A 3D-QSAR Study on the Antitrypanosomal and Cytotoxic Activities of Steroid Alkaloids by Comparative Molecular Field Analysis

 , and
, and 
Abstract
:1. Introduction
2. Results and Discussion
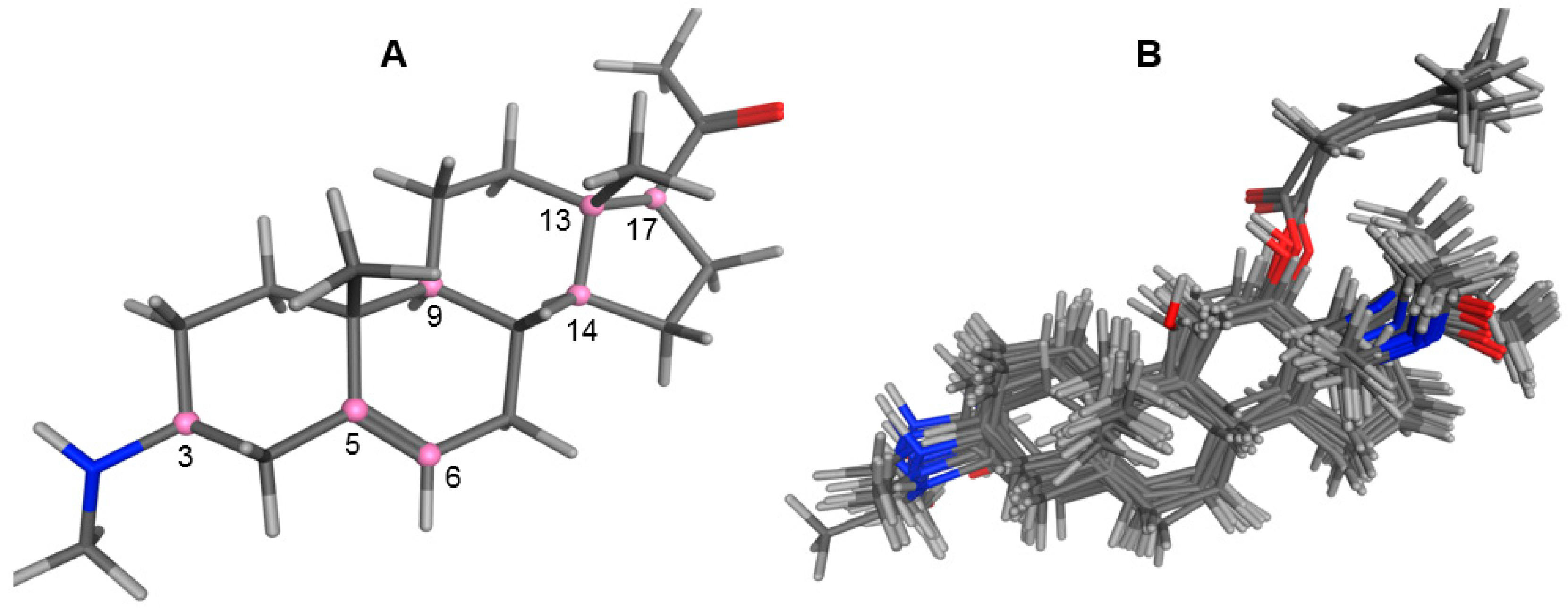
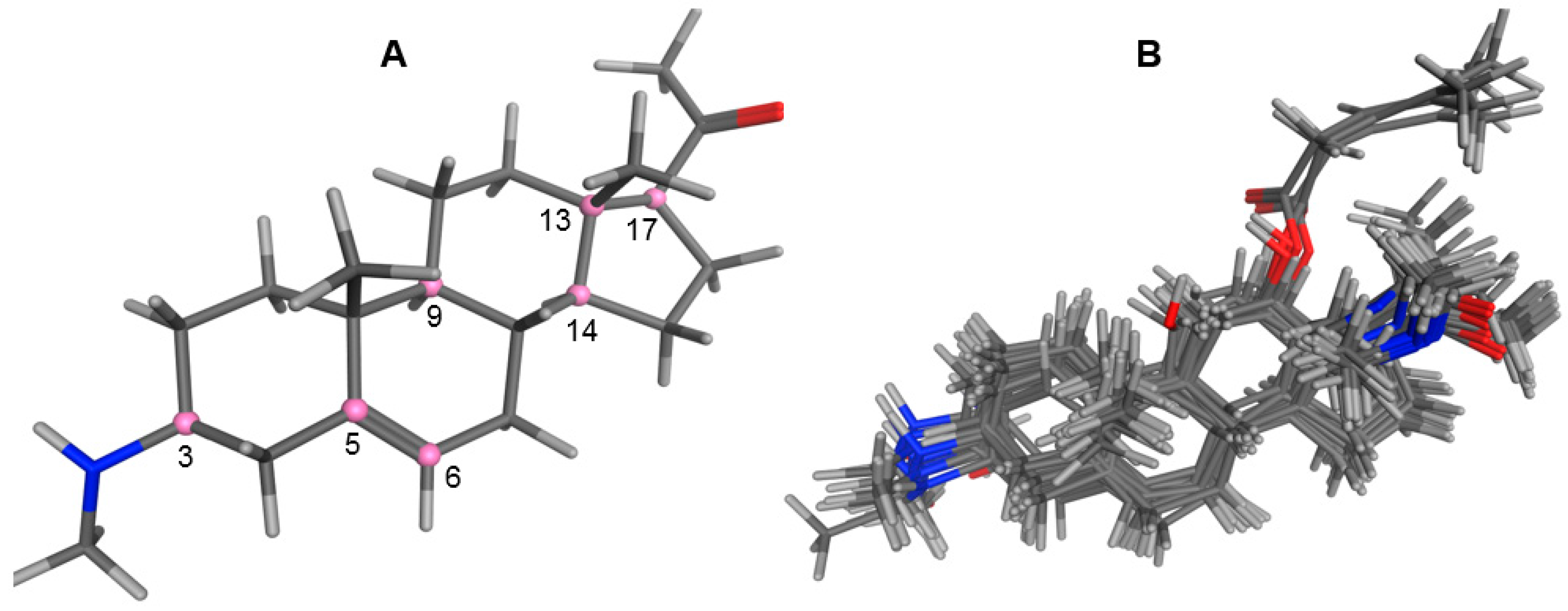
2.1. Modeling and Alignment of Molecular Structures
2.2. CoMFA Modelling
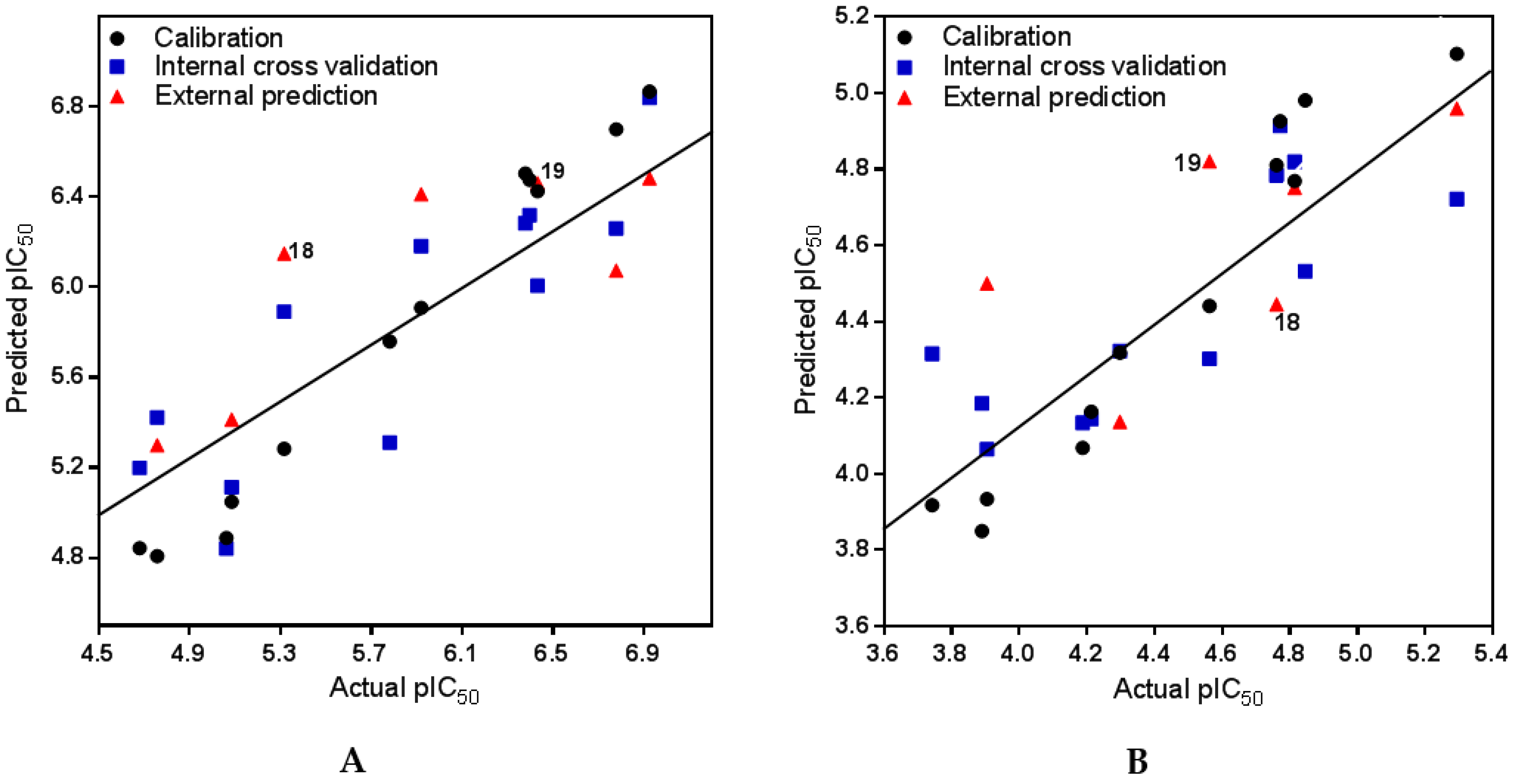
2.2.1. PLS and Model Statistics for Anti-Tbr Activities of Steroid Alkaloids
2.2.2. PLS and Model Statistics for L6 Cytotoxic Activities of Steroid Alkaloids
2.3. Analysis of the CoMFA Contour Maps
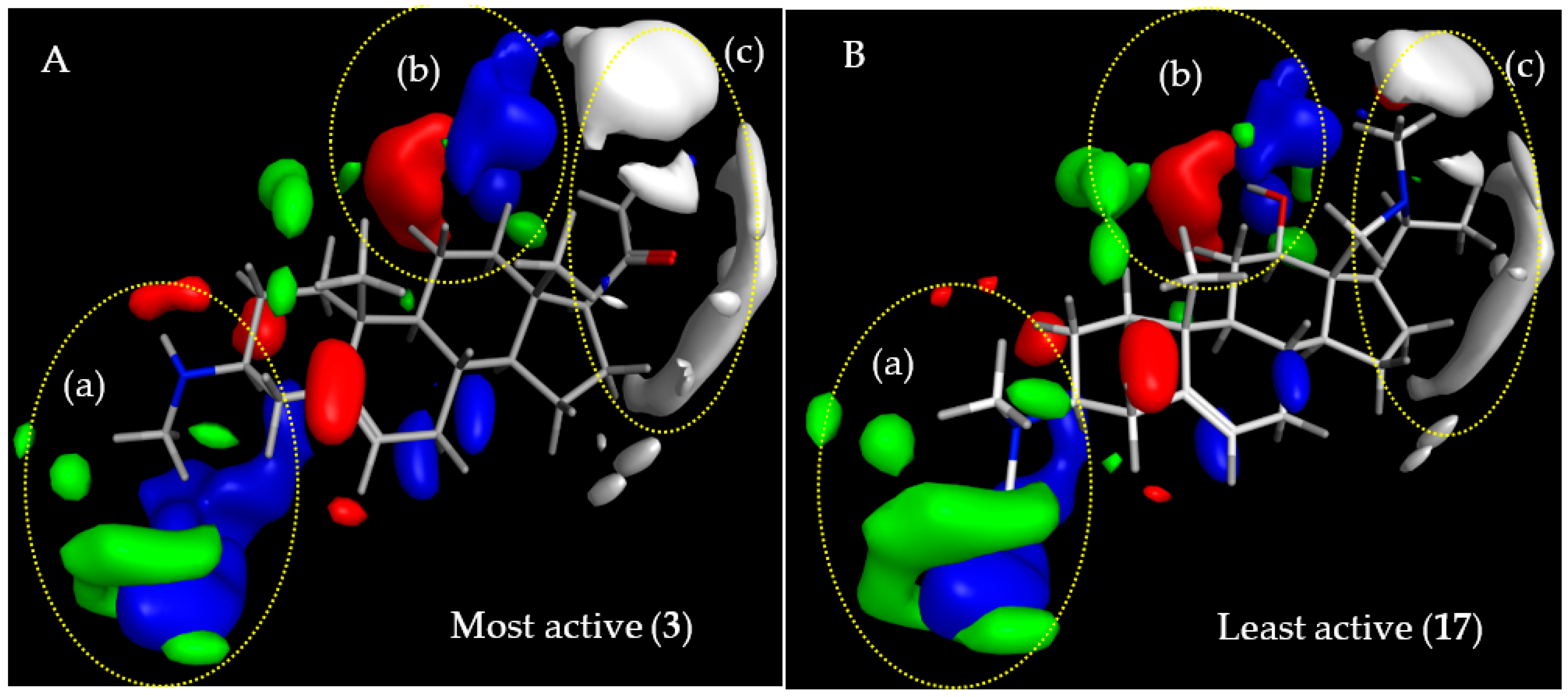
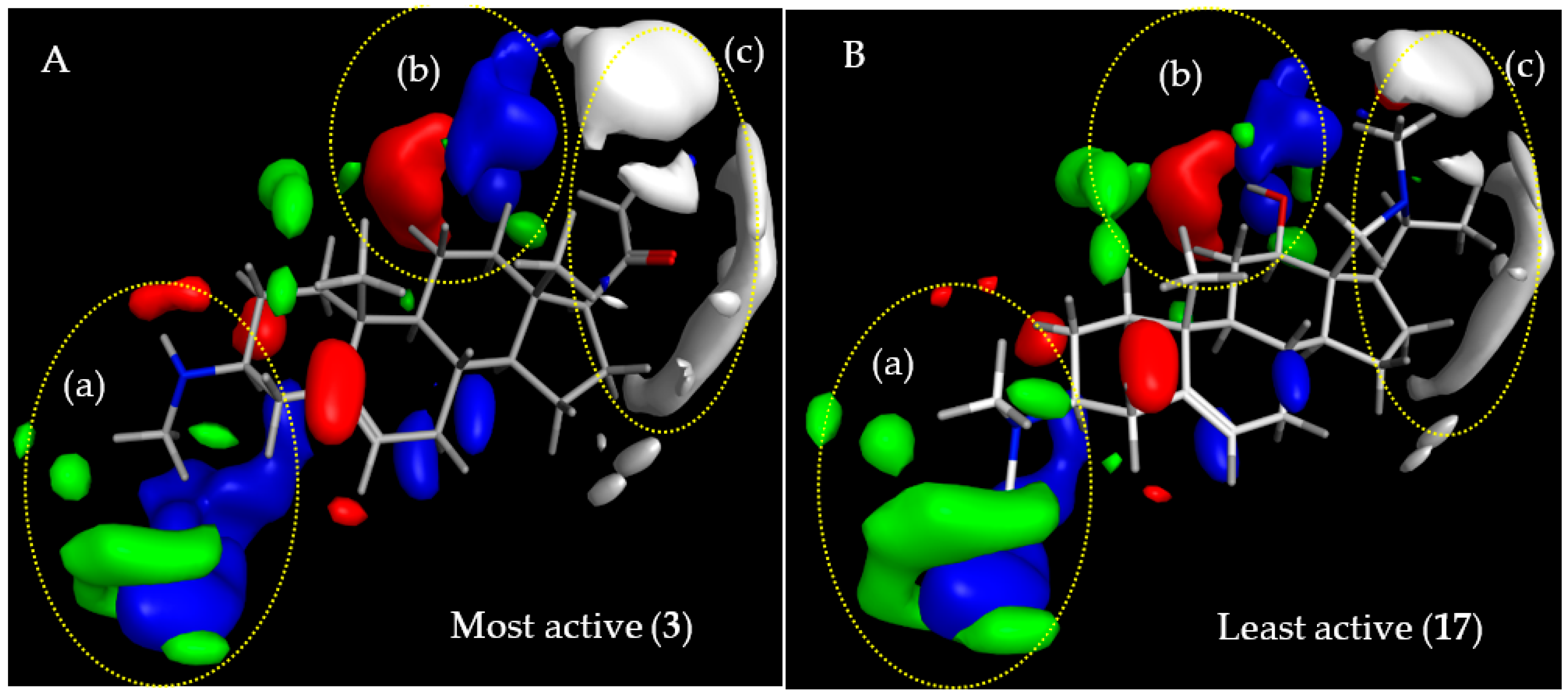
2.3.1. CoMFA Model for Antitrypanosomal Activity of Steroid Alkaloids
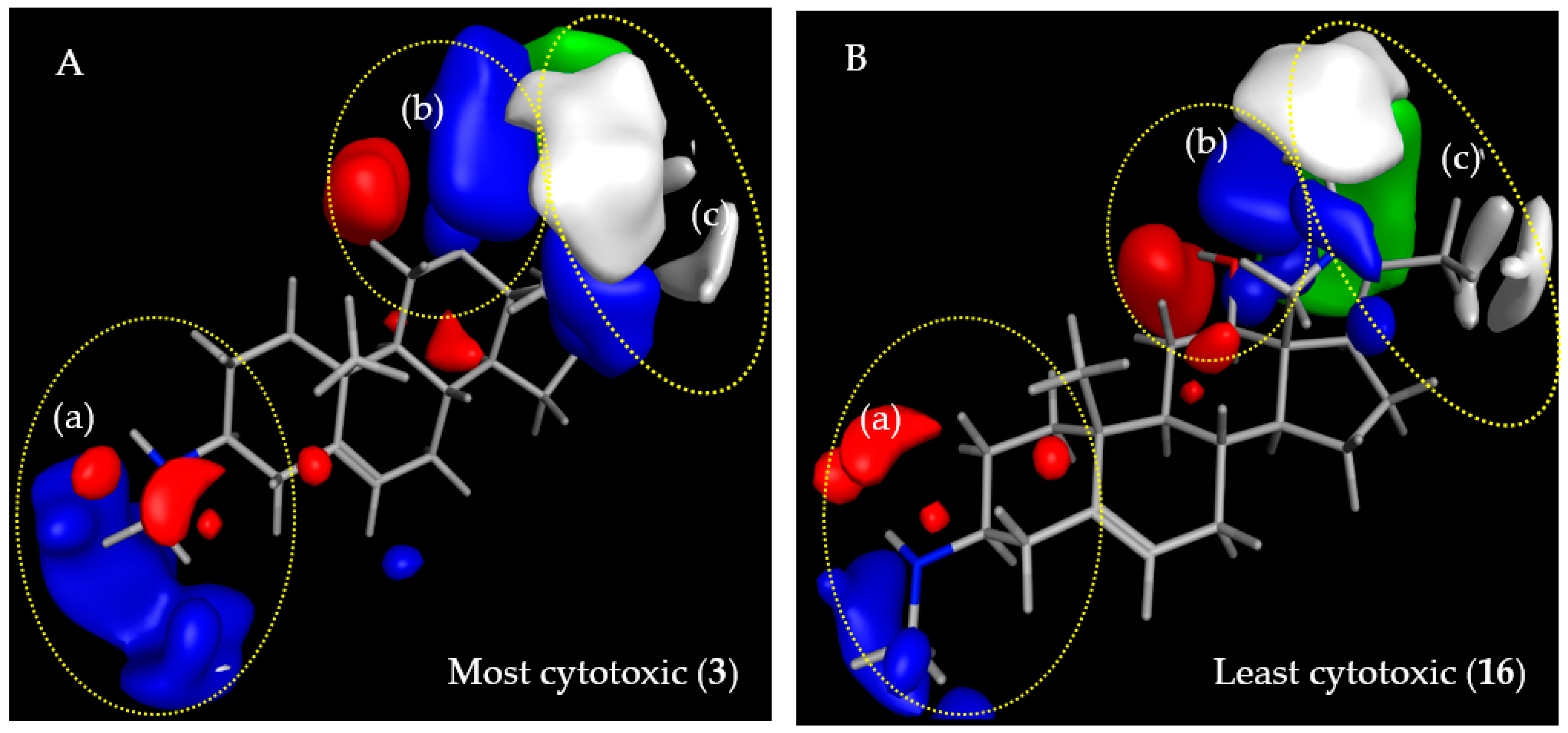
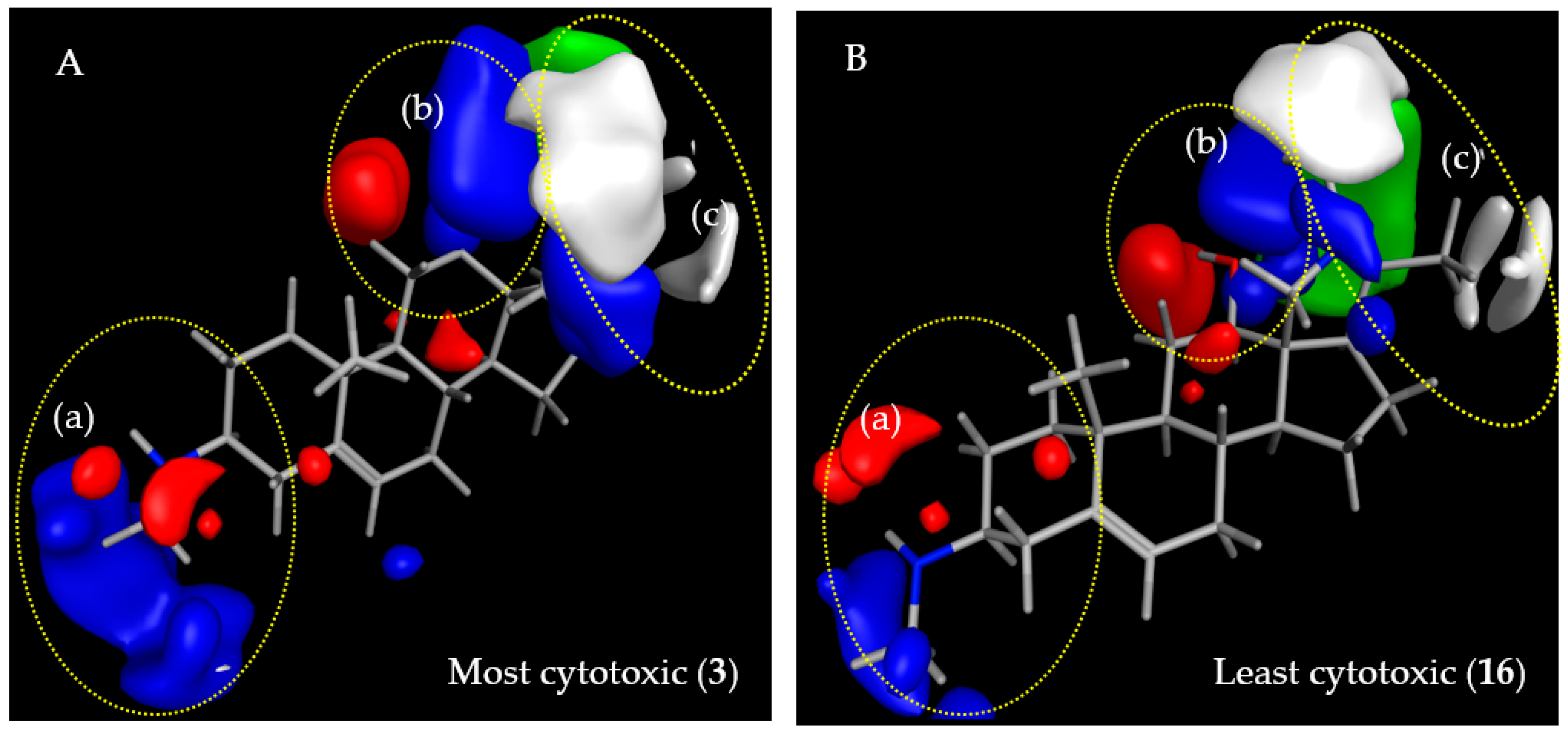
2.3.2. CoMFA Model for L6 Cytotoxic Activity of Steroid Alkaloids
2.3.3. Comparison of CoMFA Models and Considerations on Antitrypanosomal Selectivity of Steroid Alkaloids
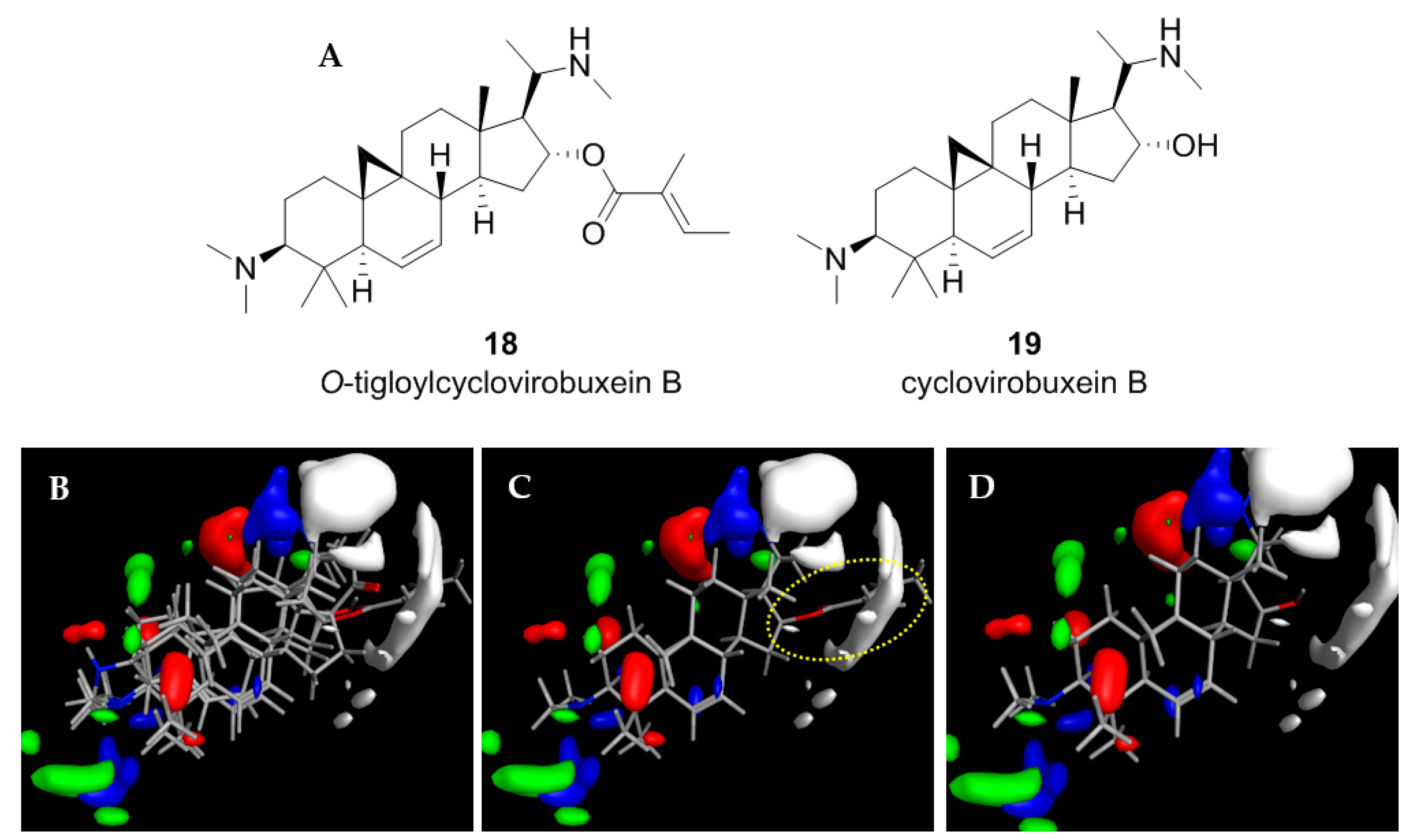
