The Double Face of Mucin-Type O-Glycans in Lectin-Mediated Infection and Immunity


{kind=link}
{kind=link}
{kind=link}
Abstract
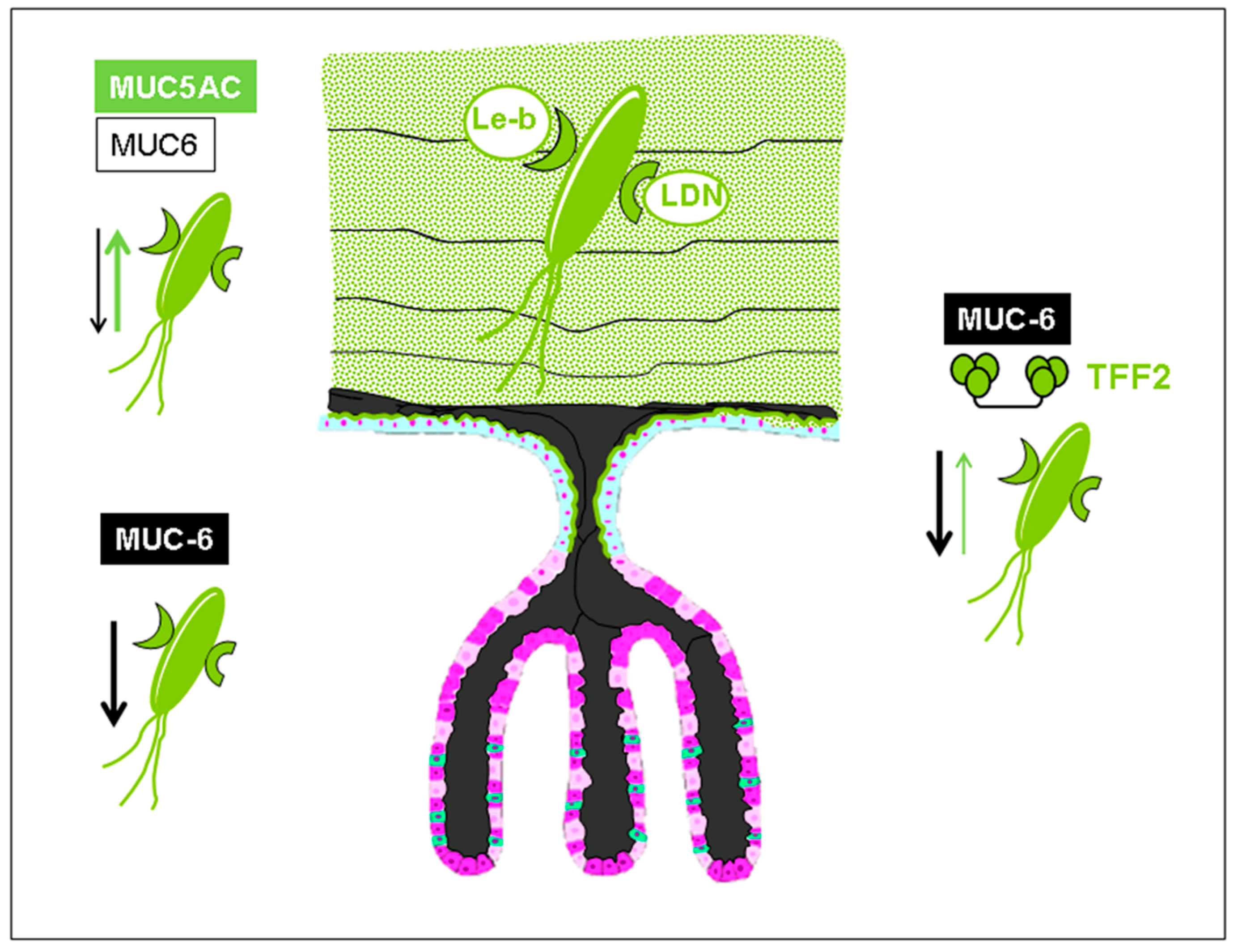
1. Trefoil Family Factor 2 (TFF2), a Lectin with Probiotic Activity?
2. The Lectin Domain of hTFF2
3. Lectin-Based Host–Pathogen Interactions in Helicobacter pylori Colonization and Infection
4. Adaption of the Helicobacter Lectin BabA to Host Glycome Polymorphisms
5. Mucin-Type O-Glycan Interactions in Viral Infections
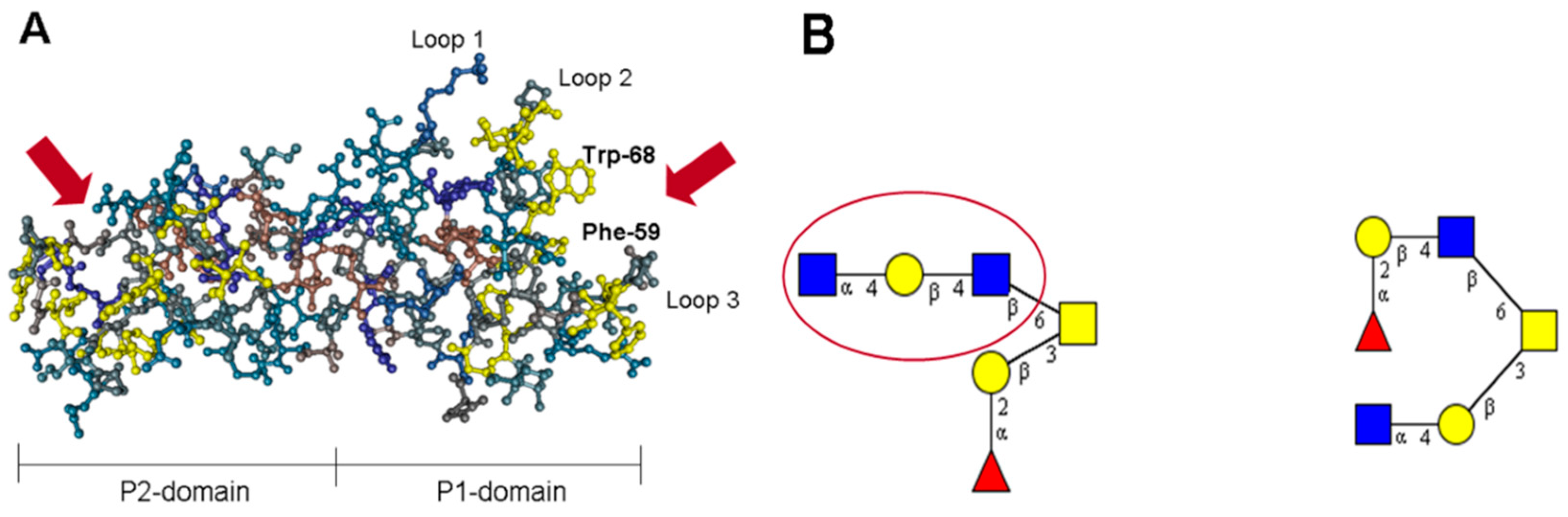
6. Norovirus Capsid Proteins Exhibit Lectin-Like Functions
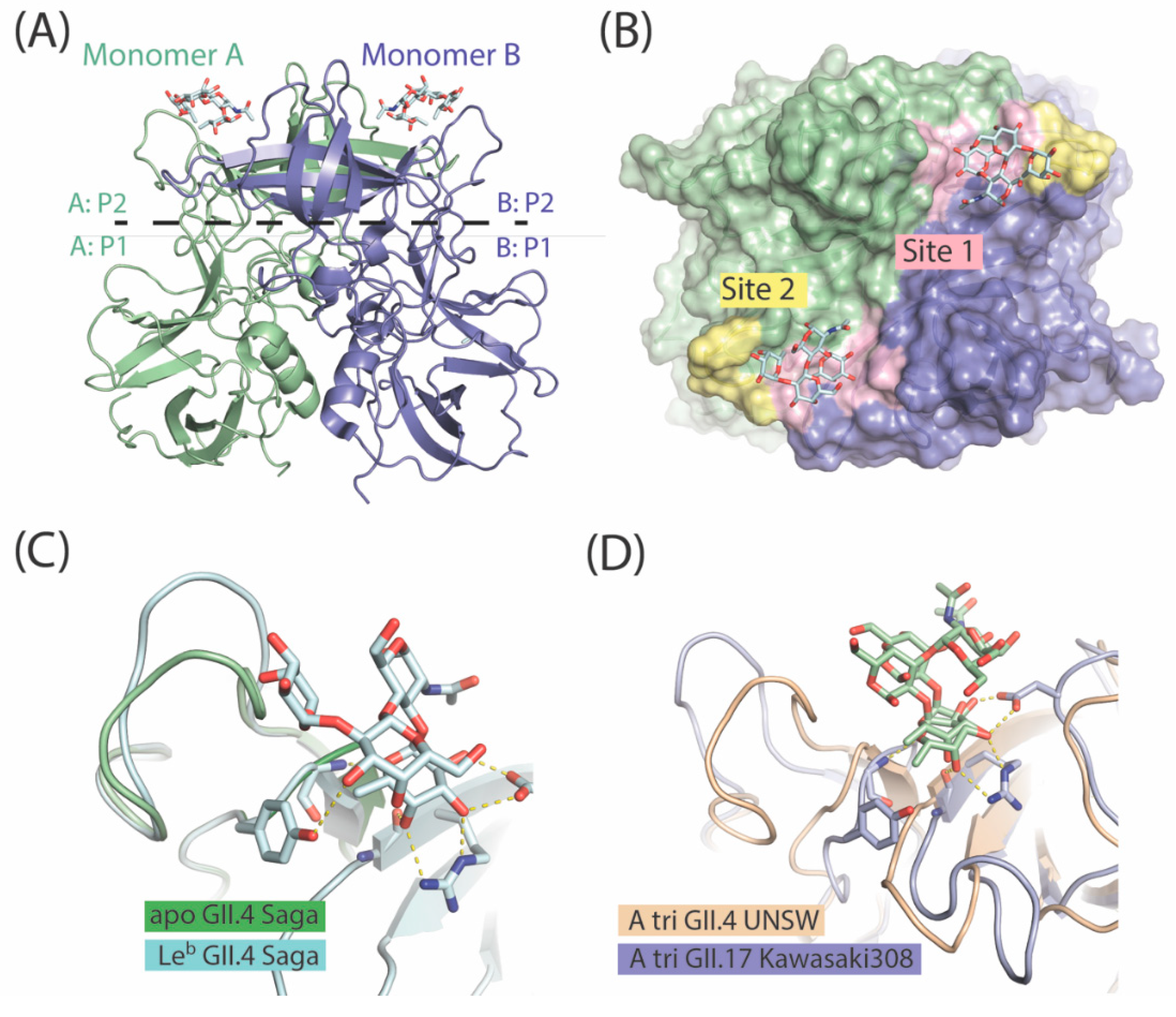
7. The P Domain of GII.4 Noroviruses
8. The P Domain of GII.17 Noroviruses
9. Specificity vs. Valency Aspects in Carbohydrate Recognition by Noroviral Lectins
