
Synthesis of New Glycosylated Flavonoids with Inhibitory Activity on Cell Growth

 ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
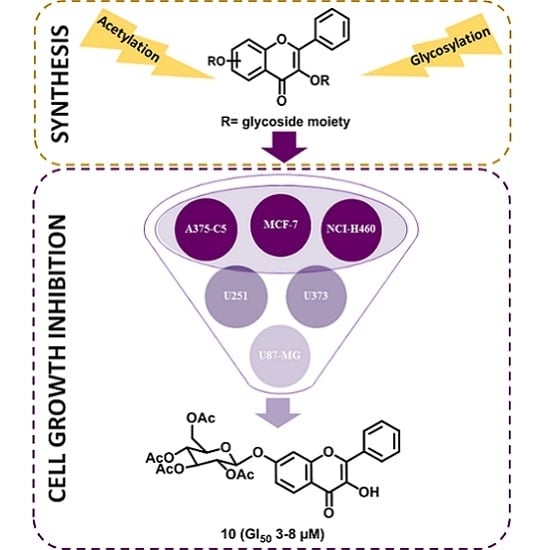
1. Introduction
2. Results and Discussion
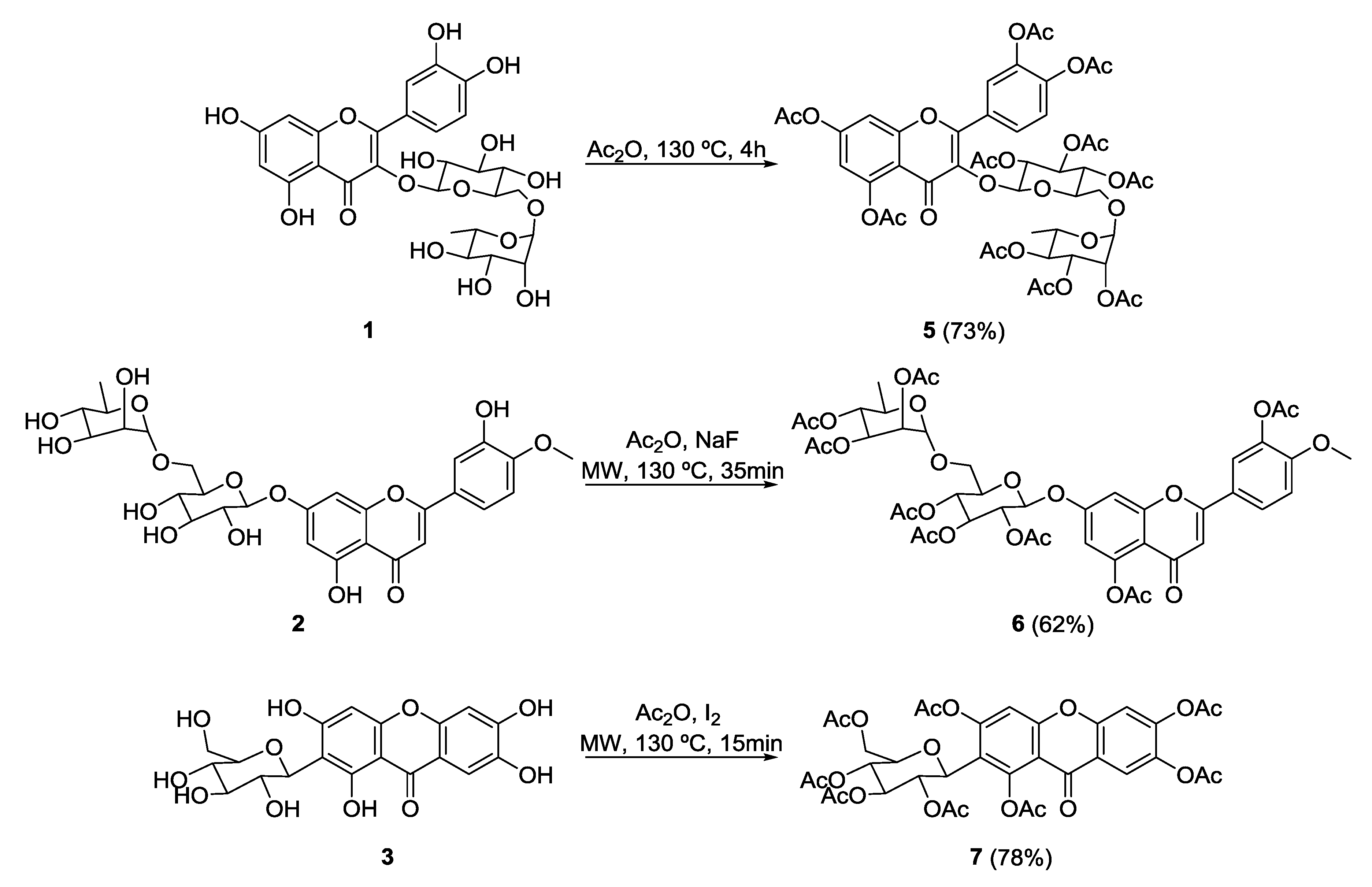
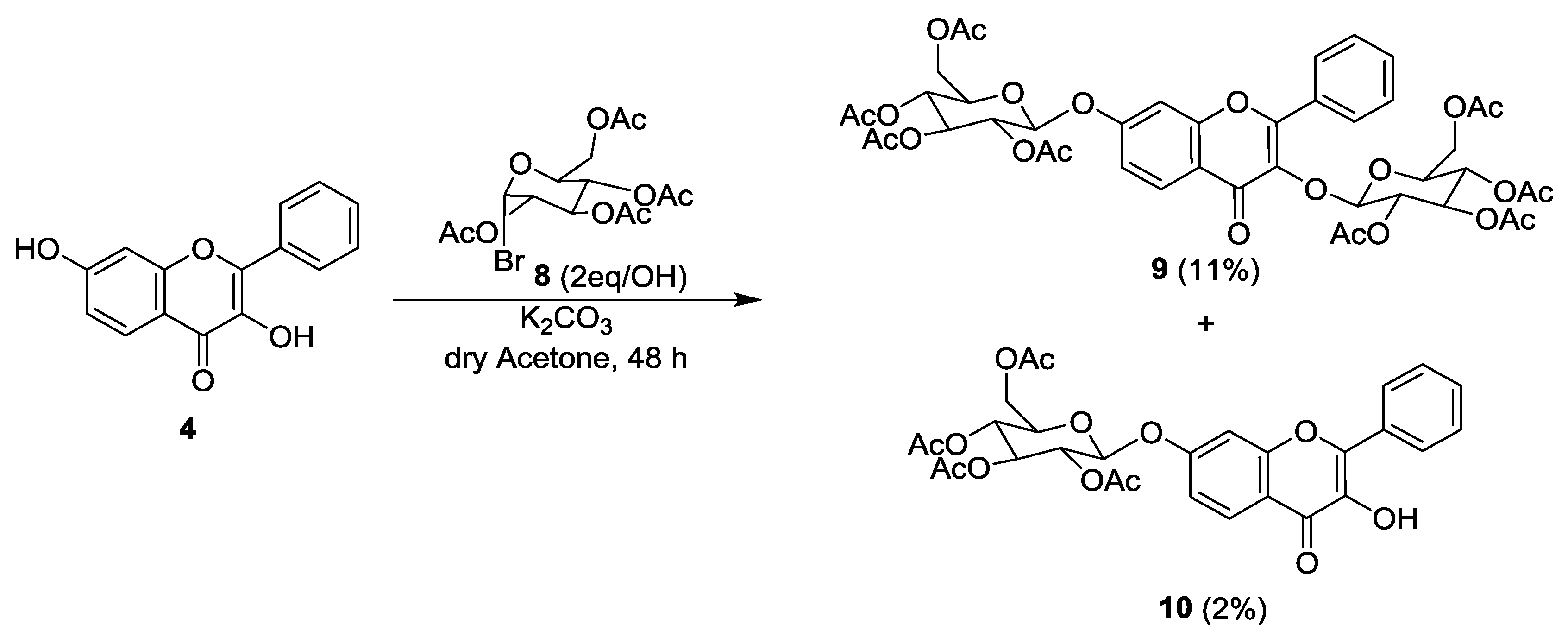
2.1. Synthesis
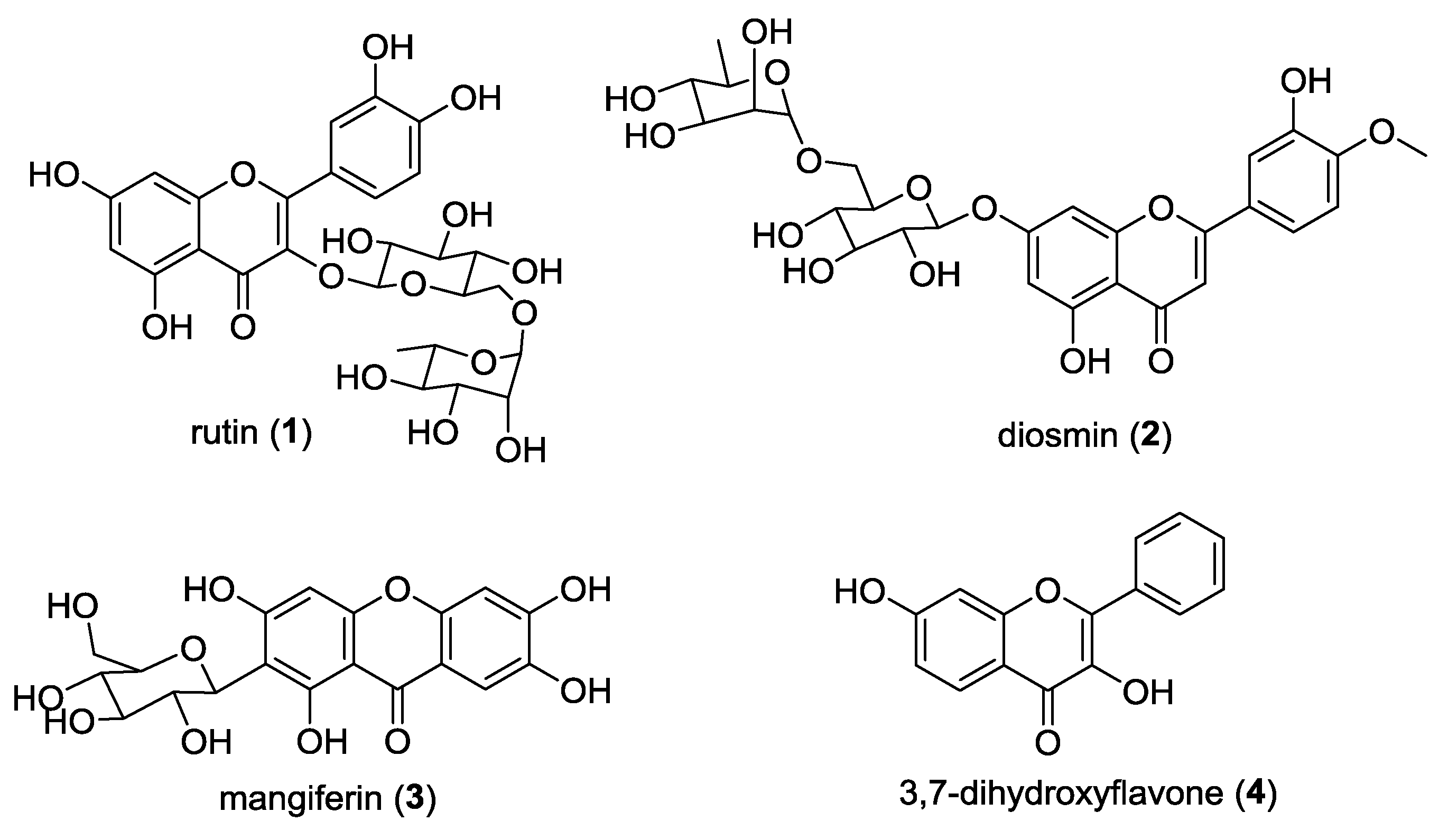
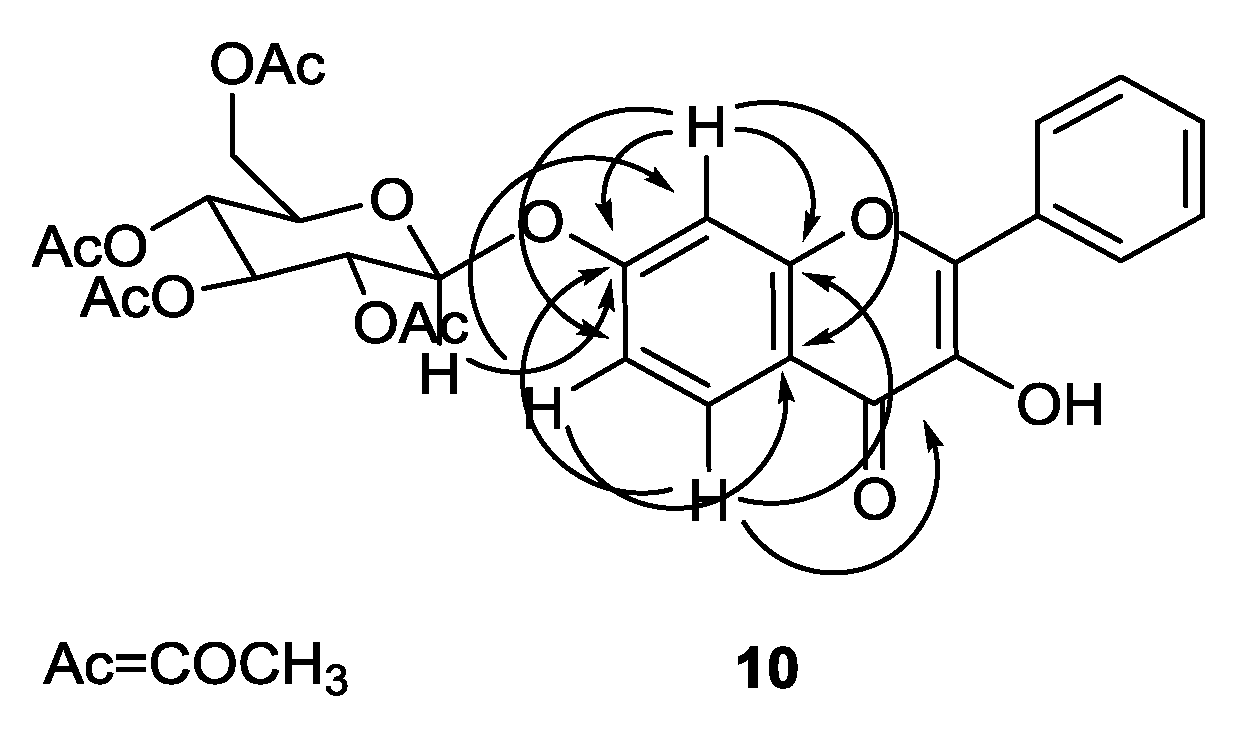
2.2. Structure Elucidation
2.3. Growth Inhibition Activity in Human Tumor Cell Lines
3. Materials and Methods
3.1. General Information
3.2. Chemistry
3.3. Tumor Cell Growth Assay and Selectivity Index
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pinto, M.M.M.; Sousa, M.E.; Nascimento, M.S.J. Xanthone derivatives: New insights in biological activities. Curr. Med. Chem. 2005, 12, 2517–2538. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Pandey, A.K. Chemistry and biological activities of flavonoids: An overview. Sci. World J. 2013, 2013, 162750. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, D.; Kaiser, M.; Brun, R.; Yardley, V.; Schmidt, T.J.; Tosun, F.; Rüedi, P. Antitrypanosomal and antileishmanial activities of flavonoids and their analogues: In vitro, in vivo, structure-activity relationship, and quantitative structure-activity relationship studies. Antimicrob. Agents Chemother. 2006, 50, 1352–1364. [Google Scholar] [CrossRef] [PubMed]