2.4. Application of the CoMFA Model to Cycloartenoid Alkaloids from Buxus Sempervirens
3. Materials and Methods
3.1. Data Set
3.2. Building of Molecular Models
3.3. Alignment Procedure
3.4. Comparative Molecular Field Analysis (CoMFA)
3.4.1. Data Pretreatment
3.4.2. PLS Regression and Model Validation
3.5. Contour Mapping of Steric and Electrostatic Fields
3.6. Isolation, Characterization, and Biological Testing of Cyclovirobuxein B (19)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nnadi, C.O.; Nwodo, N.J.; Kaiser, M.; Brun, R.; Schmidt, T.J. Steroid Alkaloids from Holarrhena africana with Strong Activity against Trypanosoma brucei rhodesiense. Molecules 2017, 22, 1129. [Google Scholar] [CrossRef] [PubMed]
- Babu, S.; Sohn, H.; Madhavan, T. Computational analysis of CRTh2 receptor antagonist: A ligand-based CoMFA and CoMSIA approach. Comput. Biol. Chem. 2015, 56, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Lill, M.A. Multi-dimensional QSAR in drug discovery. Drug Discov. Today 2007, 12, 1013–1017. [Google Scholar] [CrossRef] [PubMed]
- Wellsow, J.; Machulla, H.J.; Kovar, K.A. 3D QSAR of serotonin transporter ligands: CoMFA and CoMSIA studies. Quant. Struct. Act. Relat. 2002, 21, 577–589. [Google Scholar] [CrossRef]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecular field analysis (CoMFA). 1. Effects of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef] [PubMed]
- Cook, C.E.; Wani, M.C.; Lee, Y.W.; Fail, P.A.; Petrow, V. Reversal of activity profile in analogs of the antiprogestin RU 486: Effect of a 16α-substituent on progestational (agonist) activity. Life Sci. 1993, 52, 155–162. [Google Scholar] [CrossRef]
- Polanski, J.; Gieleciak, R.; Bak, A. Probability issues in molecular design: Predictive and modeling ability in 3D-QSAR schemes. Comb. Chem. High Throughput Screen. 2004, 7, 793–807. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Li, Q.; Yao, X.; Fan, B.; Yuan, S.; Panaye, A.; Doucet, J.P. 3D-QSAR and Docking Study of the Binding Mode of Steroids to Progesterone Receptor in Active Site. Mol. Inf. 2003, 22, 604–613. [Google Scholar] [CrossRef]
