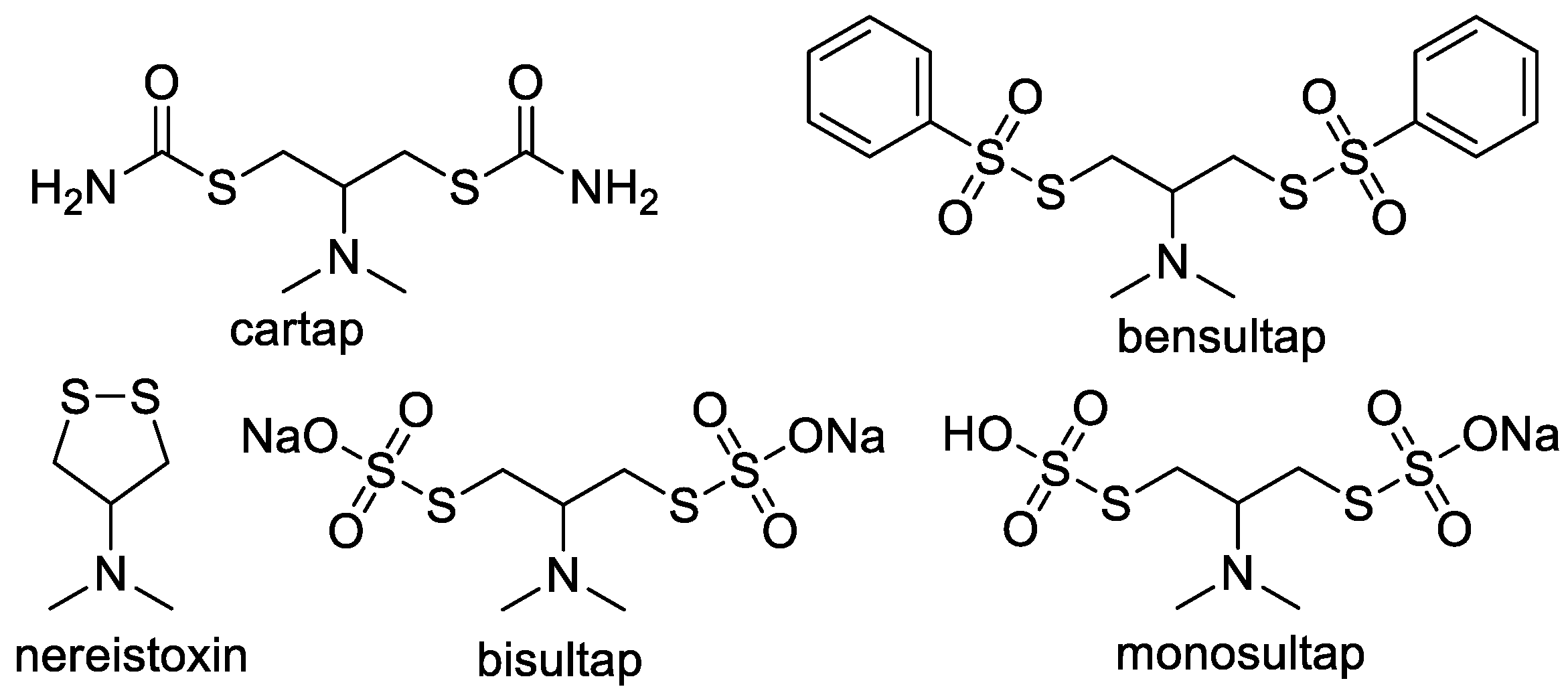
Rapid Colorimetric Detection of Cartap Residues by AgNP Sensor with Magnetic Molecularly Imprinted Microspheres as Recognition Elements


Abstract
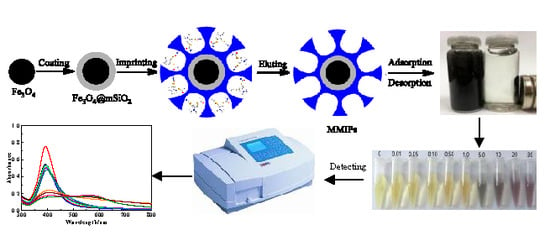
:
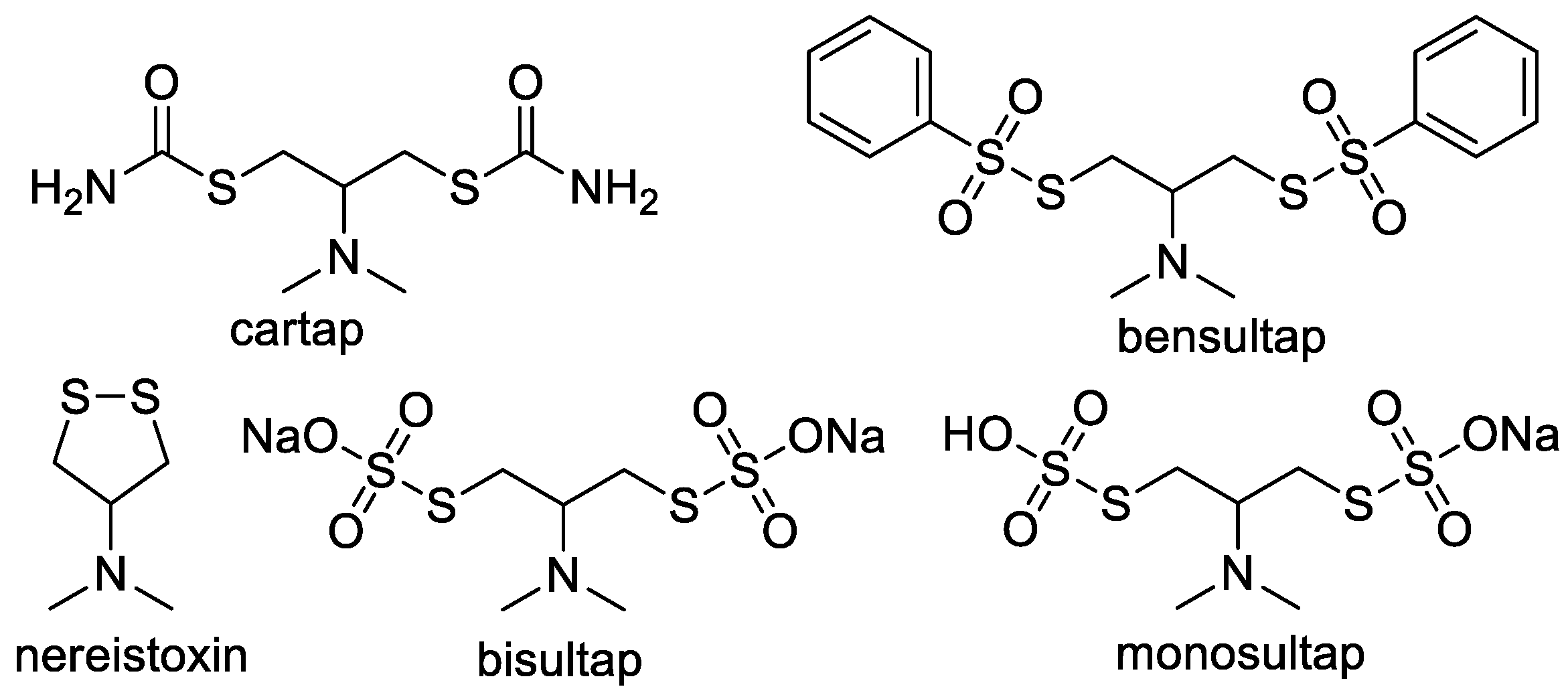
1. Introduction
2. Results and Discussion
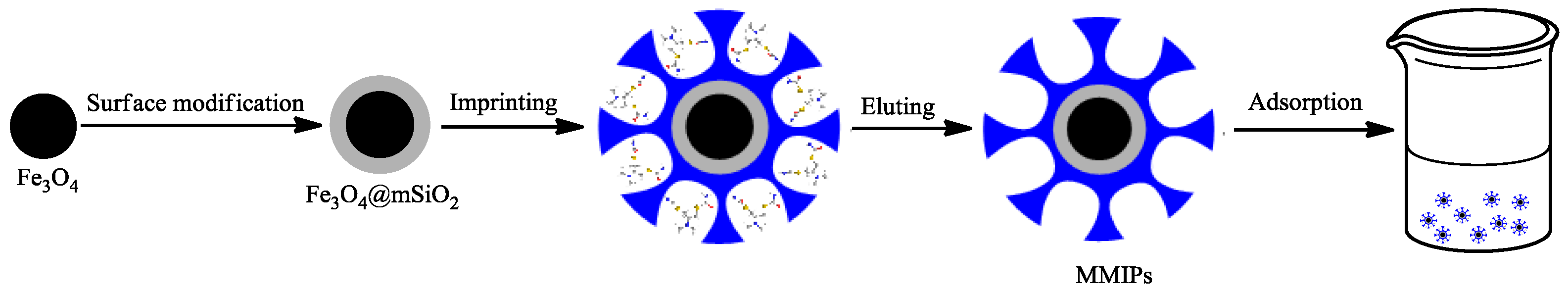
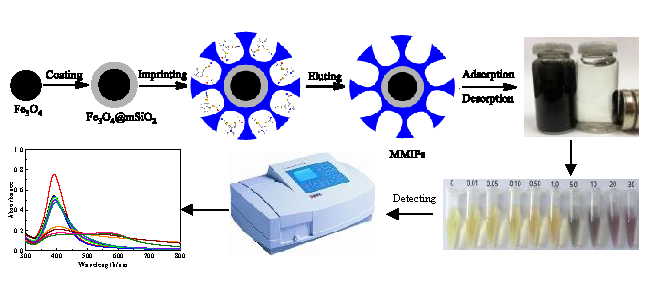
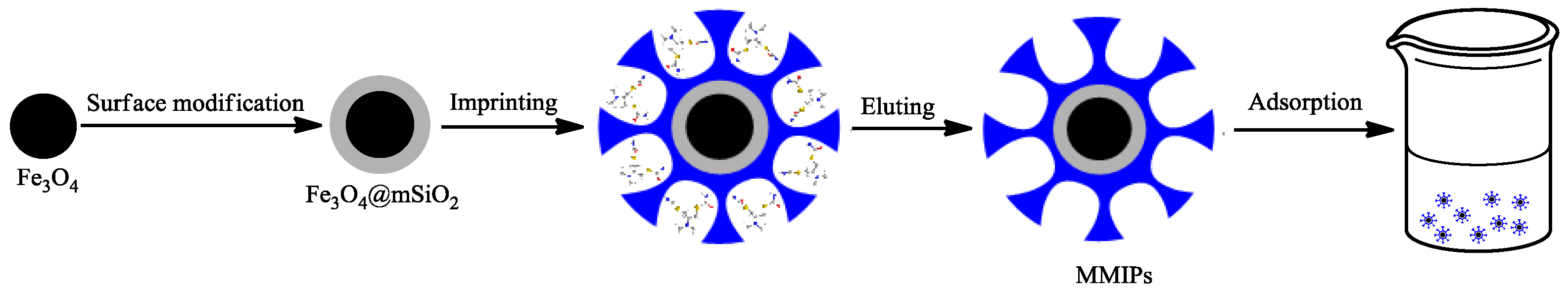
2.1. Preparation of Fe3O4@mSiO2@MIPs
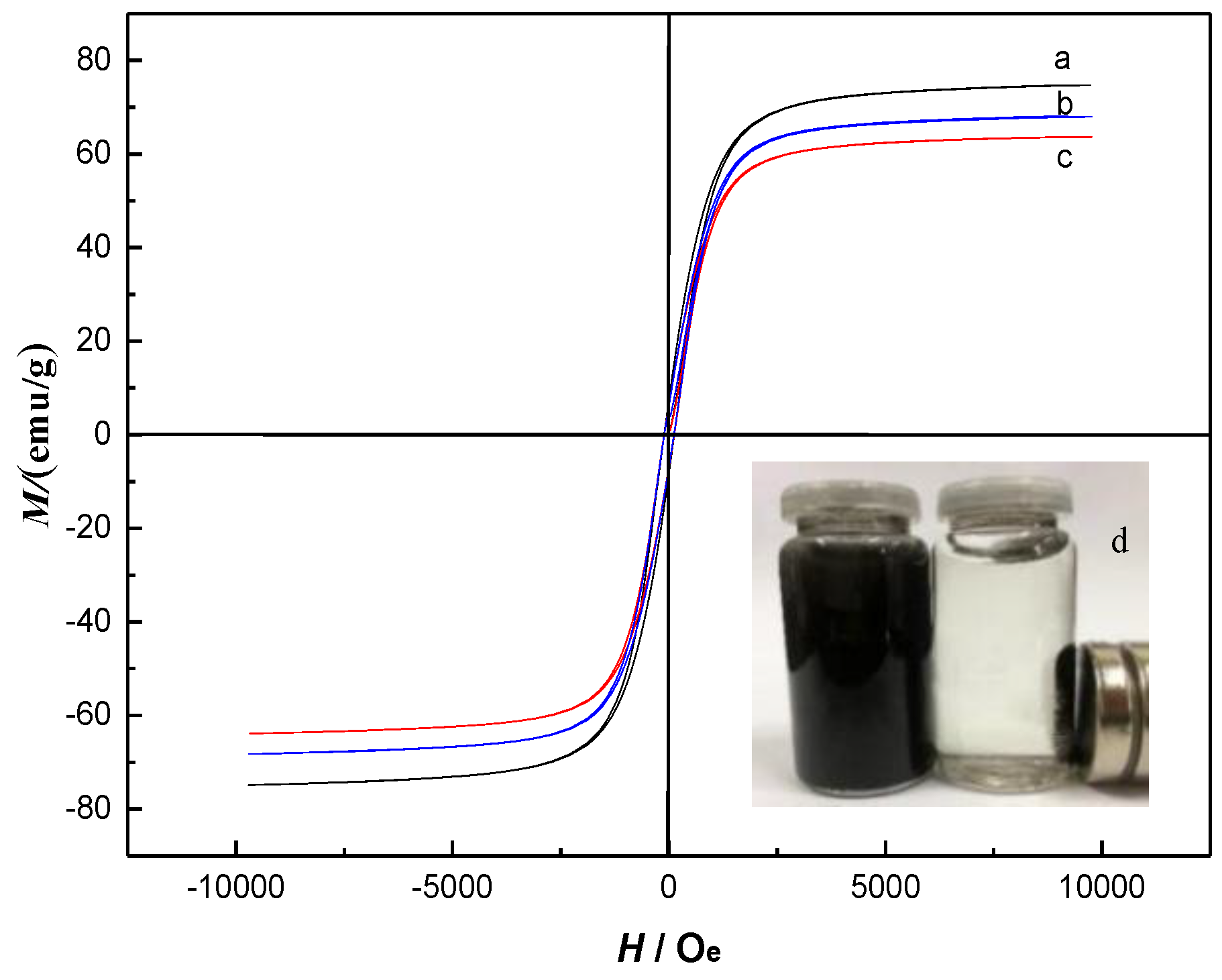
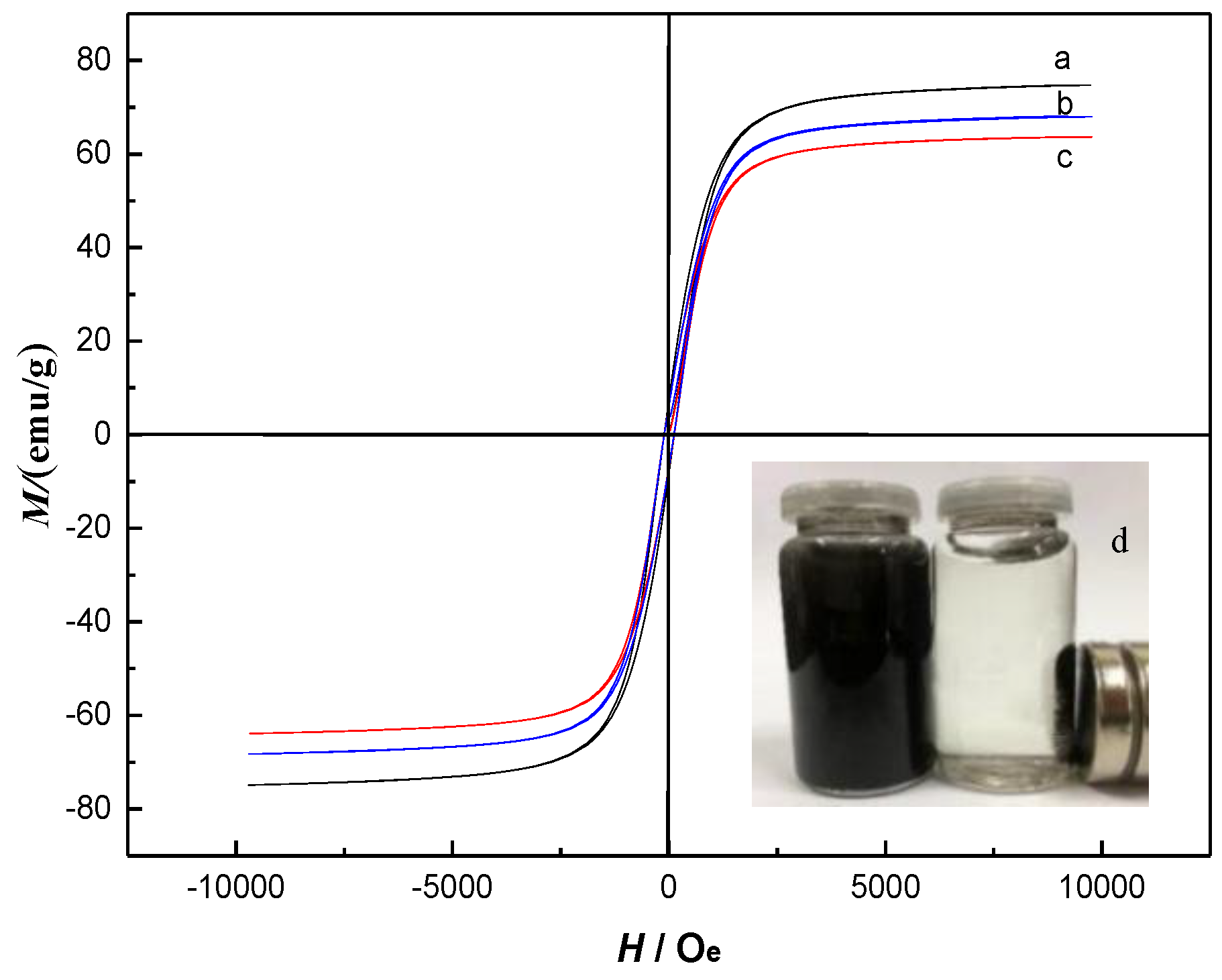
2.2. Characterization of Fe3O4@mSiO2@MIPs
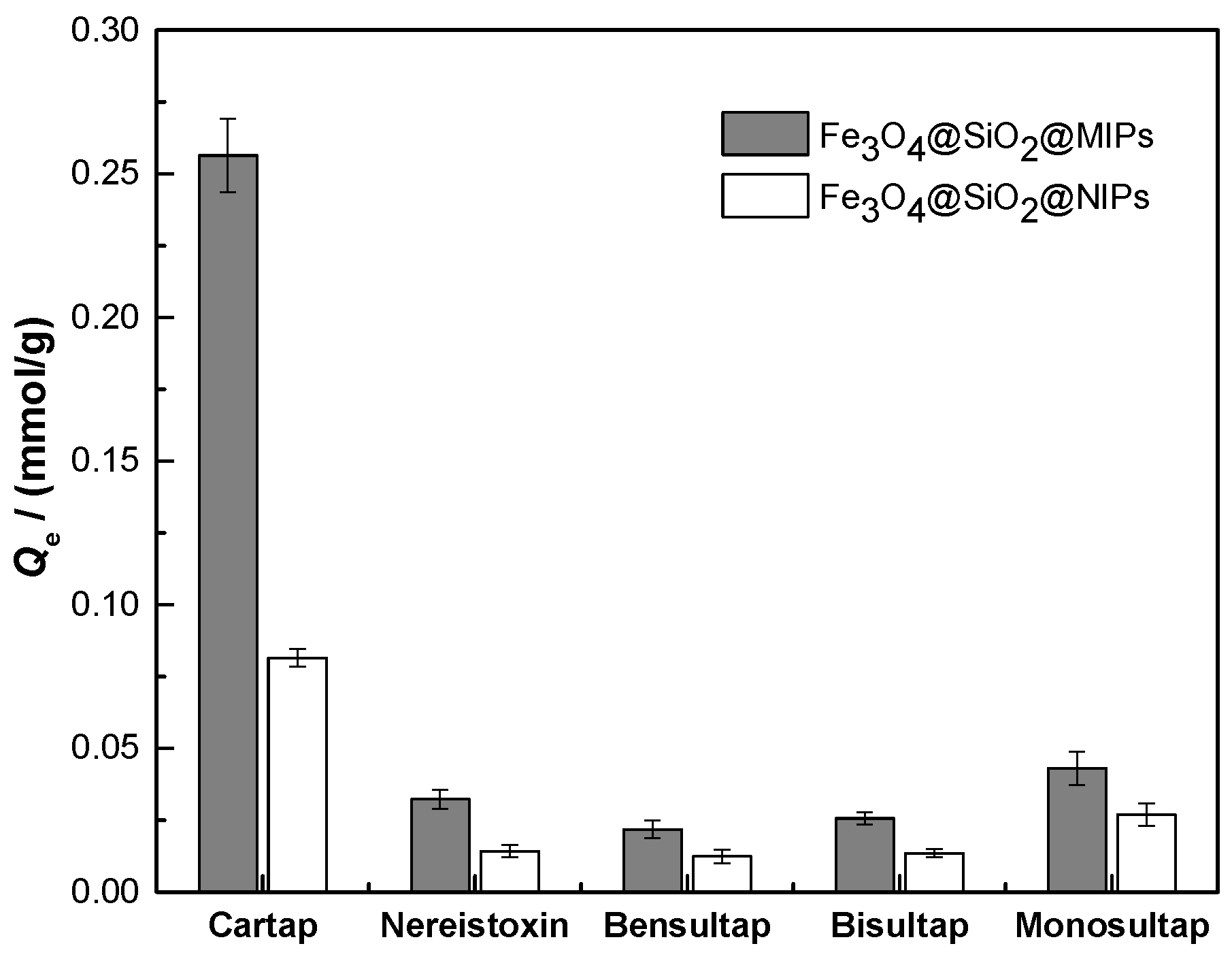
2.3. Adsorption Characteristics
2.4. Evaluation of Selectivity for Recognition
2.5. Reusability of Fe3O4@mSiO2@MIPs
2.6. Selective Enrichment of Cartap from Spiked Tea Beverages
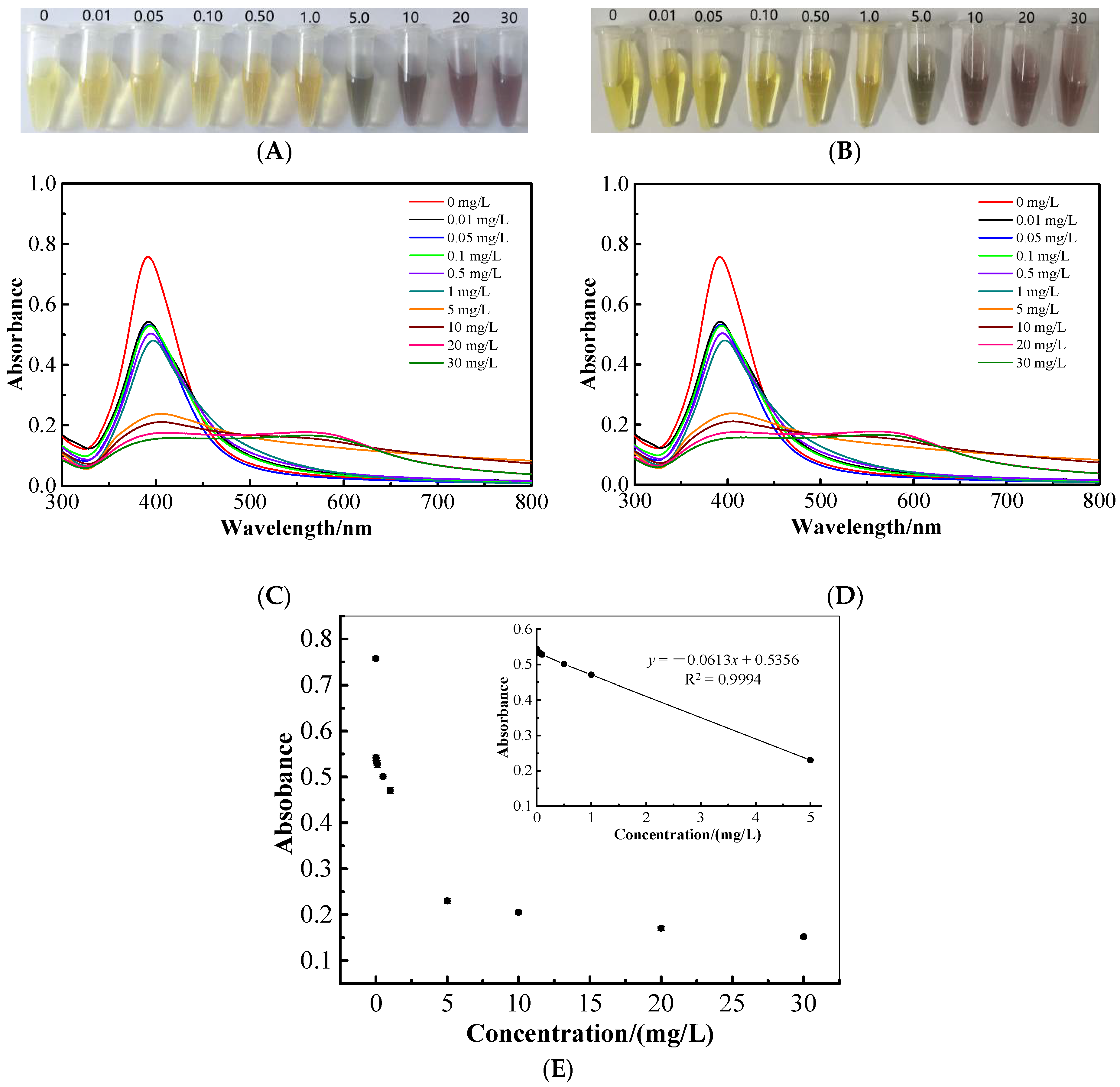
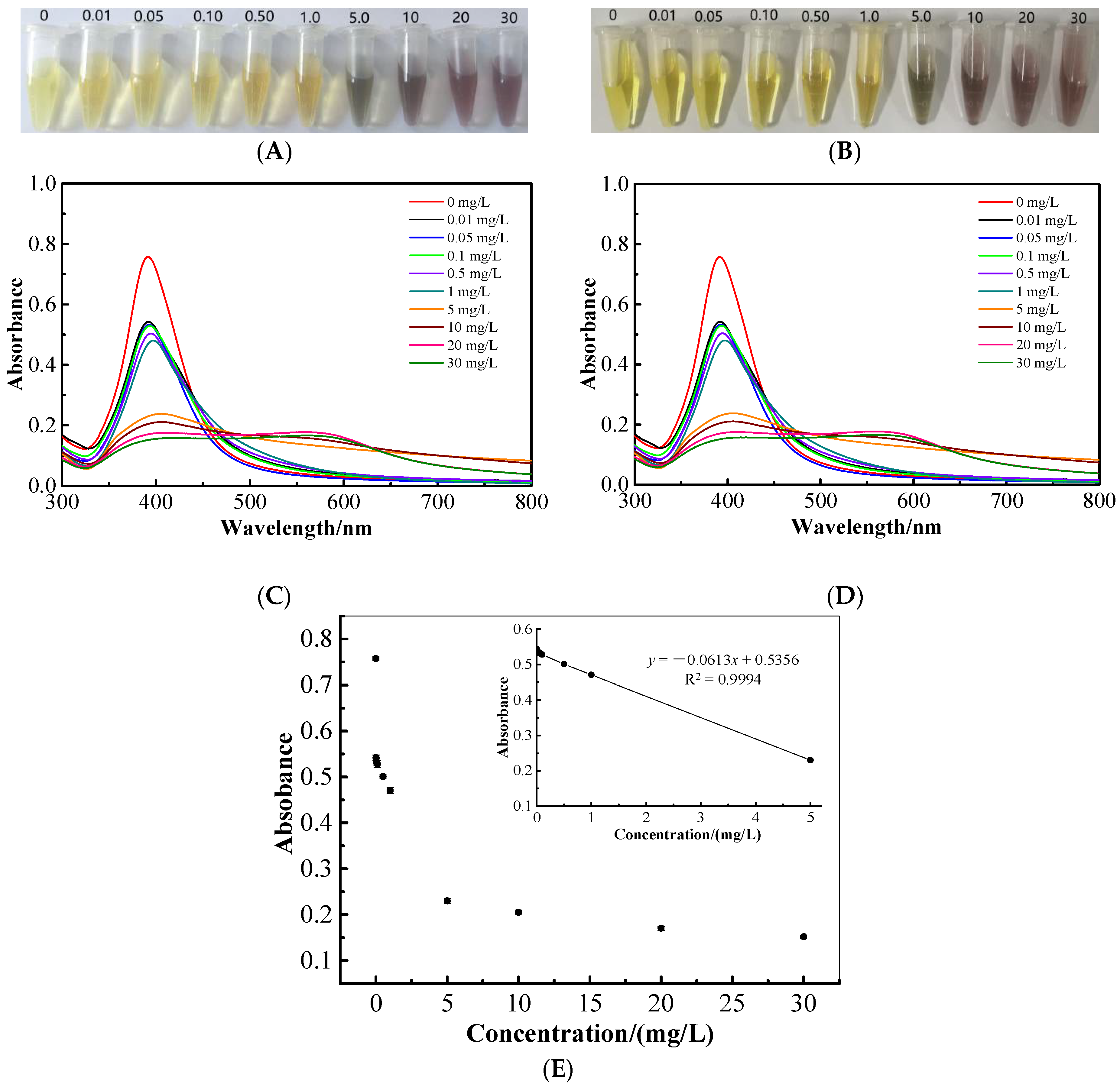
2.7. Colorimetric and UV-Vis Spectroscopic Determination of Cartap
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Apparatus and Conditions
3.3. Preparation of Fe3O4@mSiO2@MIPs
3.4. Adsorption Experiments
3.5. Regeneration and Reused Experiments
3.6. Selective Recognition of Cartap from Tea Beverages by Fe3O4@mSiO2@MIPs
3.7. Colorimetric, UV-Vis Spectroscopic and HPLC Determination of Cartap
3.8. Statistical Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liao, J.W.; Kang, J.J.; Jeng, C.R.; Chang, S.K.; Kuo, M.J.; Wang, S.C.; Liu, M.R.S.; Pang, V.F. Cartap-induced cytotoxicity in mouse C2C12 myoblast cell line and the roles of calcium ion and oxidative stress on the toxic effects. Toxicology 2006, 219, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Berg, H. Pesticide use in rice and rice-fish farms in the Mekong Delta, Vietnam. Crop Protect. 2001, 20, 897–905. