Novel Guanidine Compound against Multidrug-Resistant Cystic Fibrosis-Associated Bacterial Species

 ,
,  and
and
Abstract

1. Introduction
2. Materials and Methods
2.1. Reagents and Equipment
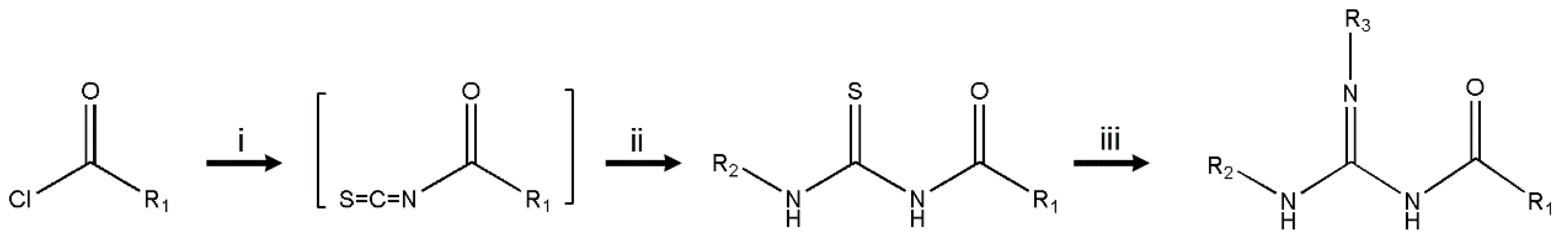
2.2. Synthesis of Compounds
2.3. Bacterial Strains
2.4. Antimicrobial Activity Assays
2.5. Checkerboard Assay
2.6. Cytotoxicity Assays
3. Results and Discussion
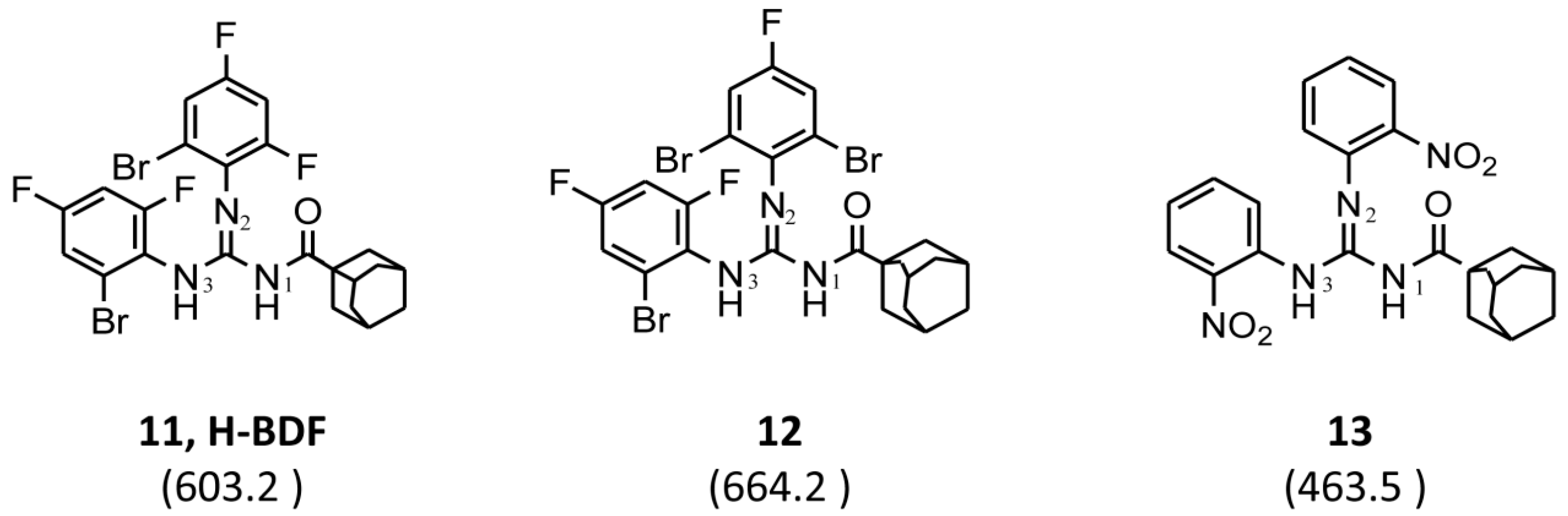
3.1. Chemistry
3.2. Biological Activity
3.2.1. Antimicrobial Evaluation of Newly Synthesized Compounds
3.2.2. Cytotoxic Evaluation of H-BDF
3.2.3. Synergistic Effects between H-BDF and Conventional Antibiotics
3.2.4. Activity of Compound H-BDF against Multidrug-Resistant Clinical Isolates Recovered from Respiratory Samples of CF Patients
4. Conclusions
5. Patents
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Livermore, D.M. Has the era of untreatable infections arrived? J. Antimicrob. Chemother. 2009, 64, i29–i36. [Google Scholar] [CrossRef] [PubMed]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad bugs, no drugs: No ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.R. Emerging carbapenemases: A global perspective. Int. J. Antimicrob. Agents 2010, 36, S8–S14. [Google Scholar] [CrossRef]
- McGowan, J.E., Jr. Resistance in nonfermenting gram-negative bacteria: Multidrug resistance to the maximum. Am. J. Med. 2006, 119, S29–S36. [Google Scholar] [CrossRef] [PubMed]
- Speert, D.P.; Henry, D.; Vandamme, P.; Corey, M.; Mahenthiralingam, E. Epidemiology of Burkholderia cepacia complex in patients with cystic fibrosis, Canada. Emerg. Infect. Dis. 2002, 8, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Schloss, P.D.; Kalikin, L.M.; Carmody, L.A.; Foster, B.K.; Petrosino, J.F.; Cavalcoli, J.D.; VanDevanter, D.R.; Murray, S.; Li, J.Z.; et al. Decade-long bacterial community dynamics in cystic fibrosis airways. Proc. Natl. Acad. Sci. USA 2012, 109, 5809–5814. [Google Scholar] [CrossRef] [PubMed]
- Falagas, M.E.; Bliziotis, I.A. Pandrug-resistant Gram-negative bacteria: The dawn of the post-antibiotic era? Int. J. Antimicrob. Agents 2007, 29, 630–636. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. The Antibiotic Resistance Crisis: Part 1: Causes and Threats. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- Delcour, A.H. Outer Membrane Permeability and Antibiotic Resistance. Biochim. Biophys. Acta 2009, 1794, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Batra, S. Thiourea and guanidine derivatives as antimalarial and antimicrobial agents. Curr. Top. Med. Chem. 2013, 13, 2011–2025. [Google Scholar] [CrossRef] [PubMed]