- Reutrakul, V.; Ningnuek, N.; Pohmakotr, M.; Yoosook, C.; Napaswad, C.; Kasisit, J.; Santisuk, T.; Tuchinda, P. Anti HIV-1 flavonoid glycosides from ochna integerrima. Planta Med. 2007, 73, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Orhan, D.D.; Özçelik, B.; Özgen, S.; Ergun, F. Antibacterial, antifungal, and antiviral activities of some flavonoids. Microbiol. Res. 2010, 165, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Leong, C.N.A.; Tako, M.; Hanashiro, I.; Tamaki, H. Antioxidant flavonoid glycosides from the leaves of Ficus pumila L. Food Chem. 2008, 109, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Ahmad, A.; Rawat, P.; Khan, M.F.; Rasheed, N.; Gupta, P.; Sathiamoorthy, B.; Bhatia, G.; Palit, G.; Maurya, R. Antioxidant flavonoid glycosides from evolvulus alsinoides. Fitoterapia 2010, 81, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Zhao, Y.; Jiang, Y.; Yu, L.; Zeng, X.; Yang, J.; Tian, M.; Liu, H.; Yang, B. Identification of a flavonoid c-glycoside as potent antioxidant. Free Radic. Biol. Med. 2017, 110, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Abd El-kader, A.M.; Ahmed, A.S.; Nafady, A.M.; Ibraheim, Z.Z. Xanthone and lignan glycosides from the aerial parts of polygonum bellardii all growing in egypt. Pharmacogn. Mag. 2013, 9, 135–143. [Google Scholar] [PubMed]
- Li, S.; Dong, P.; Wang, J.; Zhang, J.; Gu, J.; Wu, X.; Wu, W.; Fei, X.; Zhang, Z.; Wang, Y.; et al. Icariin, a natural flavonol glycoside, induces apoptosis in human hepatoma smmc-7721 cells via a ros/jnk-dependent mitochondrial pathway. Cancer Lett. 2010, 298, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Huang, J.; Yang, B.; Xiang, T.; Yin, X.; Peng, W.; Cheng, W.; Wan, J.; Luo, F.; Li, H.; et al. Mangiferin exerts antitumor activity in breast cancer cells by regulating matrix metalloproteinases, epithelial to mesenchymal transition, and beta-catenin signaling pathway. Toxicol. Appl. Pharmacol. 