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, D.; Tang, Y.; Wang, Y.; Yan, F.; Li, Z.; Wang, J.; Zhou, H.S. Highly sensitive and selective colorimetric detection of cartap residue in agricultural products. Talanta 2012, 101, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Ministry of Agriculture of People’s Republic of China. National Food Safety Standard: Maximum Residue Limits for Pesticides in Food; Ministry of Agriculture of People’s Republic of China: Beijing, China, 2016; GB 2763-2016; p. 26.
- Wang, Z.; Wu, L.; Shen, B.; Jiang, Z. Highly sensitive and selective cartap nanosensor based on luminescence resonance energy transfer between NaYF4:Yb,Ho nanocrystals and gold nanoparticles. Talanta 2013, 114, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Zhou, R.; Jiang, D.; Fu, H.; Su, R.; Liu, Y.; Su, H. Rapid detection of pesticide residues in Chinese herbal medicines by Fourier transform infrared spectroscopy coupled with partial least squares regression. J. Spectrosc. 2016, 2016, 9492030. [Google Scholar] [CrossRef]
- Bhatia, J.; Sharma, J.D. Thin-layer chromatographic detection of carbosulfan by 4-aminoantipyrine reagent. J. Planar Chromatogr. Mod. TLC 2011, 24, 545–546. [Google Scholar] [CrossRef]
- Wu, G.; Yu, H.; Bao, X.; Chen, H.; Ye, Q. Determination of cartap residues in tea by GC/micro-ECD. Chin. J. Chromatogr. 2007, 25, 288–289. (In Chinese) [Google Scholar]
- Vivek, C.; Veeraiah, K.; Padmavathi, P.; Rao, H.D.; Bramhachari, P.V. Acute toxicity and residue analysis of cartap hydrochloride pesticide: Toxicological implications on the fingerlings of fresh water fish Labeo rohita. Biocatal. Agric. Biotechnol. 2016, 7, 193–201. [Google Scholar] [CrossRef]