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Pinho, S.S.; Carvalho, S.; Marcos-Pinto, R.; Magalhães, A.; Oliveira, C.; Gu, J.; Dinis-Ribeiro, M.; Carneiro, F.; Seruca, R.; Reis, C.A. Gastric cancer: Adding glycosylation to the equation. Trends Mol. Med. 2013, 19, 664–676. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.; Oertli, M.; Arnold, I.C. H. pylori exploits and manipulates innate and adaptive immune cell signaling pathways to establish persistent infection. Cell Commun. Signal. 2011, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Kawakubo, M.; Ito, Y.; Okimura, Y.; Kobayashi, M.; Sakura, K.; Kasama, S.; Fukuda, M.N.; Fukuda, M.; Katsuyama, T.; Nakayama, J. Natural antibiotic function of a human gastric mucin against Helicobacter pylori infection. Science 2004, 305, 1003–1006. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, B.; Marcos, N.T.; David, L.; Nakayama, J.; Reis, C.A. Terminal alpha1,4-linked N-acetylglucosamine in Helicobacter pylori-associated intestinal metaplasia of the human stomach and gastric carcinoma cell lines. J. Histochem. Cytochem. 2006, 54, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, W.; Jagla, W. Cell type specific expression of secretory TFF peptides: Colocalization with mucins and synthesis in the brain. In International Review of Cytology; Elsevier: New York City, NY, USA, 2002; Volume 213, pp. 147e–188e. [Google Scholar]
- Hanisch, F.-G.; Bonar, D.; Schloerer, N.; Schroten, H. Human trefoil factor 2 is a lectin that binds α-GlcNAc-capped mucin glycans with antibiotic activity against Helicobacter pylori. J. Biol. Chem. 2014, 289, 27363–27375. [Google Scholar] [CrossRef] [PubMed]
- Borkowski, J.; Schroten, H.; Hanisch, F.-G. Unpublished work.