2013, 272, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Vieira, L.M.; Kijjoa, A. Naturally-occurring xanthones: Recent developments. Curr. Med. Chem. 2005, 12, 2413–2446. [Google Scholar] [CrossRef] [PubMed]
- Delazar, A.; Celik, S.; Göktürk, R.S.; Unal, O.; Nahar, L.; Sarker, S.D. Two acylated flavonoid glycosides from stachys bombycina, and their free radical scavenging activity. Pharmazie 2005, 60, 878–880. [Google Scholar] [CrossRef] [PubMed]
- Uriarte-Pueyo, I.; Calvo, M.I. Structure–activity relationships of acetylated flavone glycosides from Galeopsis ladanum L. (lamiaceae). Food Chem. 2010, 120, 679–683. [Google Scholar] [CrossRef]
- Browning, A.M.; Walle, U.K.; Walle, T. Flavonoid glycosides inhibit oral cancer cell proliferation-role of cellular uptake and hydrolysis to the aglycones. J. Pharm. Pharmacol. 2005, 57, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Poteet-Smith, C.E.; Xu, Y.; Errington, T.M.; Hecht, S.M.; Lannigan, D.A. Identification of the first specific inhibitor of p90 ribosomal S6 kinase (RSK) reveals an unexpected role for RSK in cancer cell proliferation. Cancer Res. 2005, 65, 1027–1034. [Google Scholar] [PubMed]
- Kong, C.S.; Kim, Y.A.; Kim, M.M.; Park, J.S.; Kim, J.A.; Kim, S.K.; Lee, B.J.; Nam, T.J.; Seo, Y. Flavonoid glycosides isolated from Salicornia herbacea inhibit matrix metalloproteinase in HT1080 cells. Toxicol. In Vitro 2008, 22, 1742–1748. [Google Scholar] [CrossRef] [PubMed]