- Tian, K.; Ming, C.; Dai, Y.; Ake, K.M.H. Fenton degradation of cartap hydrochloride: Identification of the main intermediates and the degradation pathway. Water Sci. Technol. 2015, 72, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Choe, S.; Lee, H.; Jo, J.; Park, Y.; Kim, E.; Pyo, J.; Jung, J.H. Advanced analytical method of nereistoxin using mixed-mode cationic exchange solid-phase extraction and GC/MS. Forensic Sci. Int. 2015, 252, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, M.A.Z.; Fakhruddin, A.N.M.; Islam, M.N.; Moniruzzaman, M.; Gan, S.H.; Alam, M.K. Detection of the residues of nineteen pesticides in fresh vegetable samples using gas chromatography-mass spectrometry. Food Control 2013, 34, 457–465. [Google Scholar] [CrossRef]
- Namera, A.; Watanabe, T.; Yashiki, M.; Kojima, T.; Urabe, T. Simple and sensitive analysis of nereistoxin and its metabolites in human serum using headspace solid-phase microextraction and gas chromatography mass spectrometry. J. Chromatogr. Sci. 1999, 37, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Fukuwatari, T.; Shibata, K. Fluorometric determination of pantothenic acid in human urine by isocratic reversed-phase ion-pair high-performance liquid chromatography with post-column derivatization. J. Chromatogr. B 2009, 877, 2168–2172. [Google Scholar] [CrossRef] [PubMed]
- Tao, C.J.; Hu, J.Y.; Li, J.Z. Determination of insecticide monosultap residues in tomato and soil by capillary gas chromatography with flame photometric detection. Can. J. Anal. Spectrosc. 2007, 52, 295–304. [Google Scholar]
- Saha, K.; Agasti, S.S.; Kim, C.; Li, X.; Rotello, V.M. Gold nanoparticles in chemical and biological sensing. Chem. Rev. 2012, 112, 2739–2779. [Google Scholar] [CrossRef] [PubMed]
- Mi, F.-L.; Wu, S.-J.; Zhong, W.-Q.; Huang, C.-Y. Preparation of a silver nanoparticle-based dual-functional sensor using a complexation-reduction method. Phys. Chem. Chem. Phys. 2015, 17, 21243–21253. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Hu, Y.; Ma, L.; Lu, X. Development of molecularly imprinted polymers-surface-enhanced Raman spectroscopy/colorimetric dual sensor for determination of chlorpyrifos in apple juice. Sens. Actuators B Chem. 2017, 241, 750–757. [Google Scholar] [CrossRef]