- Ding, S.Z.; Zheng, P.Y. Helicobacter pylori infection induced gastric cancer; advance in gastric stem cell research and the remaining challenges. Gut Pathog. 2012, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.H.; Yang, Y.; Lam, S.K.; Wong, W.M.; Leung, S.Y.; Yuen, S.T.; Elia, G.; Wright, N.A.; Wong, B.C. Aberrant epithelial expression of trefoil family factor 2 and mucin 6 in Helicobacter pylori infected gastric antrum, incisura, and body and its association with antralisation. J. Clin. Pathol. 2004, 57, 861–866. [Google Scholar] [CrossRef] [PubMed]
- Kurt-Jones, E.A.; Cao, L.; Sandor, F.; Rogers, A.B.; Whary, M.T.; Nambiar, P.R.; Cerny, A.; Bowen, G.; Yan, J.; Takaishi, S. Trefoil family factor 2 is expressed in murine gastric and immune cells and controls both gastrointestinal inflammation and systemic immune responses. Infect. Immun. 2007, 75, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.G.; Rogers, A.B.; Whary, M.T.; Ge, Z.; Ohtani, M.; Jones, E.K.; Wang, T.C. Accelerated progression of gastritis to dysplasia in the pyloric antrum of TFF2−/− C57BL6× Sv129 Helicobacter pylori-infected mice. Am. J. Pathol. 2007, 171, 1520–1528. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, F.G.; Ragge, H.; Kalinski, T.; Meyer, F.; Kalbacher, H.; Hoffmann, W. Human gastric TFF2 peptide contains an N-linked fucosylated N,N′-diacetyllactosediamine (LacdiNAc) oligosaccharide. Glycobiology 2013, 23, 2–11. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gajhede, M.; Petersen, T.N.; Henriksen, A.; Petersen, J.F.; Dauter, Z.; Wilson, K.S.; Thim, L. Pancreatic spasmolytic polypeptide: First three-dimensional structure of a member of the mammalian trefoil family of peptides. Structure 1993, 1, 253–262. [Google Scholar] [CrossRef]
- Bonar, D.; Hanisch, F.G. Trefoil factor family domains represent highly efficient conformational determinants for N-linked N,N′-di-N-acetyllactosediamine (LacdiNAc) synthesis. J. Biol. Chem. 2014, 289, 29677–29690. [Google Scholar] [CrossRef] [PubMed]
- Suerbaum, S.; Michetti, P. Helicobacter pylori infection. N. Engl. J. Med. 2002, 347, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Xia, M.; Cao, S.; Huang, P.; Farkas, T.; Meller, J.; Hegde, R.S.; Li, X.; Rao, Z.; Jiang, X. Elucidation of strain-specific interaction of a GII-4 norovirus with HBGA receptors by site-directed mutagenesis study. Virology 2008, 379, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W.; Windhager, L.; Rohrer, S.; Zeiller, M.; Karnholz, A.; Hoffmann, R.; Zimmer, R.; Haas, R. Strain-specific genes of Helicobacter pylori: Genome evolution driven by a novel type IV secretion system and genomic island transfer. Nucleic Acids Res. 2010, 38, 6089–6101. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W. Assembly and molecular mode of action of the Helicobacter pylori Cag type IV secretion apparatus. FEBS J. 2011, 278, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, M. Helicobacter pylori CagA and gastric cancer: A paradigm for hit-and-run carcinogenesis. Cell Host Microbe 2014, 15, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Ilver, D.; Arnqvist, A.; Ogren, J.; Frick, I.M.; Kersulyte, D.; Incecik, E.T.; Berg, D.E.; Covacci, A.; Engstrand, L.; Boren, T. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science 1998, 279, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Mollicone, R.; Bara, J.; Le Pendu, J.; Oriol, R. Immunohistologic pattern of type 1 (Lea, Leb) and type 2 (X, Y, H) blood group-related antigens in the human pyloric and duodenal mucosae. Lab. Investig. 1985, 53, 219–227. [Google Scholar] [PubMed]
- Aspholm-Hurtig, M.; Dailide, G.; Lahmann, M.; Kalia, A.; Ilver, D.; Roche, N.; Vikstrom, S.; Sjostrom, R.; Linden, S.; Backstrom, A.; et al. Functional adaptation of BabA, the H. pylori ABO blood group antigen binding adhesin. Science 2004, 305, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, J.; Sonden, B.; Hurtig, M.; Olfat, F.O.; Forsberg, L.; Roche, N.; Angstrom, J.; Larsson, T.; Teneberg, S.; Karlsson, K.A.; et al. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science 2002, 297, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Hakomori, S. General concept of tumor-associated carbohydrate antigens: Their chemical, physical and enzymatic basis. Gangliosides Cancer 1989, 93–102. [Google Scholar]