- Tundis, R.; Deguin, B.; Loizzo, M.R.; Bonesi, M.; Statti, G.A.; Tillequin, F.; Menichini, F. Potential antitumor agents: Flavones and their derivatives from linaria reflexa desf. Bioorg. Med. Chem. Lett. 2005, 15, 4757–4760. [Google Scholar] [CrossRef] [PubMed]
- Aligiannis, N.; Mitaku, S.; Mitrocotsa, D.; Leclerc, S. Flavonoids as cycline-dependent kinase inhibitors: Inhibition of cdc 25 phosphatase activity by flavonoids belonging to the quercetin and kaempferol series. Planta Med. 2001, 67, 468–470. [Google Scholar] [CrossRef] [PubMed]
- Viskupicova, J.; Ondrejovic, M.; Maliar, T. Enzyme-mediated preparation of flavonoid esters and their applications. In Biochemistry; Ekinci, D., Ed.; IntechOpen: London, UK, 2012. [Google Scholar]
- Dixit, S. Anticancer effect of rutin isolated from the methanolic extract of triticum aestivum straw in mice. Med. Sci. 2014, 2, 153–160. [Google Scholar] [CrossRef]
- Alonso-Castro, A.J.; Domínguez, F.; García-Carrancá, A. Rutin exerts antitumor effects on nude mice bearing sw480 tumor. Arch. Med. Res. 2013, 44, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Monasterio, A.; Urdaci, M.C.; Pinchuk, I.V.; Lopez-Moratalla, N.; Martinez-Irujo, J.J. Flavonoids induce apoptosis in human leukemia U937 cells through caspase- and caspase-calpain-dependent pathways. Nutr. Cancer 2004, 50, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Kawaii, S.; Tomono, Y.; Katase, E.; Ogawa, K.; Yano, M. Antiproliferative activity of flavonoids on several cancer cell lines. Biosci. Biotechnol. Biochem. 1999, 63, 896–899. [Google Scholar] [CrossRef] [PubMed]