- Roy, S.M.; Roy, D.R. Levofloxacin capped Ag-nanoparticles: A new highly selective sensor for cations under joint experimental and DFT investigation. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 179, 178–187. [Google Scholar] [CrossRef]
- Varun, S.; Daniel, S.C.G. K.; Gorthi, S.S. Rapid sensing of melamine in milk by interference green synthesis of silver nanoparticles. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 74, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tao, J.; Chen, X.; Yang, H. An ultrasensitive and selective “off-on” rhodamine-based colorimetric and fluorescent chemodosimeter for the detection of Cu2+. Sens. Actuators B Chem. 2017, 244, 709–716. [Google Scholar] [CrossRef]
- Polavarapu, L.; Perez-Juste, J.; Xu, Q.-H.; Liz-Marzan, L.M. Optical sensing of biological, chemical and ionic species through aggregation of plasmonic nanoparticles. J. Mater. Sci. C 2014, 2, 7460–7476. [Google Scholar] [CrossRef]
- Zhao, L.; Zhu, J.; Cheng, Y.; Xiong, Z.; Tang, Y.; Guo, L.; Shi, X.; Zhao, J. Chlorotoxin-conjugated multifunctional dendrimers labeled with radionuclide 131I for single photon emission computed tomography imaging and radiotherapy of gliomas. ACS Appl. Mater. Interfaces 2015, 7, 19798–19808. [Google Scholar] [CrossRef] [PubMed]
- Patel, G.M.; Rohit, J.V.; Singhal, R.K.; Kailasa, S.K. Recognition of carbendazim fungicide in environmental samples by using 4-aminobenzenethiol functionalized silver nanoparticles as a colorimetric sensor. Sens. Actuators B Chem. 2015, 206, 684–691. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, K.; Cui, Y.; Zhang, W.; He, L.; Guo, S.; Chen, Y.; Li, Q.X.; Liu, S.; Wang, B. Development of a monoclonal antibody-based ELISA for the detection of the novel insecticide cyantraniliprole. RSC Adv. 2015, 5, 35874–35881. [Google Scholar] [CrossRef]
