 (1 C, CSer-2), 54.50 (1 C, CLeu-2), 54.40 (1 C, CLeu-2), 54.20 (1 C, CHis-2), 53.59 (1 C, CGln-2), 52.14 (1 C, CLys-2), 43.93 (2 C, CMorph-3 + 5), 40.42 (1 C, CLeu-3), 39.61 (1 C, CLeu-3), 39.10 (2 C, C12-AD-12 + CLys-6), 38.02 (1 C, C-9), 34.14 (1 C, C12-AD-2), 33.78 (1 C,Coct-2), 31.45 (1 C, Coct-7), 31.40 (1 C, CGln-4), 30.08 (1 C, CLys-3), 29.28–28.69 (9 C, C12-AD-4 − 9 + 11, Coct-4 − 5), 27.00 (1 C, CGln-3), 26.70 (1 C, C12-AD-10), 26.54 (1 C, CHis-3), 26.49 (1 C, CLys-5), 24.60 (1 C, C12-AD-3), 24.57 (2 C, 2 CLeu-4), 24.19 (1 C, Coct-4), 22.25 (2 C, 2 CLeu-5), 22.17 (1 C, Coct-6), 22.09 (1 C, CLys-4), 22.01 (1 C, C-14), 20.31 (2 C, 2 CLeu-5) 19.35 (1 C, CH3 from CAng-2), 14.67 (1 C, CAng-4), 14.47 (1 C, C-13), 13.02 (1 C, Coct-8), 11.58 (1 C, C-15); HR-MALDI-TOF: [C82H133N13O2 + H]+ m/z: 1683.9586. Found 1684.9673.
3.4.3. 12-Aminododecanoate-(O-8)-Debutanoyl-Thapsigargin (5)
To a solution of Boc-12-aminododecanoate-(
O-8)-debutanoyl-thapsigargin (150 mg; 0.171 mmol) [
27] in DCM (0.5 mL) TFA (2 mL) was added, together with 4 drops of H
2O. The mixture was stirred at rt for 90 min. The reaction mixture was concentrated, and the was residue purified on a silica column (DCM–MeOH 10:1) to provide
5 (120 mg; 90%) as a yellowish solid.
1H NMR,
13C NMR and HR-MS were as reported [
27].
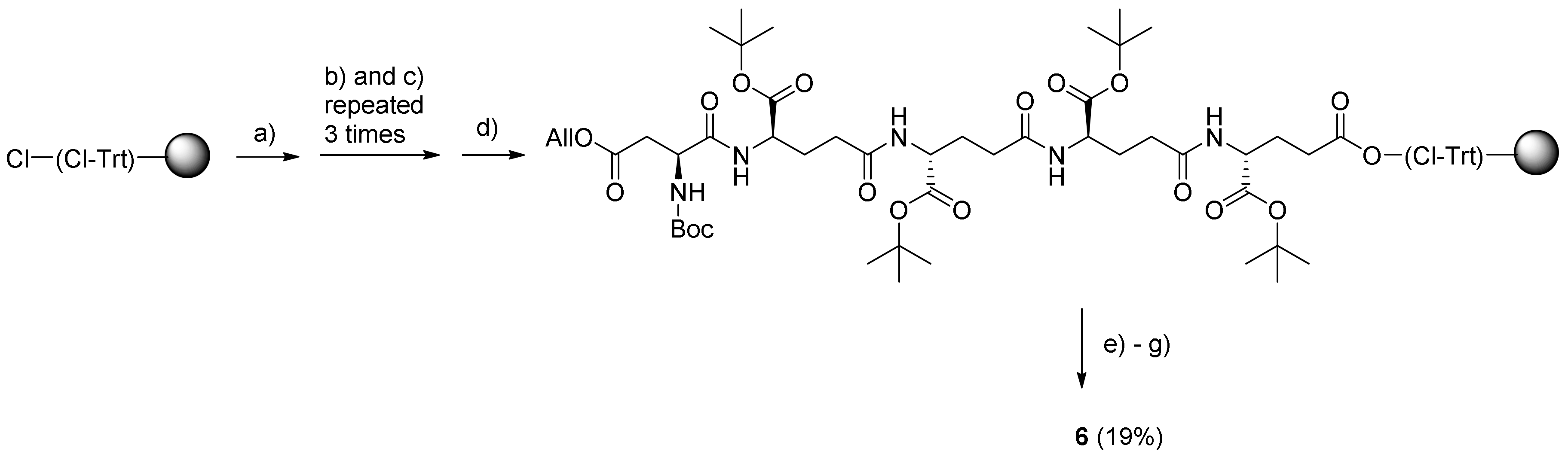
3.4.4. (γ-Glu-)4-β-Asp-12-Aminododecanoate-(O-8)-Debutanoyl-Thapsigargin (6)