- Sączewski, F.; Balewski, Ł. Biological activities of guanidine compounds, 2008–2012 update. Expert Opin. Ther. Pat. 2013, 23, 965–995. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.; Rafique, H.; Hameed, A.; Rasheed, S. Synthesis and antibacterial activity of some new 1-aroyl-3-(substituted-2-benzothiazolyl)thioureas. Pharm. Chem. J. 2008, 42, 191. [Google Scholar] [CrossRef]
- Cunha, S.; Macedo, F.C.; Costa, G.A.N.; Rodrigues, M.T.; Verde, R.B.V.; de Souza Neta, L.C.; Vencato, I.; Lariucci, C.; Sá, F.P. Antimicrobial Activity and Structural Study of Disubstituted Thiourea Derivatives. Monatsh. Chem. 2007, 138, 511–516. [Google Scholar] [CrossRef]
- Saeed, A.; Shaheen, U.; Hameed, A.; Naqvi, S.Z.H. Synthesis, characterization and antimicrobial activity of some new 1-(fluorobenzoyl)-3-(fluorophenyl)thioureas. J. Fluor. Chem. 2009, 130, 1028–1034. [Google Scholar] [CrossRef]
- Andreev, K.; Bianchi, C.; Laursen, J.S.; Citterio, L.; Hein-Kristensen, L.; Gram, L.; Kuzmenko, I.; Olsen, C.A.; Gidalevitz, D. Guanidino Groups Greatly Enhance the Action of Antimicrobial Peptidomimetics against Bacterial Cytoplasmic Membranes. Biochim. Biophys. Acta 2014, 1838, 2492–2502. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.; Belagali, S.L. Guanidinyl benzothiazole derivatives: Synthesis and structure activity relationship studies of a novel series of potential antimicrobial and antioxidants. Res. Chem. Intermediat. 2016, 42, 6195–6208. [Google Scholar] [CrossRef]
- Böhm, H.-J.; Banner, D.; Bendels, S.; Kansy, M.; Kuhn, B.; Müller, K.; Obst-Sander, U.; Stahl, M. Fluorine in Medicinal Chemistry. Chem. Biol. Chem. 2004, 5, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Filler, R.; Saha, R. Fluorine in medicinal chemistry: A century of progress and a 60-year retrospective of selected highlights. Future Med. Chem. 2009, 1, 777–791. [Google Scholar] [CrossRef] [PubMed]
- Deep, A.; Jain, S.; Sharma, P.C.; Mittal, S.K.; Phogat, P.; Malhotra, M. Synthesis, characterization and antimicrobial evaluation of 2,5-disubstituted-4-thiazolidinone derivatives. Arab. J. Chem. 2014, 7, 287–291. [Google Scholar] [CrossRef]
- Krishnanjaneyulu, I.S.; Saravanan, G.; Vamsi, J.; Supriya, P.; Bhavana, J.U.; Sunil Kumar, M.V. Synthesis, characterization and antimicrobial activity of some novel benzimidazole derivatives. J. Adv. Pharm. Technol. Res. 2014, 5, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Stefanska, J.; Nowicka, G.; Struga, M.; Szulczyk, D.; Koziol, A.E.; Augustynowicz-Kopec, E.; Napiorkowska, A.; Bielenica, A.; Filipowski, W.; Filipowska, A.; et al. Antimicrobial and anti-biofilm activity of thiourea derivatives incorporating a 2-aminothiazole scaffold. Chem. Pharm. Bull. 2015, 63, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Douglass, I.B.; Dains, F.B. Some Derivatives of Benzoyl and Furoyl Isothiocyanates and their Use in Synthesizing Heterocyclic Compounds. J. Am. Chem. Soc. 1934, 56, 719–721. [Google Scholar] [CrossRef]
