
β-Phenylalanine Ester Synthesis from Stable β-Keto Ester Substrate Using Engineered ω-Transaminases

 , ,
, ,  and
and
Abstract
:
1. Introduction
Biocatalytic Synthesis of β-Amino Acids
2. Results
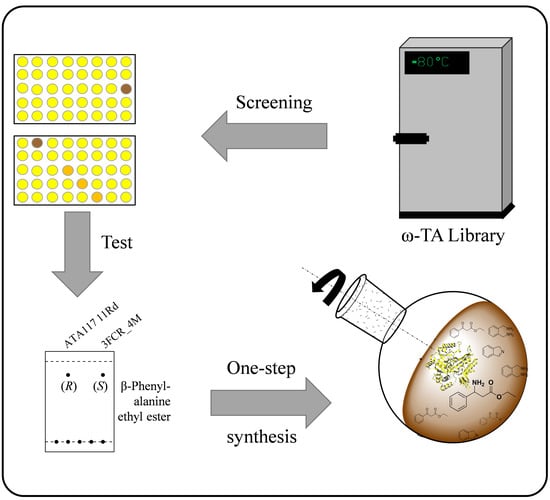
2.1. Identification of Promising ω-TA Variants
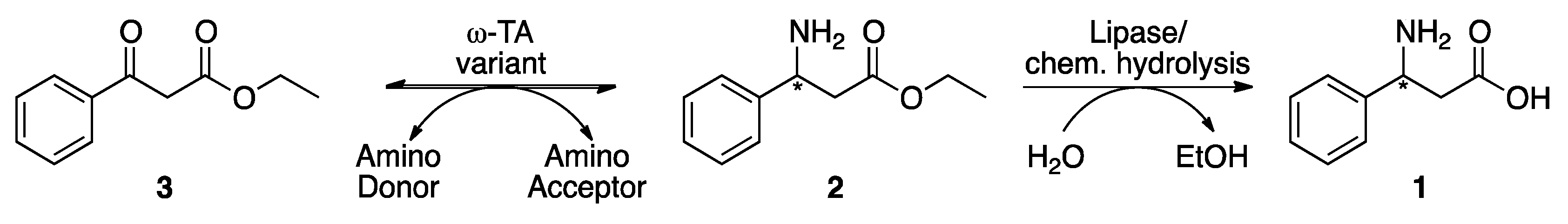
2.2. Asymmetric Synthesis of the β-Phenylalanine Ethyl Ester
2.3. Optimization of the Reaction Condition and Larger Scale Synthesis of 2
3. Discussion
4. Materials and Methods
4.1. Protein Expression and Purification
4.2. Screening of the ω-TA Library
4.3. TLC-(MS) Analysis
4.4. HPLC Analysis
4.5. Small Scale Enzymatic Synthesis
4.6. Design of Experiments
4.7. GC Analysis
4.8. Determination of Water Activity
4.9. Synthesis Scale-up of (S)-phenylalanine Ethyl Ester and Product Purification
4.10. Analytical NMR Data of (S)-Phenylalanine Ethyl Ester
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Midelfort, K.S.; Kumar, R.; Han, S.; Karmilowicz, M.J.; McConnell, K.; Gehlhaar, D.K.; Mistry, A.; Chang, J.S.; Anderson, M.; Villalobos, A.; et al. Redesigning and characterizing the substrate specificity and activity of Vibrio fluvialis aminotransferase for the synthesis of imagabalin. Protein Eng. Des. Sel. 2013, 26, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Berglund, P. Transaminase Biocatalysis: Optimization and Application. Green Chem. 2016, 333–360. [Google Scholar] [CrossRef]
- Henson, C.P.; Cleland, W.W. Kinetic Studies of Glutamic Oxaloacetic Transaminase Isozymes *. Biochemistry 1964, 3, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Gomm, A.; O’Reilly, E. Transaminases for chiral amine synthesis. Curr. Opin. Chem. Biol. 2018, 43, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Seebach, D.; Gardiner, J. β-Peptidic Peptidomimetics. Acc. Chem. Res. 2008, 41, 1366–1375. [Google Scholar] [CrossRef] [PubMed]
- Steer, D.L.; Lew, R.A.; Perlmutter, P.; Smith, A.I.; Aguilar, M.-I. β-amino acids: versatile peptidomimetics. Curr. Med. Chem. 2002, 9, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, S.B. Taxol (paclitaxel): mechanisms of action. Ann. Oncol. 1994, 5, 3–6. [Google Scholar]
- Juaristi, E.; Soloshonok, V. Enantioselective Synthesis of Beta-Amino Acids; John Wiley & Sons, Inc.: London, UK, 2005; pp. 1–18. ISBN 0471467383. [Google Scholar]
- Magriotis, P.A. Recent Progress in the Enantioselective Synthesis of beta-Lactams: Development of the First Catalytic Approaches. Angew. Chem. Int. Ed. Engl. 2001, 40, 4377–4379. [Google Scholar] [CrossRef]
- Tao, F.; Zhou, Z.; Zhang, T.; Xu, H.; Yang, J.; Ding, W.; Guo, Y. Method for Preparation of Tobacco Flavor with Honey and Aromatic β-amino Acid. CN Patent 2016-10793745, 31 August 2017. Available online: https://patents.google.com/patent/CN106244322A/en (accessed on 16 May 2018).