- Kapou, A.; Benetis, N.P.; Durdagi, S.; Nikolaropoulos, S.; Mavromoustakos, T. 3D QSAR/CoMFA and CoMSIA studies on antileukemic steroidal esters coupled with conformationally flexible nitrogen mustards. J. Chem. Inf. Model. 2008, 48, 2254–2264. [Google Scholar] [CrossRef] [PubMed]
- Loughney, D.A.; Schwender, C.F. A comparison of progestin and androgen receptor binding using the CoMFA technique. J. Comput.-Aided Mol. Des. 1992, 6, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Norinder, U. Experimental design based 3D QSAR analysis of steroid-protein interactions: Application to human CBG complexes. J. Comput.-Aided Mol. Des. 1990, 4, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Norinder, U. 3-D QSAR analysis of steroid/protein interactions: The use of difference maps. J. Comput.-Aided Mol. Des. 1991, 5, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Norinder, U. 3D-QSAR investigation of the tripos benchmark steroids and some protein-tyrosine kinase inhibitors of styrene type using the TDQ approach. J. Chemom. 1996, 10, 533–545. [Google Scholar] [CrossRef]
- Chemical Computing Group. Molecular Operating Environment (MOE) rel. 2011.10; Chemical Computing Group Inc.: Montreal, QC, Canada, 2014. [Google Scholar]
- Tosco, P.; Balle, T. Open3DQSAR: A new open-source software aimed at high throughput chemometric analysis of molecular interaction fields. J. Mol. Model. 2011, 17, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Althaus, J.B.; Jerz, G.; Winterhalter, P.; Kaiser, M.; Brun, R.; Schmidt, T.J. Antiprotozoal activity of Buxus sempervirens and activity-guided isolation of O-tigloylcyclovirobuxeine-B as the main constituent active against Plasmodium falciparum. Molecules 2014, 19, 6184–6201. [Google Scholar] [CrossRef] [PubMed]
- Althaus, J.B. Natural Products with Antiprotozoal Activity: Alkamides Isolated from Asteraceae and Triterpene Alkaloids from Buxus sempervirens L. Doctoral Thesis (Dr. rer. nat.), IPBP, University of Münster, Münster, Germany, 2015. [Google Scholar]