- Rossez, Y.; Maes, E.; Lefebvre Darroman, T.; Gosset, P.; Ecobichon, C.; Joncquel Chevalier Curt, M.; Boneca, I.G.; Michalski, J.-C.; Robbe-Masselot, C. Almost all human gastric mucin O-glycans harbor blood group A, B or H antigens and are potential binding sites for Helicobacter pylori. Glycobiology 2012, 22, 1193–1206. [Google Scholar] [CrossRef] [PubMed]
- Madrid, J.F.; Ballesta, J.; Castells, M.T.; Hernandez, F. Glycoconjugate distribution in the human fundic mucosa revealed by lectin- and glycoprotein-gold cytochemistry. Histochemistry 1990, 95, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Magalhães, A.; Marcos-Pinto, R.; Nairn, A.V.; dela Rosa, M.; Ferreira, R.M.; Junqueira-Neto, S.; Freitas, D.; Gomes, J.; Oliveira, P.; Santos, M.R.; et al. Helicobacter pylori chronic infection and mucosal inflammation switches the human gastric glycosylation pathways. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2015, 1852, 1928–1939. [Google Scholar] [CrossRef] [PubMed]
- Marcos, N.T.; Magalhães, A.; Ferreira, B.; Oliveira, M.J.; Carvalho, A.S.; Mendes, N.; Gilmartin, T.; Head, S.R.; Figueiredo, C.; David, L.; et al. Helicobacter pylori induces β3GnT5 in human gastric cell lines, modulating expression of the SabA ligand sialyl–Lewis x. J. Clin. Investig. 2008, 118, 2325–2336. [Google Scholar] [CrossRef] [PubMed]
- Alper, J. Searching for Medicine’s Sweet Spot; American Association for the Advancement of Science: Washington, DC, USA, 2001. [Google Scholar]
- Rossez, Y.; Gosset, P.; Boneca, I.G.; Magalhaes, A.; Ecobichon, C.; Reis, C.A.; Cieniewski-Bernard, C.; Joncquel Chevalier Curt, M.; Leonard, R.; Maes, E.; et al. The lacdiNAc-specific adhesin LabA mediates adhesion of Helicobacter pylori to human gastric mucosa. J. Infect. Dis. 2014, 210, 1286–1295. [Google Scholar] [CrossRef] [PubMed]
- Backert, S.; Clyne, M.; Tegtmeyer, N. Molecular mechanisms of gastric epithelial cell adhesion and injection of CagA by Helicobacter pylori. Cell Commun. Signal. 2011, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Moonens, K.; Remaut, H. Evolution and structural dynamics of bacterial glycan binding adhesins. Curr. Opin. Struct. Biol. 2017, 44, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Moonens, K.; Gideonsson, P.; Subedi, S.; Bugaytsova, J.; Romaõ, E.; Mendez, M.; Nordén, J.; Fallah, M.; Rakhimova, L.; Shevtsova, A. Structural insights into polymorphic ABO glycan binding by Helicobacter pylori. Cell Host Microbe 2016, 19, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Heggelund, J.E.; Varrot, A.; Imberty, A.; Krengel, U. Histo-blood group antigens as mediators of infections. Curr. Opin. Struct. Biol. 2017, 44, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Nell, S.; Kennemann, L.; Schwarz, S.; Josenhans, C.; Suerbaum, S. Dynamics of Lewis b binding and sequence variation of the babA adhesin gene during chronic Helicobacter pylori infection in humans. MBio 2014, 5, e02281-14. [Google Scholar] [CrossRef] [PubMed]
- Bugaytsova, J.A.; Bjornham, O.; Chernov, Y.A.; Gideonsson, P.; Henriksson, S.; Mendez, M.; Sjostrom, R.; Mahdavi, J.; Shevtsova, A.; Ilver, D.; et al. Helicobacter pylori Adapts to Chronic Infection and Gastric Disease via pH-Responsive BabA-Mediated Adherence. Cell Host Microbe 2017, 21, 376–389. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, M. A Sour Relationship between BabA and Lewis b. Cell Host Microbe 2017, 21, 318–320. [Google Scholar] [CrossRef] [PubMed]
- Tassaneetrithep, B.; Burgess, T.H.; Granelli-Piperno, A.; Trumpfheller, C.; Finke, J.; Sun, W.; Eller, M.A.; Pattanapanyasat, K.; Sarasombath, S.; Birx, D.L. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J. Exp. Med. 2003, 197, 823–829. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, C.; Manya, H.; Ouellet, M.; Clark, G.F.; Endo, T.; Tremblay, M.J.; Sato, S. Host-soluble galectin-1 promotes HIV-1 replication through a direct interaction with glycans of viral gp120 and host CD4. J. Virol. 2011, 85, 11742–11751. [Google Scholar] [CrossRef] [PubMed]
- Geijtenbeek, T.B.; Kwon, D.S.; Torensma, R.; van Vliet, S.J.; van Duijnhoven, G.C.; Middel, J.; Cornelissen, I.L.; Nottet, H.S.; KewalRamani, V.N.; Littman, D.R. DC-SIGN, a dendritic cell–specific HIV-1-binding protein that enhances trans-infection of T cells. Cell 2000, 100, 587–597. [Google Scholar] [CrossRef]