In a glass vessel (300 mL) for manual SPPS (Peptides International, Louisville, KY, USA) 2-chlorotrityl chloride resin (1.6 mmol/g; 3.125 g; 5 mmol) was swelled in DCM (8 mL), and upon draining, a solution of Fmoc-Glu-OtBu (425 mg; 1 mmol) and DIPEA (2.1 mL; 12 mmol) in DCM (15 mL) was added to the reaction vessel. After shaking for 3 h at rt, the resin was drained, washed with DCM, and then capped with DIPEA–MeOH–DCM 5:15:80 (8 mL; 2 × 5 min), and finally, it was washed with DMF, MeOH, and DCM (each 3 times for 3 min). The residual solvent was removed on a freeze-dryer. Test cleavage of a dry sample of the loaded resin showed a loading of ca. 0.30 mmol/g.
By using a large syringe (with a polypropylene filter bottom), the peptide was assembled on the above resin preloaded with Fmoc-Glu-OtBu (0.304 mmol/g; 1.5 g), starting with three sequential coupling cycles, each comprising Fmoc removal with 20% piperidine–DMF (8 mL; 2 × 20 min), washing of the resin with DMF, MeOH, and DCM (each 3 times for 3 min), the addition of DMF (3 mL) and DCM (2 mL), and the addition of a solution of Fmoc-Glu-OtBu (390 mg; 0.95 mmol) pre-activated (10 min) with PyBOP (711 mg; 1.37 mmol) and DIPEA (0.475 mL; 2.72 mmol) in DMF (3 mL). Each coupling was performed under shaking for 5 h at rt, and then the resin was washed with DMF, MeOH, and DCM (each 3 times for 3 min). Boc-Asp(All)-OH (350 mg; 1.28 mmol) was coupled by using the same conditions. Allyl deprotection was performed on-resin by adding DCM (5 mL) followed by a solution of Me2N·BH3 in DCM (2 mL) and 0.02 M Pd(PPh3)4 (185 mg) in DCM (8 mL). The mixture was shaken for 6 h under argon at rt, after which the resin was washed with DMF, MeOH, and DCM (each 3 times for 3 min).
