Electrochemically Triggered Co-Conformational Switching in a [2]catenane Comprising a Non-Symmetric Calix[6]arene Wheel and a Two-Station Oriented Macrocycle

 , and
, and
Abstract
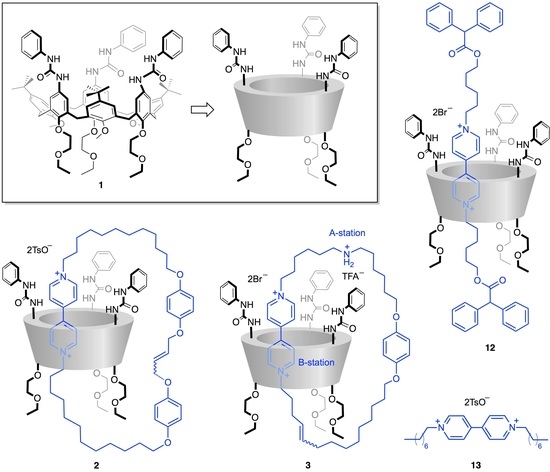
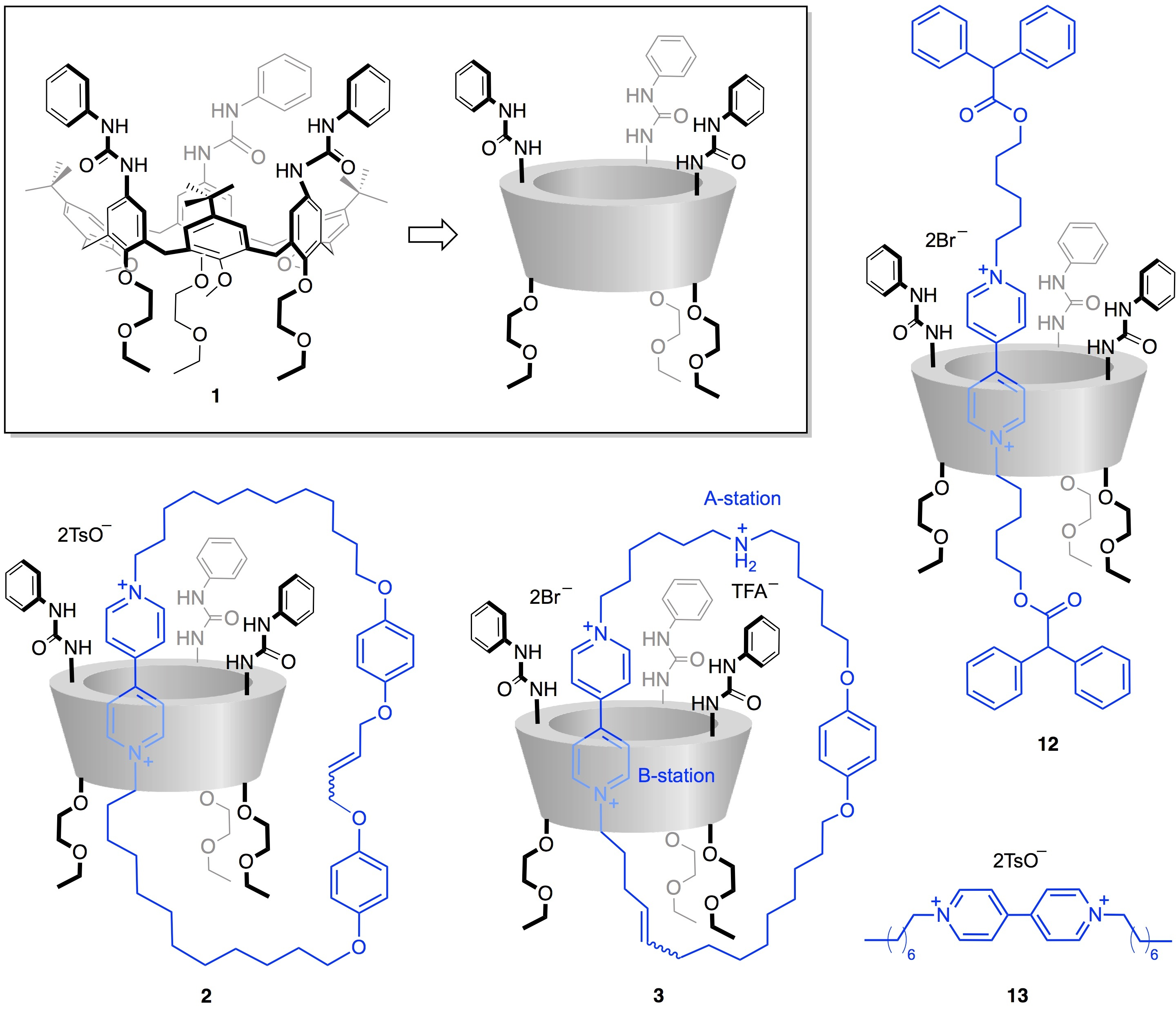{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
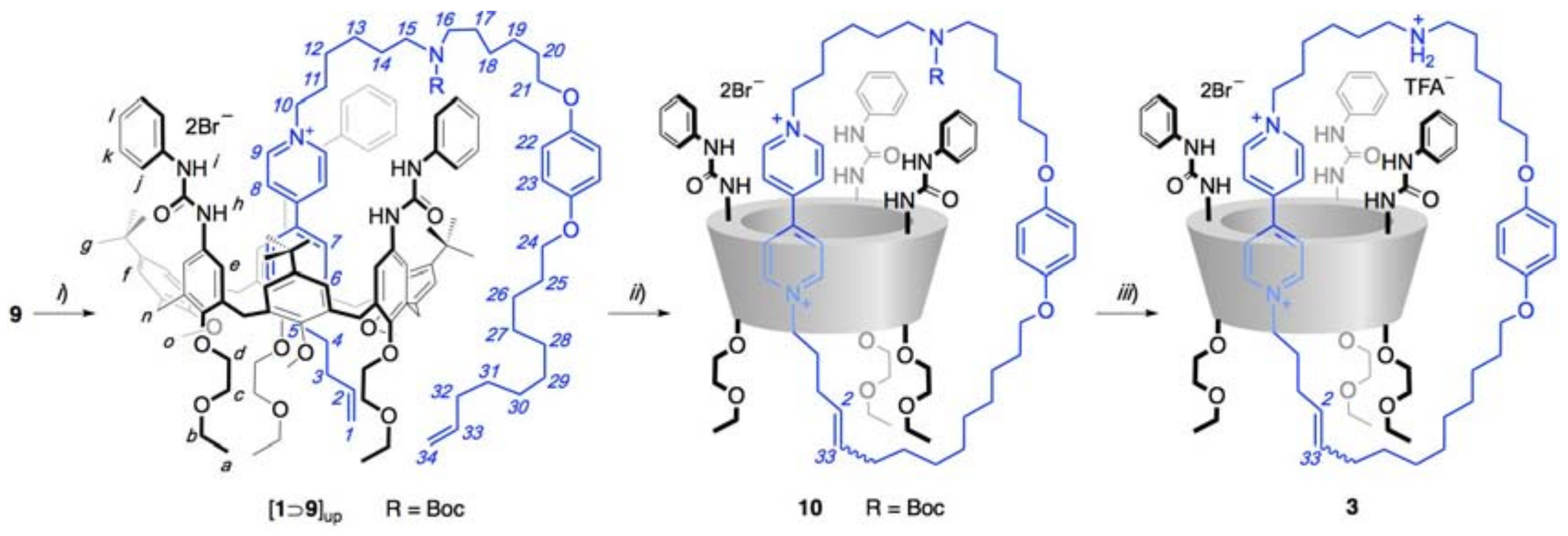{kind=link}
1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterization of Calix[6]arene-Based [2]catenanes
2.2. Electrochemical Measurements
3. Materials and Methods
3.1. Synthesis
3.2. Electrochemical Measurements
3.3. Molecular Modelling
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Sauvage, J.-P.; Dietrich-Buchecker, C. (Eds.) Molecular Catenanes, Rotaxanes and Knots. A Journey Through the World of Molecular Topology; Wiley-VCH: Weinheim, Germany, 1999; ISBN 978-3-527-29572-2. [Google Scholar]
- Bruns, C.J.; Stoddart, J.F. The Chemistry of the Mechanical Bond: From Molecules to Machines; Wiley: Hoboken, NJ, USA, 2016; ISBN 978-1-119-04400-0. [Google Scholar]
- Gil-Ramírez, G.; Leigh, D.A.; Stephens, A.J. Catenanes: Fifty Years of Molecular Links. Angew. Chem. Int. Ed. 2015, 54, 6110–6150. [Google Scholar] [CrossRef] [PubMed]
- Gibbs-Hall, I.C.; Vermeulen, N.A.; Dale, E.J.; Henkelis, J.J.; Blackburn, A.K.; Barnes, J.C.; Stoddart, J.F. Catenation through a Combination of Radical Templation and Ring-Closing Metathesis. J. Am. Chem. Soc. 2015, 137, 15640–15643. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, E. The Preparation of Interlocking Rings: A Catenane. J. Am. Chem. Soc. 1960, 82, 4433–4434. [Google Scholar] [CrossRef]
- Ashton, P.R.; Goodnow, T.T.; Kaifer, A.E.; Reddington, M.V.; Slawin, A.M.Z.; Spencer, N.; Stoddart, J.F.; Vicent, C.; Williams, D.J. A [2]Catenane Made to Order. Angew. Chem. Int. Ed. Engl. 1989, 28, 1396–1399. [Google Scholar] [CrossRef]
- Ballardini, R.; Balzani, V.; Credi, A.; Brown, C.L.; Gillard, R.E.; Montalti, M.; Philp, D.; Stoddart, J.F.; Venturi, M.; White, A.J.P.; et al. Controlling Catenations, Properties and Relative Ring-Component Movements in Catenanes with Aromatic Fluorine Substituents. J. Am. Chem. Soc. 1997, 51, 12503–12513. [Google Scholar] [CrossRef]