- Bok, K.; Green, K.Y. Norovirus gastroenteritis in immunocompromised patients. N. Engl. J. Med. 2012, 367, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Doerflinger, S.Y.; Weichert, S.; Koromyslova, A.; Chan, M.; Schwerk, C.; Adam, R.; Jennewein, S.; Hansman, G.S.; Schroten, H. Human Norovirus Evolution in a Chronically Infected Host. mSphere 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Glass, R.I.; Parashar, U.D.; Estes, M.K. Norovirus gastroenteritis. N. Engl. J. Med. 2009, 361, 1776–1785. [Google Scholar] [CrossRef] [PubMed]
- Prasad, B.V.; Hardy, M.E.; Dokland, T.; Bella, J.; Rossmann, M.G.; Estes, M.K. X-ray crystallographic structure of the Norwalk virus capsid. Science 1999, 286, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti-Ciarlet, A.; White, L.J.; Chen, R.; Prasad, B.V.; Estes, M.K. Structural requirements for the assembly of Norwalk virus-like particles. J. Virol. 2002, 76, 4044–4055. [Google Scholar] [CrossRef] [PubMed]
- Vinje, J. Advances in laboratory methods for detection and typing of norovirus. J. Clin. Microbiol. 2015, 53, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Siebenga, J.J.; Vennema, H.; Renckens, B.; de Bruin, E.; van der Veer, B.; Siezen, R.J.; Koopmans, M. Epochal evolution of GGII.4 norovirus capsid proteins from 1995 to 2006. J. Virol. 2007, 81, 9932–9941. [Google Scholar] [CrossRef] [PubMed]
- Hansman, G.S.; Biertumpfel, C.; Georgiev, I.; McLellan, J.S.; Chen, L.; Zhou, T.; Katayama, K.; Kwong, P.D. Crystal structures of GII.10 and GII.12 norovirus protruding domains in complex with histo-blood group antigens reveal details for a potential site of vulnerability. J. Virol. 2011, 85, 6687–6701. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Farkas, T.; Zhong, W.; Tan, M.; Thornton, S.; Morrow, A.L.; Jiang, X. Norovirus and histo-blood group antigens: Demonstration of a wide spectrum of strain specificities and classification of two major binding groups among multiple binding patterns. J. Virol. 2005, 79, 6714–6722. [Google Scholar] [CrossRef] [PubMed]
- Taube, S.; Rubin, J.R.; Katpally, U.; Smith, T.J.; Kendall, A.; Stuckey, J.A.; Wobus, C.E. High-resolution x-ray structure and functional analysis of the murine norovirus 1 capsid protein protruding domain. J. Virol. 2010, 84, 5695–5705. [Google Scholar] [CrossRef] [PubMed]
- Morozov, V.; Hansman, G.; Hanisch, F.G.; Schroten, H.; Kunz, C. Human Milk Oligosaccharides as Promising Antivirals. Mol. Nutr. Food Res. 2018, 62, e1700679. [Google Scholar] [CrossRef] [PubMed]
- Schroten, H.; Hanisch, F.G.; Hansman, G.S. Human Norovirus Interactions with Histo-Blood Group Antigens and Human Milk Oligosaccharides. J. Virol. 2016, 90, 5855–5859. [Google Scholar] [CrossRef] [PubMed]
- Ettayebi, K.; Crawford, S.E.; Murakami, K.; Broughman, J.R.; Karandikar, U.; Tenge, V.R.; Neill, F.H.; Blutt, S.E.; Zeng, X.L.; Qu, L.; et al. Replication of human noroviruses in stem cell-derived human enteroids. Science 2016, 353, 1387–1393. [Google Scholar] [CrossRef] [PubMed]
- Lopman, B.A.; Trivedi, T.; Vicuna, Y.; Costantini, V.; Collins, N.; Gregoricus, N.; Parashar, U.; Sandoval, C.; Broncano, N.; Vaca, M.; et al. Norovirus Infection and Disease in an Ecuadorian Birth Cohort: Association of Certain Norovirus Genotypes With Host FUT2 Secretor Status. J. Infect. Dis. 2015, 211, 1813–1821. [Google Scholar] [CrossRef] [PubMed]
- Larsson, M.M.; Rydell, G.E.; Grahn, A.; Rodriguez-Diaz, J.; Akerlind, B.; Hutson, A.M.; Estes, M.K.; Larson, G.; Svensson, L. Antibody prevalence and titer to norovirus (genogroup II) correlate with secretor (FUT2) but not with ABO phenotype or Lewis (FUT3) genotype. J. Infect. Dis. 2006, 194, 1422–1427. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.K.; Watanabe, M.; Zhu, S.; Graves, C.L.; Keyes, L.R.; Grau, K.R.; Gonzalez-Hernandez, M.B.; Iovine, N.M.; Wobus, C.E.; Vinje, J.; et al. Enteric bacteria promote human and mouse norovirus infection of B cells. Science 2014, 346, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Bok, K.; Abente, E.J.; Realpe-Quintero, M.; Mitra, T.; Sosnovtsev, S.V.; Kapikian, A.Z.; Green, K.Y. Evolutionary dynamics of GII.4 noroviruses over a 34-year period. J. Virol. 2009, 83, 11890–11901. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.C.; Lee, N.; Hung, T.N.; Kwok, K.; Cheung, K.; Tin, E.K.; Lai, R.W.; Nelson, E.A.; Leung, T.F.; Chan, P.K. Rapid emergence and predominance of a broadly recognizing and fast-evolving norovirus GII.17 variant in late 2014. Nat. Commun. 2015, 6, 10061. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.F.; Huang, Q.; Long, Y.; Jiang, X.; Zhang, T.; Tan, M.; Zhang, Q.L.; Huang, Z.Y.; Li, Y.H.; Ding, Y.Q.; et al. An outbreak caused by GII.17 norovirus with a wide spectrum of HBGA-associated susceptibility. Sci. Rep. 2015, 5, 17687. [Google Scholar] [CrossRef] [PubMed]