- Nanduri, S.; Nyavanandi, V.K.; Sanjeeva Rao Thunuguntla, S.; Kasu, S.; Pallerla, M.K.; Sai Ram, P.; Rajagopal, S.; Ajaya Kumar, R.; Ramanujam, R.; Moses Babu, J.; et al. Synthesis and structure–activity relationships of andrographolide analogues as novel cytotoxic agents. Bioorg. Med. Chem. Lett. 2004, 14, 4711–4717. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Chu, J.; Wang, H.; Fu, X.; Quan, D.; Ding, H.; Yao, Q.; Yu, P. Regioselective iodination of flavonoids by n-iodosuccinimide under neutral conditions. Tetrahedron Lett. 2013, 54, 6345–6348. [Google Scholar] [CrossRef]
- Mogilaiah, K.; Rani, J.U.; Vidya, K.; Sakram, B. Microwave-promoted rapid and efficient method for acetylation of phenols with acetic anhydride using naf as catalyst under solvent-free conditions. Orient. J. Chem. 2009, 25, 187–190. [Google Scholar]
- Dar, A.; Faizi, S.; Naqvi, S.; Roome, T.; Zikr-ur-Rehman, S.; Ali, M.; Firdous, S.; Moin, S.T. Analgesic and antioxidant activity of mangiferin and its derivatives: The structure activity relationship. Biol. Pharm. Bull. 2005, 28, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Liang, D.; Ning, M.; Wang, Q.; Meng, X.; Li, Z. Semi-synthesis of neomangiferin from mangiferin. Tetrahedron Lett. 2014, 55, 3083–3086. [Google Scholar] [CrossRef]
- Faizi, S.; Zikr-ur-Rehman, S.; Ali, M.; Naz, A. Temperature and solvent dependent nmr studies on mangiferin and complete nmr spectral assignments of its acyl and methyl derivatives. Magn. Reson. Chem. 2006, 44, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Lewin, G.; Maciuk, A.; Moncomble, A.; Cornard, J.