DCM (4 mL) was added to part of the resin-bound peptide (0.75 g), and a solution of PyBOP (356 mg; 0.68 mmol) and DIPEA (0.24 mL; 1.36 mmol) in DCM (4 mL) was added to pre-activate the carboxylic acid for 20 min. A solution of
5 (354 mg; 0.46 mmol) in DCM (2 mL) was added, and then the mixture was shaken for 10 h at rt. Finally, the resin was drained and washed with DMF, MeOH, and DCM (each 3 times for 3 min), and the protected conjugate was cleaved off the resin and protecting groups were removed by treatment with TFA (4 mL), DCM (0.5 mL), and H
2O (4 drops) for 2.5 h at rt. The concentration of the reaction mixture afforded a crude product, which was purified by preparative HPLC using a gradient of 30% → 100% B (20 min; retention time 13.2–13.7 min) to give the desired product, which, as assessed by analytical UHPLC, had a purity >99% when using the same gradient 30% → 100% B (10 min; retention time 7.4 min). This fraction was evaporated to give a residue, which was dissolved in dioxane–H
2O and freeze-dried to give prodrug
6 (60 mg; 18.7%) as a white solid. Characterization of prodrug
6:
1H NMR (600 MHz, methanol-
d4) δ ppm = 6.13 (qq,
J = 7.2, 1.5 Hz, 1 H, H
Ang-3), 5.65 (m, 1 H, H-6), 5.62 (m, 1 H, H-3), 5.53 (t,
J = 3.7 Hz, 1 H, H-8), 5.46 (t,
J = 2.9 Hz, 1 H, H-2), 4.45–4.37 (m, 4 H, H
Glu-α), 4.30 (m, 1 H, H-1), 4.26 (m, 1 H, H
Asp-α), 3.15 (sxt,
J = 6.6 Hz, 2 H, H
12-AD-12), 2.93 (dd,
J = 14.5, 3.5 Hz, 1 H, H
a-9), 2.87 (dd,
J = 16.5, 4.4 Hz, 1 H, H
Asp a-3), 2.71 (dd,
J = 9.0, 16.7 Hz, 1 H, H
Asp b-3), 2.41–2.32 (m, 8 H, H-γ Glu), 2.32–2.26 (m, 4 H, H
oct-2 and H
12-AD-2), 2.25 (m, 1 H, H
b-9), 2.24–2.10 (m, 6 H, H
Glu2-Glu4-β), 1.94 (dq,
J = 5.9, 1.0 Hz, 3 H, H
Ang-4), 1.91 (dd,
J = 8.8, 6.6 Hz, 2 H, H
Glu1-β), 1.88 (t,
J = 1.5 Hz, 3 H, CH
3 from C
Ang-2), 1.83 (m, 3 H, Ac), 1.81 (s, 3 H, H-15), 1.62–1.52 (m, 4 H, H
oct-3 and H
12-AD-3), 1.46 (m, 2 H, H
12-AD-11), 1.36 (s, 3 H, H-14), 1.31 (s, 3 H, H-13), 1.30–1.23 (m, 22 H, H
12-AD-4 − 10, H
oct-4 − 7), 0.86 (m, 3 H, H
oct-8);
13C NMR (600 MHz, methanol-
d4) δ ppm = 176.71 (1 C, C-12) 175.01 (1 C, C
Glu1-5), 173.68–173.60 (6 C, C
Glu1-Glu4 -1 and C
Glu2-Glu3-5), 173.43 (1 C, C
Glu4-5), 172.78 (1 C, C
oct-1), 172.35 (1 C, C
12-AD-1), 170.43 (1 C, C-1 Ac), 169.22 (1 C, C
Asp-4), 168.27 (1 C, C
Asp-1), 167.18 (1 C, C
Ang-1), 139.65 (1 C, C-5), 138.08 (1 C, C
Ang-3), 131.95 (1 C, C-4), 127.42 (1 C, C
Ang-2), 84.52 (1 C, C-10), 84.30 (1 C, C-3), 78.08 (1 C, C-11), 78.04 (1 C, C-7), 77.92 (1 C, C-2), 76.62 (1 C, C-6), 66.04 (1 C, C-8), 57.66 (1 C, C-1), 52.01 (1 C, C
Glu1-2), 51.79 (1 C, C
Glu2-2), 51.71 (1 C, C
Glu3-2), 51.67 (1 C, C
Glu4-2), 50.10 (1 C, C
Asp-2), 39.28 (1 C, C
12-AD-12), 38.01 (1 C, C-9), 35.12 (1 C, C
Asp-3), 34.14 (1 C, C
12-AD-2), 33.78 (1 C, C
oct-2), 31.56–31.51 (3 C, C
Glu2-Glu4-4), 31.45 (1 C, C
oct-7), 29.87 (1 C, C
Glu1-4), 29.30–28.65 (9 C, C
12-AD-4 − 9 and 11, C
oct-4 − 5), 27.07 (1 C, C
Glu4-3), 26.91 (1 C, C
Glu3-3), 26.86 (1 C, C
Glu2-3), 26.66 (1 C, C
12-AD-10), 26.47 (1 C, C
Glu4-3), 24.56 (1 C, C
oct-3), 24.18 (1 C, C
12-AD-3), 22.26 (1 C, C
oct-6), 21.96 (1 C, C-14), 21.30 (1 C, C-2 Ac), 19.36 (1 C, CH
3 from C
Ang-2), 14.68 (1 C, C
Ang-4), 14.45 (1 C, C-13), 13.04 (1 C, C-8), 11.57 (1 C, C-15); HR-MS was as reported [
25].