- Koromyslova, A.; Tripathi, S.; Morozov, V.; Schroten, H.; Hansman, G.S. Human norovirus inhibition by a human milk oligosaccharide. Virology 2017, 508, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Noel, J.; Fankhauser, R.; Ando, T.; Monroe, S.; Glass, R. Identification of a distinct common strain of “Norwalk-like viruses” having a global distribution. J. Infect. Dis. 1999, 179, 1334–1344. [Google Scholar] [CrossRef] [PubMed]
- Widdowson, M.-A.; Cramer, E.H.; Hadley, L.; Bresee, J.S.; Beard, R.S.; Bulens, S.N.; Charles, M.; Chege, W.; Isakbaeva, E.; Wright, J.G. Outbreaks of acute gastroenteritis on cruise ships and on land: Identification of a predominant circulating strain of norovirus—United States, 2002. J. Infect. Dis. 2004, 190, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Bull, R.A.; Tu, E.T.; McIver, C.J.; Rawlinson, W.D.; White, P.A. Emergence of a new norovirus genotype II.4 variant associated with global outbreaks of gastroenteritis. J. Clin. Microbiol. 2006, 44, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Eden, J.S.; Bull, R.A.; Tu, E.; McIver, C.J.; Lyon, M.J.; Marshall, J.A.; Smith, D.W.; Musto, J.; Rawlinson, W.D.; White, P.A. Norovirus GII.4 variant 2006b caused epidemics of acute gastroenteritis in Australia during 2007 and 2008. J. Clin. Virol. 2010, 49, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.; Wikswo, M.E.; Lopman, B.A.; Vinje, J.; Parashar, U.D.; Hall, A.J. Impact of an emergent norovirus variant in 2009 on norovirus outbreak activity in the United States. Clin. Infect. Dis. 2011, 53, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Eden, J.S.; Hewitt, J.; Lim, K.L.; Boni, M.F.; Merif, J.; Greening, G.; Ratcliff, R.M.; Holmes, E.C.; Tanaka, M.M.; Rawlinson, W.D.; et al. The emergence and evolution of the novel epidemic norovirus GII.4 variant Sydney 2012. Virology 2014, 450–451, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Debbink, K.; Lindesmith, L.C.; Donaldson, E.F.; Costantini, V.; Beltramello, M.; Corti, D.; Swanstrom, J.; Lanzavecchia, A.; Vinje, J.; Baric, R.S. Emergence of new pandemic GII.4 Sydney norovirus strain correlates with escape from herd immunity. J. Infect. Dis. 2013, 208, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Eden, J.-S.; Tanaka, M.M.; Boni, M.F.; Rawlinson, W.D.; White, P.A. Recombination within the pandemic norovirus GII.4 lineage. J. Virol. 2013, 87, 6270–6282. [Google Scholar] [CrossRef] [PubMed]
- De Rougemont, A.; Ruvoen-Clouet, N.; Simon, B.; Estienney, M.; Elie-Caille, C.; Aho, S.; Pothier, P.; Le Pendu, J.; Boireau, W.; Belliot, G. Qualitative and quantitative analysis of the binding of GII.4 norovirus variants onto human blood group antigens. J. Virol. 2011, 85, 4057–4070. [Google Scholar] [CrossRef] [PubMed]
- Lindesmith, L.C.; Donaldson, E.F.; Lobue, A.D.; Cannon, J.L.; Zheng, D.P.; Vinje, J.; Baric, R.S. Mechanisms of GII.4 norovirus persistence in human populations. PLoS Med. 2008, 5, e31. [Google Scholar] [CrossRef] [PubMed]
- Shanker, S.; Czako, R.; Sankaran, B.; Atmar, R.L.; Estes, M.K.; Prasad, B.V. Structural analysis of determinants of histo-blood group antigen binding specificity in genogroup I noroviruses. J. Virol. 2014, 88, 6168–6180. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.K.; Leuthold, M.M.; Hansman, G.S. Human noroviruses’ fondness for histo-blood group antigens. J. Virol. 2015, 89, 2024–2040. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Lou, Z.; Tan, M.; Chen, Y.; Liu, Y.; Zhang, Z.; Zhang, X.C.; Jiang, X.; Li, X.; Rao, Z. Structural basis for the recognition of blood group trisaccharides by norovirus. J. Virol. 2007, 81, 5949–5957. [Google Scholar] [CrossRef] [PubMed]
- Shanker, S.; Choi, J.M.; Sankaran, B.; Atmar, R.L.; Estes, M.K.; Prasad, B.V. Structural analysis of histo-blood group antigen binding specificity in a norovirus GII.4 epidemic variant: Implications for epochal evolution. J. Virol. 2011, 85, 8635–8645. [Google Scholar] [CrossRef] [PubMed]
- Koromyslova, A.; Morozov, V.; Hefele, L.; Hansman, G. The norovirus histo-blood group antigen pocket could be protected from temporal changes in antigenicity. Unpublished work.