- Vögtle, F.; Meier, S.; Hoss, R. One-Step Synthesis of a Fourfold Functionalized Catenane. Angew. Chem. Int. Ed. Engl. 1992, 31, 1619–1622. [Google Scholar] [CrossRef]
- Johnston, A.G.; Leigh, D.A.; Pritchard, R.J.; Deegan, M.D. Facile Synthesis and Solid-State Structure of a Benzylic Amide [2]Catenane. Angew. Chem. Int. Ed. Engl. 1995, 34, 1209–1212. [Google Scholar] [CrossRef]
- Dietrich-Buchecker, C.O.; Sauvage, J.P.; Kern, J.M. Templated Synthesis of Interlocked Macrocyclic Ligands: The Catenands. J. Am. Chem. Soc. 1984, 106, 3043–3045. [Google Scholar] [CrossRef]
- Wang, K.; Yee, C.-C.; Au-Yeung, H.Y.; Lee, E.; Kang, J.-K.; Sakamoto, S.; Yamaguchi, K.; Kim, K.; Haino, T.; Williams, D.J. Facile Syntheses of [3]-, [4]- and [6]catenanes Templated by Orthogonal Supramolecular Interactions. Chem. Sci. 2016, 7, 2787–2792. [Google Scholar] [CrossRef] [PubMed]
- Evans, N.H.; Beer, P.D. Advances in anion supramolecular chemistry: From recognition to chemical applications. Angew. Chem. Int. Ed. 2014, 53, 11716–11754. [Google Scholar] [CrossRef] [PubMed]
- Balzani, V.; Credi, A.; Venturi, M. Molecular Devices and Machines—Concepts and Perspectives for the Nanoworld; Wiley-VCH: Weinheim, Germany, 2008; ISBN 978-3-527-31800-1. [Google Scholar]
- Erbas-Cakmak, S.; Leigh, D.A.; McTernan, C.T.; Nussbaumer, A.L. Artificial Molecular Machines. Chem. Rev. 2015, 115, 10081–10206. [Google Scholar] [CrossRef] [PubMed]
- Livoreil, A.; Dietrich-Buchecker, C.O.; Sauvage, J.-P. Electrochemically Triggered Swinging of a [2]-Catenate. J. Am. Chem. Soc. 1994, 116, 9399–9400. [Google Scholar] [CrossRef] [PubMed]
- Asakawa, M.; Ashton, P.R.; Balzani, V.; Boyd, S.E.; Credi, A.; Mattersteig, G.; Menzer, S.; Montalti, M.; Raymo, F.M.; Ruffilli, C.; et al. Pseudorotaxanes and catenanes containing a redox-active unit derived from tetrathiafulvalene. Eur. J. Org. Chem. 1999, 985–994. [Google Scholar] [CrossRef]