- Saeed, A.; Flörke, U.; Erben, M.F. The role of substituents in the molecular and crystal structure of 1-(adamantane-1-carbonyl)-3-(mono)- and 3,3-(di) substituted thioureas. J. Mol. Struct. 2014, 1065–1066, 150–159. [Google Scholar] [CrossRef]
- Saeed, A.; Erben, M.F.; Bolte, M. Synthesis, structural and vibrational properties of 1-(adamantane-1-carbonyl)-3-halophenyl thioureas. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2013, 102, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, S.K.; Jaber, A.A.; Saeed, S.; Ahmad, K.S.; Wong, W.T. 1-(Adamantan-1-ylcarbon-yl)-3-(2,6-difluoro-4-hy-droxy-phen-yl)thio-urea. Acta Crystallogr. Sect. E. Struct. Rep. Online 2012, 68, o1597. [Google Scholar] [CrossRef] [PubMed]
- Cunha, S.; Costa, M.s.B.; Napolitano, H.B.; Lariucci, C.; Vencato, I. Study of N-benzoyl-activation in the HgCl2-promoted guanylation reaction of thioureas. Synthesis and structural analysis of N-benzoyl-guanidines. Tetrahedron 2001, 57, 1671–1675. [Google Scholar] [CrossRef]
- Gueirard, P.; Guiso, N. Virulence of Bordetella bronchiseptica: Role of adenylate cyclase-hemolysin. Infect. Immun. 1993, 61, 4072–4078. [Google Scholar] [PubMed]
- Martina, P.; Leguizamon, M.; Prieto, C.I.; Sousa, S.A.; Montanaro, P.; Draghi, W.O.; Stammler, M.; Bettiol, M.; de Carvalho, C.; Palau, J.; et al. Burkholderia puraquae sp. nov., a novel species of the Burkholderia cepacia complex isolated from hospital settings and agricultural soils. Int. J. Syst. Evol. Microbiol. 2017, 68, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Martina, P.; Bettiol, M.; Vescina, C.; Montanaro, P.; Mannino, M.C.; Prieto, C.I.; Vay, C.; Naumann, D.; Schmitt, J.; Yantorno, O.; et al. Genetic Diversity of Burkholderia contaminans Isolates from Cystic Fibrosis Patients in Argentina. J. Clin. Microbiol. 2013, 51, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.; Mahenthiralingam, E.; Thickett, K.M.; Honeybourne, D.; Maiden, M.C.; Govan, J.R.; Speert, D.P.; Lipuma, J.J.; Vandamme, P.; Dowson, C.G. Multilocus sequence typing scheme that provides both species and strain differentiation for the Burkholderia cepacia complex. J. Clin. Microbiol. 2005, 43, 4665–4673. [Google Scholar] [CrossRef] [PubMed]
- CLSI Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically; Approved Standard M07-A9, 9th ed.; CLSI: Wayne, PA, USA, 2012. [Google Scholar]
- Isenberg, H.D. Synergism Testing: Broth Microdilution Checkerboard and Broth Macrodilution Methods. In Clinical Microbiology Procedures Handbook; American Society of Microbiology: Washington, DC, USA, 1992; pp. 1–28. [Google Scholar]