- Martin, Y.C.; Willett, P.; Heller, S.R. Designing Bioactive Molecules: Three-Dimensional Techniques and Applications (Computer Applications in Chemistry Collection), 1st ed.; American Chemical Society: Washington, DC, USA, 1998; pp. 181–194. ISBN 9780841234901. [Google Scholar]
- Kupchan, S.M.; Ohta, G. Buxus alkaloids. X. The Isolation and Constitution of Cyclovirobuxeine-B. J. Org. Chem. 1966, 31, 608–610. [Google Scholar] [CrossRef]
- Kupchan, S.M.; Kennedy, R.M.; Schleigh, W.R.; Ohta, G. Buxus alkaloids-XII. Benzamide alkaloids from Buxus sempervirens L. Tetrahedron 1967, 23, 4563–4586. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | pIC50 (Tbr) | pIC50 (L6) | Compounds | pIC50 (Tbr) | pIC50 (L6) |
|---|---|---|---|---|---|
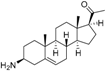 1 | 6.3958 | 5.2928 | 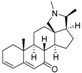 10 | 5.1326 * | 4.3800 * |
 2 | 5.3159 | 4.8135 | 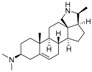 11 | 6.7781 | 4.2972 |
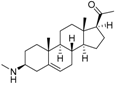 3 | 7.1249 * | 5.6057 * | 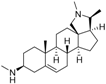 12 | 6.7781 * | 4.5618 |