- Duan, Y.; Wang, L.; Gao, Z.; Wang, H.; Zhang, H.; Li, H. An aptamer-based effective method for highly sensitive detection of chloramphenicol residues in animal-sourced food using real-time fluorescent quantitative PCR. Talanta 2017, 165, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Wang, D.-D.; Zhang, Q.; Yang, F.-Q.; Xia, Z.-N.; Zhang, Q.-H.; Yuan, C.-S. In vivo selective capture and rapid identification of luteolin and its metabolites in rat livers by molecularly imprinted solid-phase microextraction. J. Agric. Food Chem. 2017, 65, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Chen, L.; Pan, X.; Wang, S. Synthesis of magnetic molecularly imprinted polymers by reversible addition fragmentation chain transfer strategy and its application in the Sudan dyes residue analysis. J. Chromatogr. A 2015, 1405, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Lu, H.; Xu, S. Preparation of Magnetic Hollow Molecularly Imprinted Polymers for Detection of Triazines in Food Samples. J. Agric. Food Chem. 2016, 64, 5110–5116. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lu, J.; Qiao, X.; Xu, Z. A study on biomimetic immunoassay-capillary electrophoresis method based on molecularly imprinted polymer for determination of trace trichlorfon residue in vegetables. Food Chem. 2017, 221, 1285–1290. [Google Scholar] [CrossRef] [PubMed]
- Kittlaus, S.; Lipinski, J.; Speer, K. New approaches for determination of glyphosate and aminomethylphosphonic acid from different tea samples-prospects and limits of cleanup with molecularly imprinted polymer and titanium dioxide. J. AOAC Int. 2009, 92, 703–714. [Google Scholar] [PubMed]