- Berenbaum, M.C. Correlations between methods for measurement of synergy. J. Infect. Dis. 1980, 142, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Forcet, C.; Ye, X.; Granger, L.; Corset, V.; Shin, H.; Bredesen, D.E.; Mehlen, P. The dependence receptor DCC (deleted in colorectal cancer) defines an alternative mechanism for caspase activation. Proc. Natl. Acad. Sci. USA 2001, 98, 3416–3421. [Google Scholar] [CrossRef] [PubMed]
- Orme, I. Search for new drugs for treatment of tuberculosis. Antimicrob. Agents Chemother. 2001, 45, 1943–1946. [Google Scholar] [PubMed]
- Orzeszko, A.; Kaminska, B.; Starosciak, B.J. Synthesis and antimicrobial activity of new adamantane derivatives III. Farmaco 2002, 57, 619–624. [Google Scholar] [CrossRef]
- Orzeszko, B.; Fedorynski, M.; Laudy, A.E.; Starosciak, B.J.; Orzeszko, A. Synthesis and antibacterial activity of 5-adamantan-1-yl-methyl analogues of trimethoprim. Acta Pol. Pharm. 2006, 63, 374–377. [Google Scholar] [PubMed]
- Orzeszko, A.; Kaminska, B.; Orzeszko, G.; Starosciak, B.J. Synthesis and antimicrobial activity of new adamantane derivatives II. Farmaco 2000, 55, 619–623. [Google Scholar] [CrossRef]
- Orzeszko, A.; Gralewska, R.; Starosciak, B.J.; Kazimierczuk, Z. Synthesis and antimicrobial activity of new adamantane derivatives I. Acta Biochim. Pol. 2000, 47, 87–94. [Google Scholar] [CrossRef]
- Al-Abdullah, E.S.; Al-Tuwaijri, H.M.; Hassan, H.M.; Al-Alshaikh, M.A.; Habib, E.E.; El-Emam, A.A. Synthesis, Antimicrobial and Hypoglycemic Activities of Novel N-(1-Adamantyl)carbothioamide Derivatives. Molecules 2015, 20, 8125–8143. [Google Scholar] [CrossRef] [PubMed]
- Levallet, C.; Lerpiniere, J.; Ko, S.Y. The HgCl2-promoted guanylation reaction: The scope and limitations. Tetrahedron 1997, 53, 5291–5304. [Google Scholar] [CrossRef]
- Saeed, A.; Erben, M.F.; Abbas, N.; Flörke, U. Synthesis, crystal X-ray diffraction structure, vibrational properties and quantum chemical calculations on 1-(4-(4-Fluorobenzamido)phenyl)-3-(4-fluorobenzoyl)thiourea. J. Mol. Struct. 2010, 984, 240–245. [Google Scholar] [CrossRef]
- Saeed, A.; Erben, M.F.; Shaheen, U.; Flörke, U. Synthesis, structural and vibrational properties of 1-(4-Fluorobenzoyl)-3-(isomeric fluorophenyl)thioureas. J. Mol. Struct. 2011, 1000, 49–57. [Google Scholar] [CrossRef]
- Nzula, S.; Vandamme, P.; Govan, J.R.W. Influence of taxonomic status on the in vitro antimicrobial susceptibility of the Burkholderia cepacia complex. J. Antimicrob. Chemother. 2002, 50, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Stover, C.K.; Pham, X.Q.; Erwin, A.L.; Mizoguchi, S.D.; Warrener, P.; Hickey, M.J.; Brinkman, F.S.; Hufnagle, W.O.; Kowalik, D.J.; Lagrou, M.; et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 2000, 406, 959–964. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.L. Nosocomial infections in adult intensive-care units. Lancet 2003, 361, 2068–2077. [Google Scholar] [CrossRef]