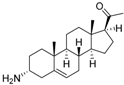 4 | 6.4320 | 4.7993 * | 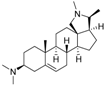 13 | 6.3778 | 4.2130 |
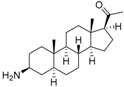 5 | 6.1726 * | 4.7707 | 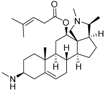 14 | 6.9245 | 4.8444 |
 6 | 5.9190 | 4.7603 |  15 | 5.7807 | 4.0103 * |
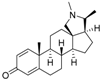 7 | 4.8282 * | n.t | 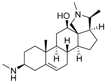 16 | 5.0856 | 3.7426 |
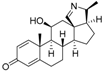 8 | 5.0624 | 4.1816 | 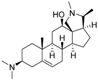 17 | 4.6798 | 3.9045 |
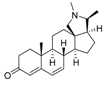 9 | 4.7568 | 3.8897 |
| Model Statistics | Anti-Tbr, PC = 3 | L6 Cytotoxic, PC = 2 |
|---|---|---|
| R2 ± SDEC | 0.995 ± 0.056 | 0.940 ± 0.111 |
| Q2 ± SDEP | 0.83 ± 0.33 | 0.64 ± 0.28 |
| P2 ± SDEP | 0.79 ± 0.51 | 0.59 ± 0.42 |
| F-ratio | 482.639 (4.066) | 70.452 (4.256) |
| Equation of regression trendline | y = 0.621x + 2.206 | y = 0.674x + 1.420 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nnadi, C.O.; Althaus, J.B.; Nwodo, N.J.; Schmidt, T.J. A 