- Spruell, J.M.; Paxton, W.F.; Olsen, J.-C.; Benítez, D.; Tkatchouk, E.; Stern, C.L.; Trabolsi, A.; Friedman, D.C.; Goddard, W.A.; Stoddart, J.F. A Push-Button Molecular Switch. J. Am. Chem. Soc. 2009, 131, 11571–11580. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Amelia, M.; Klivansky, L.M.; Koshkakaryan, G.; Khan, S.I.; Semeraro, M.; Silvi, S.; Venturi, M.; Credi, A.; Liu, Y. Probing Donor-Acceptor Interactions and Co-Conformational Changes in Redox Active Desymmetrized [2]Catenanes. J. Am. Chem. Soc. 2010, 132, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Han, Y.; Wang, L.-N.; Xiang, J.-F.; He, S.-G.; Chen, C.-F. Stepwise Motion in a Multivalent [2](3)Catenane. J. Am. Chem. Soc. 2015, 137, 9739–9745. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, J.V.; Kay, E.R.; Leigh, D.A. A Reversible Synthetic Rotary Molecular Motor. Science 2004, 306, 1532–1537. [Google Scholar] [CrossRef] [PubMed]
- Leigh, D.A.; Wong, J.K.Y.; Dehez, F.; Zerbetto, F. Unidirectional Rotation in a Mechanically Interlocked Molecular Rotor. Nature 2003, 424, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.R.; Solà, J.; Carlone, A.; Goldup, S.M.; Lebrasseur, N.; Leigh, D.A. An autonomous chemically fuelled small-molecule motor. Nature 2016, 534, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Erbas-Cakmak, S.; Fielden, S.D.P.; Karaca, U.; Leigh, D.A.; McTernan, C.T.; Tetlow, D.J.; Wilson, M.R. Rotary and linear molecular motors driven by pulses of a chemical fuel. Science 2017, 358, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Gaeta, C.; Talotta, C.; Mirra, S.; Margarucci, L.; Casapullo, A.; Neri, P. Catenation of Calixarene Annulus. Org. Lett. 2013, 15, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Orlandini, G.; Zanichelli, V.; Secchi, A.; Arduini, A.; Ragazzon, G.; Credi, A.; Venturi, M.; Silvi, S. Synthesis by Ring Closing Metathesis and Properties of an Electroactive calix[6]arene [2]catenane. Supramol. Chem. 2016, 28, 427–435. [Google Scholar] [CrossRef]
- Arduini, A.; Orlandini, G.; Secchi, A.; Credi, A.; Silvi, S.; Venturi, M. Calixarene Threading by Viologen-Based Axles. In Calixarenes and Beyond; Neri, P., Sessler, J.L., Wang, M.-X., Eds.; Springer: Cham, Switzerland, 2016; pp. 761–781. [Google Scholar]