- Fiege, B.; Leuthold, M.; Parra, F.; Dalton, K.P.; Meloncelli, P.J.; Lowary, T.L.; Peters, T. Epitope mapping of histo blood group antigens bound to norovirus VLPs using STD NMR experiments reveals fine details of molecular recognition. Glycoconj. J. 2017, 34, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Shirato, H.; Ogawa, S.; Ito, H.; Sato, T.; Kameyama, A.; Narimatsu, H.; Xiaofan, Z.; Miyamura, T.; Wakita, T.; Ishii, K.; et al. Noroviruses distinguish between type 1 and type 2 histo-blood group antigens for binding. J. Virol. 2008, 82, 10756–10767. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, F.G.; Hansman, G.S.; Morozov, V.; Kunz, C.; Schroten, H. Type 1 blood group h and lewis-b antigens and their valency on high-mass human milk oligosaccharides are potent norovirus binding targets. Unpublished work.
- Nasir, W.; Frank, M.; Kunze, A.; Bally, M.; Parra, F.; Nyholm, P.G.; Höök, F.; Larson, G. Histo-Blood Group Antigen Presentation Is Critical for Binding of Norovirus VLP to Glycosphingolipids in Model Membranes. ACS Chem. Biol. 2017, 12, 1288–1296. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, Y.; Ishikawa, M.; Shimizu, T.; Komane, A.; Kasuo, S.; Shinohara, M.; Nagasawa, K.; Kimura, H.; Ryo, A.; Okabe, N.; et al. Genetic analyses of GII.17 norovirus strains in diarrheal disease outbreaks from December 2014 to March 2015 in Japan reveal a novel polymerase sequence and amino acid substitutions in the capsid region. Eurosurveillance 2015, 20, 21173. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Feng, Y.; Chen, S.Y.; Tsai, C.N.; Lai, M.W.; Chiu, C.H. Emerging norovirus GII.17 in Taiwan. Clin. Infect. Dis. 2015, 61, 1762–1764. [Google Scholar] [PubMed]
- Fu, J.; Ai, J.; Jin, M.; Jiang, C.; Zhang, J.; Shi, C.; Lin, Q.; Yuan, Z.; Qi, X.; Bao, C.; et al. Emergence of a new GII.17 norovirus variant in patients with acute gastroenteritis in Jiangsu, China, September 2014 to March 2015. Eurosurveillance 2015, 20, 21157. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.K.; Koromyslova, A.; Hefele, L.; Gurth, C.; Hansman, G.S. Structural Evolution of the Emerging 2014–2015 GII.17 Noroviruses. J. Virol. 2015, 90, 2710–2715. [Google Scholar] [CrossRef] [PubMed]
- Weichert, S.; Koromyslova, A.; Singh, B.K.; Hansman, S.; Jennewein, S.; Schroten, H.; Hansman, G.S. Structural Basis for Norovirus Inhibition by Human Milk Oligosaccharides. J. Virol. 2016, 90, 4843–4848. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Lu, F.; Wei, X.; Zhao, R. Fucoidan: Structure and bioactivity. Molecules 2008, 13, 1671–1695. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morozov, V.; Borkowski, J.; Hanisch, F.-G. The Double Face of Mucin-Type O-Glycans in Lectin-Mediated Infection and Immunity. Molecules 2018, 23, 1151. https://doi.org/10.3390/molecules23051151
Morozov V, Borkowski J, Hanisch F-G. The Double Face of Mucin-Type O-Glycans in Lectin-Mediated Infection and Immunity. Molecules. 2018; 23(5):1151. https://doi.org/10.3390/molecules23051151
Chicago/Turabian StyleMorozov, Vasily, Julia Borkowski, and Franz-Georg Hanisch. 2018. "The Double Face of Mucin-Type O-Glycans in Lectin-Mediated Infection and Immunity" Molecules 23, no. 5: 1151. https://doi.org/10.3390/molecules23051151
APA StyleMorozov, V., Borkowski, J., & Hanisch, F.-G. (2018). The Double Face of Mucin-Type O-Glycans in Lectin-Mediated Infection and Immunity. Molecules, 23(5), 1151. https://doi.org/10.3390/molecules23051151