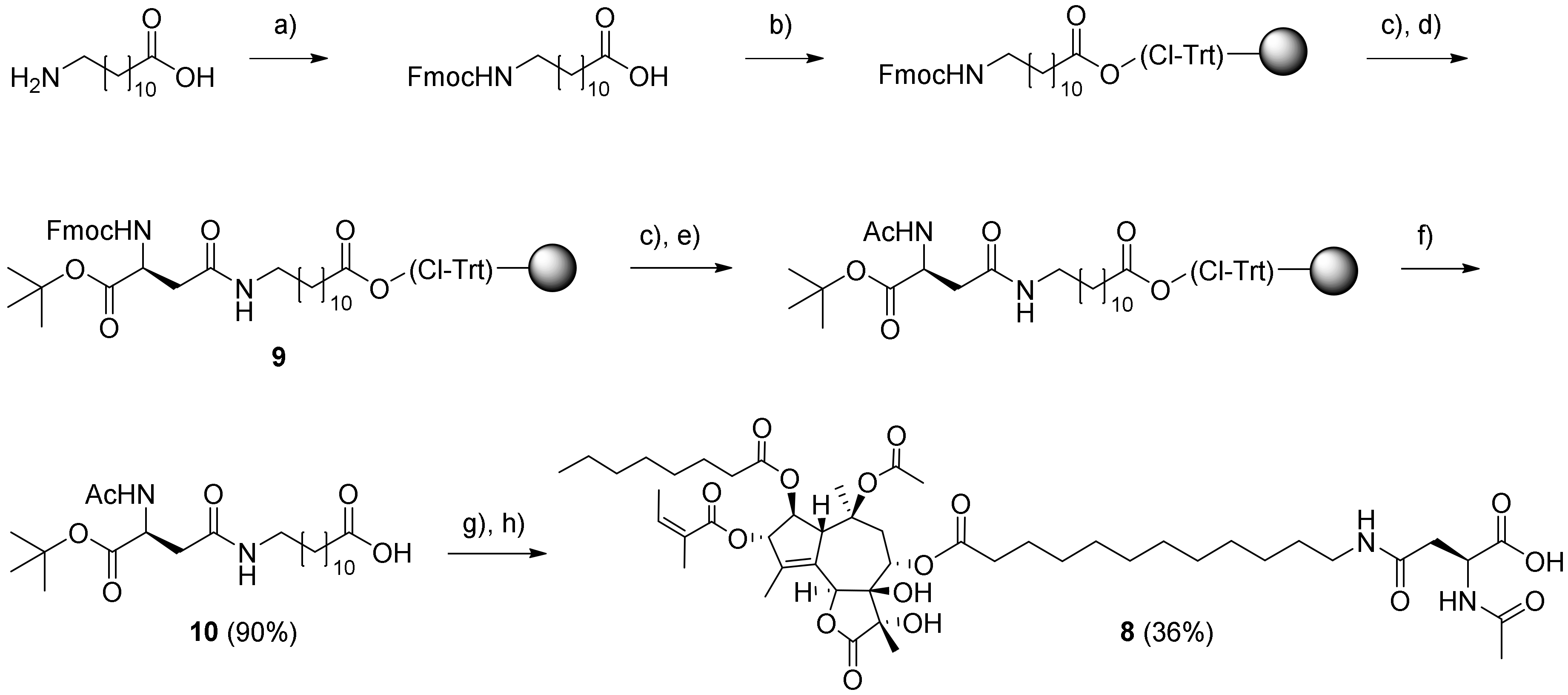
3.4.5. Ac-Asp-12-Aminododecanoate-(O-8)-Debutanoyl-Thapsigargin (8)
Fmoc-OSu (6.55 g; 21.9 mmol) and NaHCO
3 (2.39 g; 28.5 mmol) were added to a solution of 12-aminododecanoic acid (4.71 g; 21.9 mmol) in acetone (90 mL) and water (90 mL), and then the mixture was stirred at rt for 20 h. The reaction mixture was quenched with concentrated HCl until pH 4–5, and the resulting precipitate was extracted into with EtOAc (3 × 50 mL). The resulting combined organic phases were dried with Na
2SO
4, filtered, and evaporated. The crude product was purified on a silica column (Hexane–EtOAc 3:1) to provide Fmoc-12-aminododecanoic acid (4.20 g; 43%) as a white solid.
1H NMR and
13C NMR were as reported [
28].
2-Chlorotrityl resin (2.58 g; loading: 1.6 mmol/g) was swelled in DCM (5 mL) in a teflon reaction vessel fitted with a polypropylene (PP) filter, and a solution of Fmoc-12-aminododecanoic acid (0.60 g; 1.37 mmol) and DIPEA (2.38 ml; 13.7 mmol) in DCM (3 mL) was added to the resin. The mixture was shaken for 3 h at rt. Then, the resin was drained and washed twice with DCM followed by capping with DIPEA–MeOH–DCM 5:15:80 (8 mL; 2 × 5 min). The