-P. Enhancement of the water solubility of flavone glycosides by disruption of molecular planarity of the aglycone moiety. J. Nat. Prod. 2013, 76, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Quintin, J.; Roullier, C.; Thoret, S.; Lewin, G. Synthesis and anti-tubulin evaluation of chromone-based analogues of combretastatins. Tetrahedron 2006, 62, 4038–4051. [Google Scholar] [CrossRef]
- Mei, Q.; Wang, C.; Zhao, Z.; Yuan, W.; Zhang, G. Synthesis of icariin from kaempferol through regioselective methylation and para-claisen–cope rearrangement. Beilstein J. Org. Chem. 2015, 11, 1220–1225. [Google Scholar] [CrossRef] [PubMed]
- Needs, P.W.; Williamson, G. Syntheses of daidzein-7-yl β-d-glucopyranosiduronic acid and daidzein-4′,7-yl di-β-d-glucopyranosiduronic acid. Carbohydr. Res. 2001, 330, 511–515. [Google Scholar] [CrossRef]
- Smith, J.A.; Maloney, D.J.; Clark, D.E.; Xu, Y.; Hecht, S.M.; Lannigan, D.A. Influence of rhamnose substituents on the potency of SL0101, an inhibitor of the Ser/Thr kinase, RSK. Bioorg. Med. Chem. Lett. 2006, 14, 6034–6042. [Google Scholar] [CrossRef] [PubMed]
- Ngameni, B.; Patnam, R.; Sonna, P.; Ngadjui, B.T.; Roy, R.; Abegaz, B.M. Hemisynthesis and spectroscopic characterization of three glycosylated 4-hydrocylonchocarpins from dorstenia barteri bureau. ARKIVOC 2008, 2008, 152–159. [Google Scholar]
- Christensen, H.M.; Oscarson, S.; Jensen, H.H. Common side reactions of the glycosyl donor in chemical glycosylation. Carbohydr. Res. 2015, 408, 51–95. [Google Scholar] [CrossRef] [PubMed]