- Wani, M.C.; Taylor, H.L.; Wall, M.E.; Coggon, P.; McPhail, A.T. Plant antitumor agents. VI. The isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. [Google Scholar] [CrossRef] [PubMed]
- Takayama, T.; Shibata, T.; Shiozawa, F.; Kawabe, K.; Shimizu, Y.; Hamada, M.; Hiratate, A.; Takahashi, M.; Ushiyama, F.; Oi, T.; et al. Preparation of Partially Saturated Nitrogen-Containing Heterocyclic Compounds, and PHD2 Inhibitors, EPO Formation Promoters, and Antianemic Agents Containing the Heterocyclic Compounds. WO Patent 2014021281 A1, 6 February 2014. [Google Scholar]
- Guizzetti, S.; Benaglia, M.; Bonsignore, M.; Raimondi, L. Triclorosilane-mediated stereoselective synthesis of β-amino esters and their conversion to highly enantiomerically enriched β-lactams. Org. Biomol. Chem. 2011, 9, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Luo, Y.; Li, Z. Method for Preparing Aminophenylpropanoic Acid-Terminated Polyoxyethylene Acid or Activated Ester, and Application Thereof. CN 102964588, 13 March 2013. [Google Scholar]
- Crawforth, J.M.; Glossop, P.A.; Hamper, B.C.; Huang, W.; Mantell, S.J.; Neal, B.E.; Olson, K.; Thorarensen, A.; Turner, S.R. Preparation of Nicotinamide Derivatives for the Treatment of Allergic and Respiratory Conditions. WO 2009153721, 23 December 2009. [Google Scholar]
- Rodríguez-Mata, M.; García-Urdiales, E.; Gotor-Fernández, V.; Gotor, V. Stereoselective chemoenzymatic preparation of β-amino esters: Molecular modelling considerations in lipase-mediated processes and application to the synthesis of (S)-dapoxetine. Adv. Synth. Catal. 2010, 352, 395–406. [Google Scholar] [CrossRef]
- Leflemme, N.; Freret, T.; Boulouard, M.; Dallemagne, P.; Rault, S. Synthesis and preliminary in vivo evaluation of new 2-Aryl-6-methyl-1,2-dihydro-1H-pyridin-4-ones and 2-Aryl-6-methylpiperidin-4-ols, as potential anti-amnesiant agents. J. Enzyme Inhib. Med. Chem. 2005, 20, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Grayson, J.I.; Roos, J.; Osswald, S. Development of a Commercial Process for (S)-β-Phenylalanine. Org. Process Res. Dev. 2011, 15, 1201–1206. [Google Scholar] [CrossRef]
- Mathew, S.; Jeong, S.S.; Chung, T.; Lee, S.H.; Yun, H. Asymmetric synthesis of aromatic β-amino acids using ω-transaminase: Optimizing the lipase concentration to obtain thermodynamically unstable β-keto acids. Biotechnol. J. 2016, 11, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Cheng, S.; Wei, D.; Ren, Y.; Zhang, D. Production of enantiomerically pure (S)-β-phenylalanine and (R)-β-phenylalanine by penicillin G acylase from Escherichia coli in aqueous medium. Biotechnol. Lett. 2007, 29, 1825–1830. [Google Scholar] [CrossRef] [PubMed]
- Rudat, J.; Brucher, B.R.; Syldatk, C. Transaminases for the synthesis of enantiopure beta-amino acids. AMB Express 2012, 2, 11. [Google Scholar] [CrossRef] [PubMed]
- Mathew, S.; Nadarajan, S.P.; Sundaramoorthy, U.; Jeon, H.; Chung, T.; Yun, H. Biotransformation of β-keto nitriles to chiral (S)-β-amino acids using nitrilase and ω-transaminase. Biotechnol. Lett. 2016. [Google Scholar] [CrossRef] [PubMed]