resin was washed successively with DMF, MeOH, and DCM (each 3 times with 8 mL for 3 min); the residual solvent was removed on a freeze-dryer. Test cleavage of the dry preloaded resin with 20% HFIP in DCM showed a loading of ~0.48 mmol/g.
The Fmoc protecting group was removed with 20% piperidine–DMF (8 mL; 2 × 20 min), followed by washing with DMF, MeOH, and DCM (each 3 times as above). A solution of Fmoc-Asp-OtBu (587 mg; 1.42 mmol), PyBOP (743 mg; 1.42 mmol) and DIPEA (0.25 mL; 2.62 mmol) dissolved in DMF (6 mL) was added to the resin-bound 12-aminododecanoic acid (1 g, 0.48 mmol loading). The mixture was shaken for 16 h at rt, after which the resin was washed with DMF, MeOH, and DCM (each 3 times for 3 min). Then, resin 9 was dried, and a portion (502 mg) was divided into two vessels where it was Fmoc-deprotected with 20% piperidine–DMF (8 mL; 2 × 20 min), washed twice with DMF and DCM, and finally, acetylated with Ac2O–DIPEA–NMP (1:2:3; 6 mL; 2 × 5 min). After a final standard washing of the resin, it was subjected to cleavage with 20% HFIP–DCM (3 × 6 mL; each time for 30 min), and the resulting mixture was concentrated to give intermediate 10 (110 mg; 0.22 mmol; 90%).