- Arduini, A.; Orlandini, G.; Secchi, A.; Credi, A.; Silvi, S.; Venturi, M. Calix-Based Molecular Machines and Devices. In Reference Module in Chemistry, Molecular Sciences and Chemical Engineering; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Baroncini, M.; Casimiro, L.; de Vet, C.; Groppi, J.; Silvi, S.; Credi, A. Making and Operating Molecular Machines: A Multidisciplinary Challenge. ChemistryOpen 2018, 7, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Credi, A.; Dumas, S.; Silvi, S.; Venturi, M.; Arduini, A.; Pochini, A.; Secchi, A. Viologen-Calix[6]arene Pseudorotaxanes. Ion-Pair Recognition and Threading/Dethreading Molecular Motions. J. Org. Chem. 2004, 69, 5881–5887. [Google Scholar] [CrossRef] [PubMed]
- Venturi, M.; Credi, A. Electroactive [2]Catenanes. Electrochim. Acta 2014, 140, 467–475. [Google Scholar] [CrossRef]
- Arduini, A.; Bussolati, R.; Credi, A.; Secchi, A.; Silvi, S.; Semeraro, M.; Venturi, M. Toward Directionally Controlled Molecular Motions and Kinetic Intra- and Intermolecular Self-Sorting: Threading Processes of Nonsymmetric Wheel and Axle Components. J. Am. Chem. Soc. 2013, 135, 9924–9930. [Google Scholar] [CrossRef] [PubMed]
- Kidd, T.J.; Leigh, D.A.; Wilson, A.J. Organic “Magic Rings”: The Hydrogen Bond-Directed Assembly of Catenanes under Thermodynamic Control. J. Am. Chem. Soc. 1999, 121, 1599–1600. [Google Scholar] [CrossRef]
- Weck, M.; Mohr, B.; Sauvage, J.P.; Grubbs, R.H. Synthesis of Catenane Structures via Ring-Closing Metathesis. J. Org. Chem. 1999, 64, 5463–5471. [Google Scholar] [CrossRef] [PubMed]
- Guidry, E.N.; Cantrill, S.J.; Stoddart, J.F.; Grubbs, R.H. Magic Ring Catenation by Olefin Metathesis. Org. Lett. 2005, 7, 2129–2132. [Google Scholar] [CrossRef] [PubMed]
- Arduini, A.; Bussolati, R.; Credi, A.; Faimani, G.; Garaudée, S.; Pochini, A.; Secchi, A.; Semeraro, M.; Silvi, S.; Venturi, M. Towards Controlling the Threading Direction of a Calix[6]arene Wheel by Using Nonsymmetric Axles. Chem. Eur. J. 2009, 15, 3230–3242. [Google Scholar] [CrossRef] [PubMed]
- Zanichelli, V.; Bazzoni, M.; Arduini, A.; Franchi, P.; Lucarini, M.; Ragazzon, G.; Secchi, A.; Silvi, S. Redox-switchable calix[6]arene-based isomeric rotaxanes. Chem. Eur. J. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Arduini, A.; Bussolati, R.; Credi, A.; Pochini, A.; Secchi, A.; Silvi, S.; Venturi, M. Rotaxanes with a calix[6]arene Wheel and Axles of Different Length. Synthesis, Characterization, and Photophysical and Electrochemical Properties. Tetrahedron 2008, 64, 8279–8286. [Google Scholar] [CrossRef]