- Miao, S.S.; Wu, M.S.; Zuo, H.G.; Jiang, C.; Jin, S.F.; Lu, Y.C.; Yang, H. Core–shell magnetic molecularly imprinted polymers as sorbent for sulfonylurea herbicide residues. J. Agric. Food Chem. 2015, 63, 3634–3645. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Yin, Y.; Lv, P.; Zhang, Z.; Wang, J.; Long, F. Synthesis and application of novel 3D magnetic chlorogenic acid imprinted polymers based on a graphene–carbon nanotube composite. J. Agric. Food Chem. 2016, 64, 3091–3100. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Cheng, X.; Sun, N. Synthesis of multi-core-shell magnetic molecularly imprinted microspheres for rapid recognition of dicofol in tea. J. Agric. Food Chem. 2013, 61, 2896–2901. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Guo, J.; Zhang, Y.; Hu, Y.; You, Q.; Shi, S. Novel molecular imprinted polymers over magnetic mesoporous silica microspheres for selective and efficient determination of protocatechuic acid in Syzygium aromaticum. Food Chem. 2015, 178, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Xie, L.; Guo, J.; Li, H.; Jiang, X.; Zhang, Y.; Shi, S. Hydrophilic gallic acid-imprinted polymers over magnetic mesoporous silica microspheres with excellent molecular recognition ability in aqueous fruit juices. Food Chem. 2015, 179, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Guo, J.; Zhang, Y.; Shi, S. Efficient determination of protocatechuic acid in fruit juices by selective and rapid magnetic molecular imprinted solid phase extraction coupled with HPLC. J. Agric. Food Chem. 2014, 62, 8221–8228. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Lu, Y.; Zhao, Y.; Ye, H. Fast extraction and detection of 4-methylimidazole in soy sauce using magnetic molecularly imprinted polymer by HPLC. Molecules 2017, 22, 1885. [Google Scholar] [CrossRef] [PubMed]