- Velandia, J.R.; de Carvalho, M.G.; Braz-Filho, R.; Werle, A.A. Biflavonoids and a glucopyranoside derivative from ouratea semiserrata. Phytochem. Anal. 2002, 13, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Mabry, T.; Markham, K.R.; Thomas, M.B. The NMR spectra of flavonoids. In The Systematic Identification of Flavonoids; Springer: Berlin/Heidelberg, Germany, 1970; pp. 274–343. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. Ilogp: A simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the gb/sa approach. J. Chem. Inf. Model. 2014, 54, 3284–3301. [Google Scholar] [CrossRef] [PubMed]
- Kodelia, G.; Athanasiou, K.; Kolisis, F.N. Enzymatic synthesis of butyryl-rutin ester in organic solvents and its cytogenetic effects in mammalian cells in culture. Appl. Biochem. Biotechnol. 1994, 44, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Suda, I.; Oki, T.; Masuda, M.; Nishiba, Y.; Furuta, S.; Matsugano, K.; Sugita, K.; Terahara, N. Direct absorption of acylated anthocyanin in purple-fleshed sweet potato into rats. J. Agric. Food Chem. 2002, 50, 1672–1676. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.; Lygo, B.; Procter, G. Advanced and Practical Organic Chemistry, 3rd ed.; Taylor and Francis Group, LLC: Boca Raton, FL, USA, 2013. [Google Scholar]
- Badisa, R.B.; Ayuk-Takem, L.T.; Ikediobi, C.O.; Walker, E.H. Selective anticancer activity of pure licamichauxiioic-b acid in cultured cell lines. Pharm. Biol. 2006, 44, 141–145. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 5, 6, 7 and 9 are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | GI50(μM) | iLogP | |||||
|---|---|---|---|---|---|---|---|