- Mathew, S.; Bea, H.; Nadarajan, S.P.; Chung, T.; Yun, H. Production of chiral β-amino acids using ω-transaminase from Burkholderia graminis. J. Biotechnol. 2015, 196–197, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mathew, S.; Nadarajan, S.P.; Chung, T.; Park, H.H.; Yun, H. Biochemical characterization of thermostable ω-transaminase from Sphaerobacter thermophilus and its application for producing aromatic β- and γ-amino acids. Enzyme Microb. Technol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Crismaru, C.G.; Wybenga, G.G.; Szymanski, W.; Wijma, H.J.; Wu, B.; Bartsch, S.; de Wildeman, S.; Poelarends, G.J.; Feringa, B.L.; Dijkstra, B.W.; et al. Biochemical properties and crystal structure of a β-phenylalanine aminotransferase from Variovorax paradoxus. Appl. Environ. Microbiol. 2013, 79, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Green, A.P.; Turner, N.J.; O’Reilly, E. Chiral Amine Synthesis Using ω-Transaminases: An Amine Donor that Displaces Equilibria and Enables High-Throughput Screening. Angew. Chem. Int. Ed. 2014, 53, 10714–10717. [Google Scholar] [CrossRef] [PubMed]
- Truppo, M.D.; Rozzell, J.D.; Moore, J.C.; Turner, N.J. Rapid screening and scale-up of transaminase catalysed reactions. Org. Biomol. Chem. 2009, 7, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Willies, S.C.; Galman, J.L.; Slabu, I.; Turner, N.J. A stereospecific solid-phase screening assay for colonies expressing both (R)- and (S)-selective ω-aminotransferases. Philos. Trans. A. Math. Phys. Eng. Sci. 2016, 374, 20150084. [Google Scholar] [CrossRef] [PubMed]
- Scheidt, T.; Land, H.; Anderson, M.; Chen, Y.; Berglund, P.; Yi, D.; Fessner, W.-D. Fluorescence-Based Kinetic Assay for High-Throughput Discovery and Engineering of Stereoselective ω-Transaminases. Adv. Synth. Catal. 2015, 357, 1721–1731. [Google Scholar] [CrossRef]
- Schätzle, S.; Höhne, M.; Redestad, E.; Robins, K.; Bornscheuer, U.T. Rapid and Sensitive Kinetic Assay for Characterization of ω-Transaminases. Anal. Chem. 2009, 81, 8244–8248. [Google Scholar] [CrossRef] [PubMed]
- Slabu, I.; Galman, J.L.; Lloyd, R.C.; Turner, N.J. Discovery, Engineering and Synthetic Application of Transaminase Biocatalysts. ACS Catal. 2017, 7, 8263–8284. [Google Scholar] [CrossRef]
- Shin, J.S.; Kim, B.G. Kinetic modeling of ω-transamination for enzymatic kinetic resolution of α-methylbenzylamine. Biotechnol. Bioeng. 1998, 60, 534–540. [Google Scholar] [CrossRef]
- Koszelewski, D.; Lavandera, I.; Clay, D.; Rozzell, D.; Kroutil, W. Asymmetric synthesis of optically pure pharmacologically relevant amines employing ω-transaminases. Adv. Synth. Catal. 2008, 350, 2761–2766. [Google Scholar] [CrossRef]
- Buß, O.; Jager, S.; Dold, S.-M.; Zimmermann, S.; Hamacher, K.; Schmitz, K.; Rudat, J. Statistical Evaluation of HTS Assays for Enzymatic Hydrolysis of β-Keto Esters. PLoS ONE 2016, 11, e0146104. [Google Scholar] [CrossRef] [PubMed]