- Katz, A.; Davis, M.E. Molecular imprinting of bulk, microporous silica. Nature 2000, 403, 286. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Nie, M.; Shi, S.; You, Q.; Guo, J.; Liu, L. Integration of magnetic solid phase fishing and off-line two-dimensional high-performance liquid chromatography-diode array detector-mass spectrometry for screening and identification of human serum albumin binders from Radix Astragali. Food Chem. 2014, 146, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Xu, T.; Shi, R.; Lu, N.; Zhang, J.; Jiang, C.; Zhang, C.; Zhou, J. Preparation of hollow porous molecularly imprinted polymers for N-nitrosamine adsorption. Mater. Lett. 2018, 211, 21–23. [Google Scholar] [CrossRef]
- Li, W.; Zhao, D. Extension of the Stober method to construct mesoporous SiO2 and TiO2 shells for uniform multifunctional core-shell structures. Adv. Mater. 2013, 25, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; He, X.; Chen, L.; Zhang, Y. Preparation of core-shell magnetic molecularly imprinted polymer nanoparticles for recognition of bovine hemoglobin. Chem. Asian J. 2009, 4, 286–293. [Google Scholar] [CrossRef] [PubMed]
- You, Q.; Zhang, Y.; Zhang, Q.; Guo, J.; Huang, W.; Shi, S.; Chen, X. High-capacity thermo-responsive magnetic molecularly imprinted polymers for selective extraction of curcuminoids. J. Chromatogr. A 2014, 1354, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.L.; Cao, Y.H.; Cao, G.Q. Preparation and application of core-shell magnetic imprinted nanoparticles for bisphenol A. Chin. J. Anal. Chem. 2013, 41, 1724–1728. [Google Scholar] [CrossRef]