- Al-Khodor, S.; Marshall-Batty, K.; Nair, V.; Ding, L.; Greenberg, D.E.; Fraser, I.D. Burkholderia cenocepacia J2315 escapes to the cytosol and actively subverts autophagy in human macrophages. Cell. Microbiol. 2014, 16, 378–395. [Google Scholar] [CrossRef] [PubMed]
- Chmiel, J.F.; Aksamit, T.R.; Chotirmall, S.H.; Dasenbrook, E.C.; Elborn, J.S.; LiPuma, J.J.; Ranganathan, S.C.; Waters, V.J.; Ratjen, F.A. Antibiotic Management of Lung Infections in Cystic Fibrosis. I. The Microbiome, Methicillin-Resistant Staphylococcus aureus, Gram-Negative Bacteria, and Multiple Infections. Ann. Am. Thorac. Soc. 2014, 11, 1120–1129. [Google Scholar]
- Pick, N.; Cameron, S.; Arad, D.; Av-Gay, Y. Screening of Compounds Toxicity against Human Monocytic cell line-THP-1 by Flow Cytometry. Biol. Proced. Online 2004, 6, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, E. Toxicity of orally inhaled drug formulations at the alveolar barrier: Parameters for initial biological screening. Drug Deliv. 2017, 24, 891–905. [Google Scholar] [CrossRef] [PubMed]
- George, A.M.; Jones, P.M.; Middleton, P.G. Cystic fibrosis infections: Treatment strategies and prospects. FEMS Microbiol. Lett. 2009, 300, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Gibson, R.L.; Burns, J.L.; Ramsey, B.W. Pathophysiology and Management of Pulmonary Infections in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2003, 168, 918–951. [Google Scholar] [CrossRef] [PubMed]
- Blazquez, J.; Couce, A.; Rodriguez-Beltran, J.; Rodriguez-Rojas, A. Antimicrobials as promoters of genetic variation. Curr. Opin. Microbiol. 2012, 15, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Leitao, J.H.; Sousa, S.A.; Cunha, M.V.; Salgado, M.J.; Melo-Cristino, J.; Barreto, M.C.; Sa-Correia, I. Variation of the antimicrobial susceptibility profiles of Burkholderia cepacia complex clonal isolates obtained from chronically infected cystic fibrosis patients: A five-year survey in the major Portuguese treatment center. Eur. J. Clin. Microbiol. Infect. Dis. 2008, 27, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Martina, P.; Feliziani, S.; Juan, C.; Bettiol, M.; Gatti, B.; Yantorno, O.; Smania, A.M.; Oliver, A.; Bosch, A. Hypermutation in Burkholderia cepacia complex is mediated by DNA mismatch repair inactivation and is highly prevalent in cystic fibrosis chronic respiratory infection. Int. J. Med. Microbiol. 