Intermediate 10 (50 mg; 0.13 mmol) was coupled to 11 (50 mg; 0.086 mmol) using EDC (26 mg; 0.14 mmol) and DMAP (6 mg; 0.05 mmol) in DCM (2 mL). The mixture was stirred for 23 h under argon at rt. The resulting reaction mixture was evaporated and filtered through a silica column using DCM–MeOH 30:1 as the eluent. Finally, deprotection of the crude tert-butyl ester was performed with TFA (3 mL with 4 drops of water added) for 2 h at rt. The resulting crude product was purified by preparative HPLC with a gradient of 50% → 100% B for 20 min to give 8 (retention time 18-18.5 min). Upon evaporation, the product was dissolved in dioxane–H2O and freeze-dried to provide compound 8 (29 mg; 36%) as a white solid. An analytical UHPLC with the gradient 50% → 100% B for 10 min: showed a retention time of 8.4 min and a purity of >99%. Characterization of compound 8: 1H NMR (600 MHz, methanol-d4) δ ppm = 6.18 (qd, J = 7.2, 1.5 Hz, 1 H, HAng-3), 5.70 (m, 1 H, H-6), 5.67 (m, 1 H, H-3), 5.59 (t, J = 3.7 Hz, 1 H, H-8), 5.52 (t, J = 2.9 Hz, 1 H, H-2), 4.73 (dt, J = 7.0, 5.5 Hz, 1 H, Hasp-α), 4.36 (m, 1 H, H-1), 3.16 (dt, J = 7.2, 1.5 Hz, 2 H, H12-AD-12), 2.99 (dd, J = 14.7, 3.7 Hz, 1 H, Ha-9), 2.66–2.78 (m, 2 H, HAsp-β), 2.38 (m, 2H, CHoct-2), 2.32 (m, 2 H, CH12-AD-2), 2.29 (dd, J = 11.4, 3.3 Hz, 1 H, Hb-9), 2.00 (dq, 5.9, 1.0 Hz, 3 H, HAng-4), 1.99 (s, 3 H, AcAsp), 1.93 (t, J = 1.5 Hz, 3 H, CH3 from CAng-2), 1.89 (s, 3 H, AcTg), 1.86 (s, 3 H, H-15), 1.64 (m, 2H, CHoct-3), 1.61 (m, 2H, CH12-AD-3), 1.50 (m, 2 H, H12-AD-11), 1.42 (s, 3 H, H-14), 1.36 (s, 3 H, H-13), 1.35–1.30 (m, 22 H, H12-AD-4 to H12-AD-10, Hoct-4 to Hoct-7), 0.92 (m, 3 H, Hoct-8); 13C NMR (600 MHz, methanol-d4) δ ppm = 178.71 (1 C, C-12), 174.89 (1 C, Coct-1), 174.83 (1 C, Casp-1), 174.35 (1 C, C12-AD-1), 173.64 (1 C, CAsp-4), 172.62 (1 C, CAc Tg-1), 172.43 (1 C, CAc Asp-1), 169.19 (1 C, CAng-1), 141.65 (1 C, C-4), 140.07 (1 C, CAng-3), 134.00 (1 C, C-5), 129.84 (1 C, CAng-2), 86.53 (1 C, C-10), 86.33 (1 C, C-3), 80.10 (1 C, C-11), 80.05 (1 C, C-7), 79.94 (1 C, C-2), 78.62 (1 C, C-6), 68.04 (1 C, C-8), 59.66 (1 C, C-1), 51.36 (1 C, CAsp-2), 41.12 (1 C, C12-AD-12), 40.04 (1 C, C-9), 38.99 (1 C, CAsp-3), 36.15 (1 C, C12-AD-2), 35.80 (1 C, Coct-2), 31.27–30.70 (9 C, C12-AD-4 − 9 + 11, Coct-4 − 5), 28.60 (1 C, C12-AD-10), 26.58 (1 C, C12-AD-3), 26.20 (1 C, Coct-3), 24.28 (1 C, Coct-6), 24.01 (1 C, C-14), 23.32 (1 C, CAc Tg-2), 23.10 (1 C, CAc Asp-2), 21.38 (1 C, CH3 from CAng-2), 16.69 (1 C, CAng-4), 16.46 (1 C, C-13), 15.05 (1 C, Coct-8), 13.59 (1 C, C-15); HR-MS-ESI: [C48H74N2O16Na]+ m/z: 957.4930. Found 957.4931.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