- Shen, J.; Chai, W.; Wang, K.; Zhang, F. Efficient removal of anionic radioactive pollutant from water using ordered urea-functionalized mesoporous polymeric nanoparticle. ACS Appl. Mater. Interfaces 2017, 9, 22440–22448. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples * | AgNP Colorimetric Detection after Fe3O4@mSiO2@MIPs-Pretreatment | HPLC Analysis |
|---|---|---|
| Green tea (mg/kg) | 10.112 ± 0.202 a | 10.084 ± 0.307 a |
| Black tea (mg/kg) | 9.798 ± 0.219 a | 9.831 ± 0.196 a |
| White tea (mg/kg) | 9.965 ± 0.199 a | 10.022 ± 0.287 a |
| Yellow tea (mg/kg) | 9.767 ± 0.235 b | 9.779 ± 0.109 b |
| Dark tea (mg/kg) | 10.033 ± 0.201 a | 9.974 ± 0.214 a |
| Oolong tea (mg/kg) | 10.042 ± 0.318 b | 10.013 ± 0.226 b |
| Bottled green tea (mg/L) | 4.027 ± 0.082 a | 4.008 ± 0.105 a |
| Bottled iced black tea (mg/L) | 3.985 ± 0.095 a | 4.015 ± 0.083 a |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, M.; Deng, H.; Fan, Y.; Hu, Y.; Guo, Y.; Xie, L. Rapid Colorimetric Detection of Cartap Residues by AgNP Sensor with Magnetic Molecularly Imprinted Microspheres as Recognition Elements. Molecules 2018, 23, 1443. https://doi.org/10.3390/molecules23061443
Wu M, Deng H, Fan Y, Hu Y, Guo Y, Xie L. Rapid Colorimetric Detection of Cartap Residues by AgNP Sensor with Magnetic Molecularly Imprinted Microspheres as Recognition Elements. Molecules. 2018; 23(6):1443. https://doi.org/10.3390/molecules23061443
Chicago/Turabian StyleWu, Mao, Huiyun Deng, Yajun Fan, Yunchu Hu, Yaping Guo, and Lianwu Xie. 2018. "Rapid Colorimetric Detection of Cartap Residues by AgNP Sensor with Magnetic Molecularly Imprinted Microspheres as Recognition Elements" Molecules 23, no. 6: 1443. https://doi.org/10.3390/molecules23061443
APA StyleWu, M., Deng, H., Fan, Y., Hu, Y., Guo, Y., & Xie, L. (2018). Rapid Colorimetric Detection of Cartap Residues by AgNP Sensor with Magnetic Molecularly Imprinted Microspheres as Recognition Elements. Molecules, 23(6), 1443. https://doi.org/10.3390/molecules23061443