2014, 304, 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- Chernish, R.N.; Aaron, S.D. Approach to resistant gram-negative bacterial pulmonary infections in patients with cystic fibrosis. Curr. Opin. Pulm. Med. 2003, 9, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Quon, B.S.; Aitken, M.L. Cystic Fibrosis: What to Expect now in the Early Adult Years. Paediatr. Respir. Rev. 2012, 13, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Lipuma, J.J. The changing microbial epidemiology in cystic fibrosis. Clin. Microbiol. Rev. 2010, 23, 299–323. [Google Scholar] [CrossRef] [PubMed]
- Parkins, M.D.; Floto, R.A. Emerging bacterial pathogens and changing concepts of bacterial pathogenesis in cystic fibrosis. J. Cyst. Fibros. 2015, 14, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Raczyńska, E.D.; Cyrański, M.K.; Gutowski, M.; Rak, J.; Gal, J.F.; Maria, P.C.; Darowska, M.; Duczmal, K. Consequences of proton transfer in guanidine. J. Phys. Org. Chem. 2003, 16, 91–106. [Google Scholar] [CrossRef]
- Pfeffer, F.; Henderson, L.; Li, J.; Nation, R. Dioxolane Norbornane/Norbornene Compounds Suitable as Antimicrobial Agents to Treat Bacterial Infections. Patent WO2010099573A1, 10 September 2010. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
| Entry | R1 | R2 | R3 | Molecular Weight (g/mol) | Chemical Structure | P. aeruginosa PAO1 | B. cenocepacia J2315 | ||
|---|---|---|---|---|---|---|---|---|---|
| MIC (µg/mL) | MBC (µg/mL) | MIC (µg/mL) | MBC (µg/mL) | ||||||
| 1 | C10H15 a | C6H11 | 320.19 |  | >128 | nd | >128 | nd | |
| 2 | C10H15 a | C6H5 | - | 314.45 |  | >128 | nd | >128 | nd |
| 3 | C10H15 a | 3-F-4-CH3-C6H3 | - | 385.97 |  | >128 | nd | >128 | nd |
| 4 | C10H15 a | 2-NO2-C6H4 | - | 359.44 |  | >128 | nd | >128 | nd |
| 5 | C10H15 a | 4-CH3CO-C6H4 | - | 356.47 |  | >128 | nd | >128 | nd |
| 6 | C10H15 a | 2,3-di-Cl-C6H3 | - | 383.34 |  | >128 | nd | >128 | nd |
| 7 | C10H15 a | 2-Br-4,6-di-F-C6H2 | - | 428.32 |  | >128 | nd | >128 | nd |
| 8 | C10H7 b | 2-Br-4,6-di-F-C6H2 | - | 421.97 |  | >128 | nd | >128 | nd |
| 9 | 2,4-di-Cl-C6H3 | 2-Br-4,6-di-F-C6H2 | - | 439.88 |  | >128 | nd | >128 | nd |
| 10 | 4-CH3-C6H4 | 2-Br-4,6-di-F-C6H2 | - | 385.97 |  | >128 | nd | >128 | nd |
| 11 | C10H15 a | 2-Br-4,6-di-F-C6H2 | 2-Br-4,6-di-F-C6H2 | 603.2 |  | 0.5 | 4 | 2 | 8 |
| Tobramycin | 467.51 | 2 | 2 | >128 | >128 | ||||
| Meropenem | 383.46 | 1 | 4 | 8 | 64 | ||||
| Ceftazimide | 546.57 | 2 | 2 | 16 | 128 | ||||
| Compound | 11 (H-BDF) | 12 | 13 | Tobramycin | Meropenem | Ceftazimide |
|---|---|---|---|---|---|---|
| Organism | MIC/MBC | MIC/MBC | MIC/MBC | MIC/MBC | MIC/MBC | MIC/MBC |
| Gram-negative bacteria | ||||||
| Bordetella bronchiseptica 9.73H+ | 0.5/2 | 16/64 | >128/>128 | 64/64 | 0.125/0.25 | 8/64 |
| Escherichia coli ATCC25922 | 1/2 | 64/64 | >128/>128 | 16/16 | 0.03125/0.0625 | 1/1 |