- Arduini, A.; Calzavacca, F.; Pochini, A.; Secchi, A. Unidirectional Threading of triphenylureidocalix[6]arene-Based Wheels: Oriented Pseudorotaxane Synthesis. Chem. Eur. J. 2003, 9, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Ornelas, C.; Méry, D.; Cloutet, E.; Aranzaes, J.R.; Astruc, D. Cross Olefin Metathesis for the Selective Functionalization, Ferrocenylation, and Solubilization in Water of Olefin-Terminated Dendrimers, Polymers, and Gold Nanoparticles and for a Divergent Dendrimer Construction. J. Am. Chem. Soc. 2008, 130, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem. 1999, 20, 720–729. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and Analysis of Crystal Structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zanichelli, V.; Dallacasagrande, L.; Arduini, A.; Secchi, A.; Ragazzon, G.; Silvi, S.; Credi, A. Electrochemically Triggered Co-Conformational Switching in a [2]catenane Comprising a Non-Symmetric Calix[6]arene Wheel and a Two-Station Oriented Macrocycle. Molecules 2018, 23, 1156. https://doi.org/10.3390/molecules23051156
Zanichelli V, Dallacasagrande L, Arduini A, Secchi A, Ragazzon G, Silvi S, Credi A. Electrochemically Triggered Co-Conformational Switching in a [2]catenane Comprising a Non-Symmetric Calix[6]arene Wheel and a Two-Station Oriented Macrocycle. Molecules. 2018; 23(5):1156. https://doi.org/10.3390/molecules23051156
Chicago/Turabian StyleZanichelli, Valeria, Luca Dallacasagrande, Arturo Arduini, Andrea Secchi, Giulio Ragazzon, Serena Silvi, and Alberto Credi. 2018. "Electrochemically Triggered Co-Conformational Switching in a [2]catenane Comprising a Non-Symmetric Calix[6]arene Wheel and a Two-Station Oriented Macrocycle" Molecules 23, no. 5: 1156. https://doi.org/10.3390/molecules23051156
APA StyleZanichelli, V., Dallacasagrande, L., Arduini, A., Secchi, A., Ragazzon, G., Silvi, S., & Credi, A. (2018). Electrochemically Triggered Co-Conformational Switching in a [2]catenane Comprising a Non-Symmetric Calix[6]arene Wheel and a Two-Station Oriented Macrocycle. Molecules, 23(5), 1156. https://doi.org/10.3390/molecules23051156