- Pavlidis, I.V.; Weiß, M.S.; Genz, M.; Spurr, P.; Hanlon, S.P.; Wirz, B.; Iding, H.; Bornscheuer, U.T. Identification of (S)-selective transaminases for the asymmetric synthesis of bulky chiral amines. Nat. Chem. 2016, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Weiß, M.S.; Pavlidis, I.V.; Spurr, P.; Hanlon, S.P.; Wirz, B.; Iding, H.; Bornscheuer, U.T. Protein-engineering of an amine transaminase for the stereoselective synthesis of a pharmaceutically relevant bicyclic amine. Org. Biomol. Chem. 2016, 14, 10249–10254. [Google Scholar] [CrossRef] [PubMed]
- Savile, C.K.; Janey, J.M.; Mundorff, E.C.; Moore, J.C.; Tam, S.; Jarvis, W.R.; Colbeck, J.C.; Krebber, A.; Fleitz, F.J.; Brands, J.; et al. Biocatalytic Asymmetric Synthesis of Chiral Amines from Ketones Applied to Sitagliptin Manufacture. Science 2010, 329, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Buß, O.; Buchholz, P.C.F.; Gräff, M.; Klausmann, P.; Rudat, J.; Pleiss, J. The ω-Transaminase Engineering Database (oTAED): A navigation tool in protein sequence and structure space. Proteins Struct. Funct. Bioinforma. 2018. [Google Scholar] [CrossRef] [PubMed]
- Buß, O.; Muller, D.; Jager, S.; Rudat, J.; Rabe, K.S. Improvement of the thermostability of a β-amino acid converting ω-transaminase using FoldX. ChemBioChem 2017. [Google Scholar] [CrossRef] [PubMed]
- Dörr, M.; Fibinger, M.P.C.; Last, D.; Schmidt, S.; Santos-Aberturas, J.; Böttcher, D.; Hummel, A.; Vickers, C.; Voss, M.; Bornscheuer, U.T. Fully automatized high-throughput enzyme library screening using a robotic platform. Biotechnol. Bioeng. 2016, 113, 1421–1432. [Google Scholar] [CrossRef] [PubMed]
- Brucher, B.; Rudat, J.; Syldatk, C.; Vielhauer, O. Enantioseparation of Aromatic β3-Amino acid by Precolumn Derivatization with o-Phthaldialdehyde and N-Isobutyryl-l-cysteine. Chromatographia 2010, 71, 1063–1067. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Amino Donor | 3FCR_4M | ATA117 11Rd | ||||
|---|---|---|---|---|---|---|
| Amine | Concentration | Eq. | Conversion 2 | % ee (S) | Conversion 2 | % ee (R) |
| 7a | 50 mM | 5 | 1% | 99 | <0.5% | n.d. |
| Alanine 1 | 250 mM | 25 | 6% | 99 | n.d. | n.d. |
| 4 | 10 mM | 1 | 32% | 99 | 13% | 92 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buß, O.; Voss, M.; Delavault, A.; Gorenflo, P.; Syldatk, C.; Bornscheuer, U.; Rudat, J. β-Phenylalanine Ester Synthesis from Stable β-Keto Ester Substrate Using Engineered ω-Transaminases. Molecules 2018, 23, 1211. https://doi.org/10.3390/molecules23051211
Buß O, Voss M, Delavault A, Gorenflo P, Syldatk C, Bornscheuer U, Rudat J. β-Phenylalanine Ester Synthesis from Stable β-Keto Ester Substrate Using Engineered ω-Transaminases. Molecules. 2018; 23(5):1211. https://doi.org/10.3390/molecules23051211
Chicago/Turabian StyleBuß, Oliver, Moritz Voss, André Delavault, Pascal Gorenflo, Christoph Syldatk, Uwe Bornscheuer, and Jens Rudat. 2018. "β-Phenylalanine Ester Synthesis from Stable β-Keto Ester Substrate Using Engineered ω-Transaminases" Molecules 23, no. 5: 1211. https://doi.org/10.3390/molecules23051211
APA StyleBuß, O., Voss, M., Delavault, A., Gorenflo, P., Syldatk, C., Bornscheuer, U., & Rudat, J. (2018). β-Phenylalanine Ester Synthesis from Stable β-Keto Ester Substrate Using Engineered ω-Transaminases. Molecules, 23(5), 1211. https://doi.org/10.3390/molecules23051211