| Pseudomonas aeruginosa PAO1 | 0.5/4 | 32/>128 | >128/>128 | 2/2 | 1/4 | 2/2 |
| Burkholderia cenocepacia J2315 | 2/8 | 64/128 | >128/>128 | >128/>128 | 8/64 | 16/128 |
| Pandorea apista DSM16535 | 1/2 | 64/128 | >128/>128 | 32/128 | >128/nd | 128/nd |
| Gram-positive bacteria | ||||||
| Staphyloccocus aureus ATCC6538 | 0.25/1 | 8/64 | >128/>128 | 2/2 | <0.125/<0.25 | 8/8 |
| Bacillus cereus ATCC10876 | 2/2 | 64/64 | >128/>128 | 8/32 | <0.125/<0.25 | 1/1 |
| Cells | ||
|---|---|---|
| Organisms | A549 | THP-1 |
| Gram-negative bacteria | ||
| Bordetella bronchiseptica 9.73H+ | 76.8 | 30.9 |
| Escherichia coli ATCC25922 | 38.4 | 15.45 |
| Pseudomonas aeruginosa PAO1 | 76.8 | 30.9 |
| Burkholderia cenocepacia J2315 | 19.2 | 7.7 |
| Pandorea apista DSM16535 | 38.4 | 15.45 |
| Gram-positive bacteria | ||
| Staphyloccocus aureus ATCC6538 | 153.6 | 61.8 |
| Bacillus cereus ATCC10876 | 19.2 | 7.7 |
| H-BDF | Tobramycin | Meropenem | Ceftazidime | |||||
|---|---|---|---|---|---|---|---|---|
| Clinical Isolates a | MIC (µg/mL) | MBC (µg/mL) | MIC (µg/mL) | MBC (µg/mL) | MIC (µg/mL) | MBC (µg/mL) | MIC (µg/mL) | MBC (µg/mL) |
| Achromobacter xylosoxidans | ||||||||
| A. xylosoxidans HNA 001 | 0.125 | 0.25 | R | nd | S | 8 | S | nd |
| Burkholderia cenocepacia | ||||||||
| B. cenocepacia CAMPA 669 | 0.25 | 2 | S | nd | S | nd | R | nd |
| B. cenocepacia CAMPA 1533 | 4 | 16 | R | nd | R | 64 | S | 16 |
| B. cenocepacia CAMPA 1194 | 2 | 4 | R | nd | R | nd | R | nd |
| B. cenocepacia CAMPA 544 | 2 | 8 | R | nd | R | nd | S | 8 |
| B. cenocepacia CAMPA 1771 | 8 | 16 | R | nd | I | 32 | R | nd |
| B. cenocepacia CAMPA 817 | 2 | 8 | R | nd | R | nd | S | 8 |
| B. cenocepacia CAMPA 548 | 2 | 4 | R | nd | R | nd | S | 8 |
| B. cenocepacia CAMPA 825 (CBC 033) b | 4 | 16 | R | nd | I | nd | S | 32 |
| B. cenocepacia CAMPA538 (CBC 035) b | 2 | 4 | R | nd | I | 16 | S | 16 |
| B. cenocepacia CAMPA 817 | 2 | 8 | R | nd | R | nd | S | 16 |
| B.cenocepacia CAMPA 531 | 1 | 4 | R | nd | S | nd | S | nd |
| B.cenocepacia CAMPA 993 (CBC 024) b | 1 | 4 | R | nd | S | nd | S | nd |
| B.cenocepacia HE001 | 4 | 64 | R | nd | R | nd | R | nd |
| Burkholderia cepacia | ||||||||
| B. cepacia CAMPA 545 | 4 | 16 | R | nd | R | nd | S | 16 |
| B. cepacia CAMPA 233 (CBC 012) b | 2 | 4 | R | nd | S | 8 | S | 16 |
| B. cepacia CAMPA 260 | 32 | nd | R | nd | R | 32 | R | nd |
| B. cepacia CAMPA 914 | 32 | nd | R | nd | R | 32 | R | 64 |
| B. cepacia CAMPA 886 | 32 | nd | R | nd | R | 32 | R | 128 |
| B. cepacia CAMPA 998 | 32 | nd | R | nd | R | 64 | S | 32 |
| B. cepacia CAMPA 1039 | 64 | nd | R | nd | R | 32 | R | nd |
| B. cepacia CAMPA 853 (CBC 001) b | 32 | nd | R | nd | I | 64 | I | 64 |
| B. cepacia CAMPA 860 (CBC 007) b | 64 | nd | R | nd | I | 64 | R | 64 |