3D-QSAR Study on the Antitrypanosomal and Cytotoxic Activities of Steroid Alkaloids by Comparative Molecular Field Analysis. Molecules 2018, 23, 1113. https://doi.org/10.3390/molecules23051113
Nnadi CO, Althaus JB, Nwodo NJ, Schmidt TJ. A 3D-QSAR Study on the Antitrypanosomal and Cytotoxic Activities of Steroid Alkaloids by Comparative Molecular Field Analysis. Molecules. 2018; 23(5):1113. https://doi.org/10.3390/molecules23051113
Chicago/Turabian StyleNnadi, Charles Okeke, Julia Barbara Althaus, Ngozi Justina Nwodo, and Thomas Jürgen Schmidt. 2018. "A 3D-QSAR Study on the Antitrypanosomal and Cytotoxic Activities of Steroid Alkaloids by Comparative Molecular Field Analysis" Molecules 23, no. 5: 1113. https://doi.org/10.3390/molecules23051113
APA StyleNnadi, C. O., Althaus, J. B., Nwodo, N. J., & Schmidt, T. J. (2018). A 3D-QSAR Study on the Antitrypanosomal and Cytotoxic Activities of Steroid Alkaloids by Comparative Molecular Field Analysis. Molecules, 23(5), 1113. https://doi.org/10.3390/molecules23051113