| B. cepacia CAMPA 660 | 4 | 8 | R | nd | S | 4 | R | nd |
| B. cepacia CAMPA 721 (CBC 011) b | 2 | 32 | R | nd | S | 64 | R | nd |
| Burkholderia contaminans | ||||||||
| B. contaminans HNBC001 | 0.25 | 1 | R | nd | R | nd | S | nd |
| Burkholderia multivorans | ||||||||
| B. multivorans CAMPA 661(CBC 015) b | 2 | 4 | R | nd | S | 4 | S | 8 |
| B. multivorans CAMPA 1530 | 2 | 8 | R | nd | R | nd | S | 4 |
| B. multivorans CAMPA 647 (CBC 017) b | 4 | 4 | R | nd | S | 4 | S | 8 |
| B. multivorans CAMPA 653 (CBC 018) b | 2 | 8 | R | nd | S | 4 | S | 8 |
| B. multivorans CAMPA 623(CBC 019) b | 2 | 8 | R | nd | S | 8 | R | nd |
| B. multivorans CAMPA 832 (CBC 020) b | 4 | 16 | R | nd | S | 32 | R | nd |
| B. multivorans CAMPA 987 (CBC 021) b | 2 | 4 | R | nd | S | 8 | R | nd |
| B. multivorans CAMPA 997 (CBC 022) b | 4 | 8 | R | nd | S | 8 | R | nd |
| Burkholderia seminalis | ||||||||
| B. seminalis CAMPA 231 | 32 | nd | R | nd | I | nd | R | 32 |
| B. seminalis CAMPA 261 (CBC 039) b | 32 | nd | R | nd | S | 16 | S | 16 |
| B. seminalis CAMPA 475 (CBC 040) b | 4 | 8 | R | nd | I | nd | R | nd |
| B. seminalis CAMPA 227 | 1 | 8 | R | nd | R | nd | R | nd |
| Burkholderia vietnamiensis | ||||||||
| B. vietnamiensis CAMPA 992 (CBC 038) b | 32 | nd | R | nd | S | 8 | S | 16 |
| Staphylococcus aureus | ||||||||
| S. aureus CAMPA 1909 | 2 | 16 | 128 | nd | >128 | nd | >128 | nd |
| S. aureus CAMPA 1908 | 1 | 4 | 32 | >128 | >128 | nd | >128 | nd |
| Stenotrophomonas maltophilia | ||||||||
| S. maltophilia CAMPA 1911 | 2 | 16 | >128 | nd | >128 | nd | >128 | nd |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saeed, A.; Bosch, A.; Bettiol, M.; Nossa González, D.L.; Erben, M.F.; Lamberti, Y. Novel Guanidine Compound against Multidrug-Resistant Cystic Fibrosis-Associated Bacterial Species. Molecules 2018, 23, 1158. https://doi.org/10.3390/molecules23051158
Saeed A, Bosch A, Bettiol M, Nossa González DL, Erben MF, Lamberti Y. Novel Guanidine Compound against Multidrug-Resistant Cystic Fibrosis-Associated Bacterial Species. Molecules. 2018; 23(5):1158. https://doi.org/10.3390/molecules23051158
Chicago/Turabian StyleSaeed, Aamer, Alejandra Bosch, Marisa Bettiol, Diana L. Nossa González, Mauricio Federico Erben, and Yanina Lamberti. 2018. "Novel Guanidine Compound against Multidrug-Resistant Cystic Fibrosis-Associated Bacterial Species" Molecules 23, no. 5: 1158. https://doi.org/10.3390/molecules23051158
APA StyleSaeed, A., Bosch, A., Bettiol, M., Nossa González, D. L., Erben, M. F., & Lamberti, Y. (2018). Novel Guanidine Compound against Multidrug-Resistant Cystic Fibrosis-Associated Bacterial Species. Molecules, 23(5), 1158. https://doi.org/10.3390/molecules23051158