| A375-C5 | MCF-7 | NCI-H460 | U251 | U373 | U87-MG | ||
| 1 | >150 | >150 | >150 | ND | ND | ND | 2.43 |
| 2 | >150 | >150 | >150 | ND | ND | ND | 2.03 |
| 3 | >150 | >150 | >150 | ND | ND | ND | 1.42 |
| 4 | 10.81 ± 1.42 | 12.55 ± 5.23 | 12.80 ± 1.83 | 9.89 ± 0.53 | 10.24 ± 1.01 | 18.87 ± 1.36 | 2.01 |
| 5 | 9.65 ± 1.92 | 20.49 ± 2.14 | 14.32 ± 4.60 | 24.46 ± 4.08 | 20.17 ± 1.92 | 14.28 ± 1.62 | 5.44 |
| 6 | >150 | >150 | >150 | ND | ND | ND | 4.03 |
| 7 | 58.06 ± 2.74 | 88.49 ± 0.72 | 99.90 ± 5.81 | >150 | >150 | >150 | 3.08 |
| 9 | 10.44 ± 0.72 | >150 | >150 | 32.86 ± 0.042 | 109.16 ± 1.19 | 100.37 ± 9.61 | 4.35 |
| 10 | 7.34 ± 0.93 | 2.67 ± 0.49 | 7.61 ± 0.55 | 5.41 ± 0.83 | 6.42 ± 2.01 | 6.48 ± 0.67 | 3.55 |
| Doxorubicin | 0.014 ± 0.002 | 0.009 ± 0.001 | 0.009 ± 0.002 | 0.011 ± 0.004 | 0.009 ± 0.001 | 0.010 ± 0.002 | - |
| Compounds | HPAEpiC (GI50, µM) | Selectivity Index | |||||
|---|---|---|---|---|---|---|---|
| A375-C5 | MCF-7 | NCI-H460 | U251 | U373 | U87-MG | ||
| 4 | 16.03 ± 1.38 | 1.48 | 1.28 | 1.25 | 1.62 | 1.57 | 0.85 |
| 5 | 14.13 ± 5.06 | 1.46 | 0.69 | 0.99 | 0.58 | 0.70 | 0.99 |
| 7 | 110 ± 7.07 | 1.89 | 1.24 | 1.10 | - | - | - |
| 9 | 91.40 ± 0.85 | 8.75 | 2.78 | 0.84 | 0.91 | ||
| 10 | 31.71 ± 1.63 | 4.32 | 11.88 | 4.17 | 5.68 | 4.94 | 4.89 |
| Doxorubicin | 0.004 ± 0.09 | 0.29 | 0.44 | 0.44 | 0.36 | 0.44 | 0.40 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neves, A.R.; Correia-da-Silva, M.; Silva, P.M.A.; Ribeiro, D.; Sousa, E.; Bousbaa, H.; Pinto, M. Synthesis of New Glycosylated Flavonoids with Inhibitory Activity on Cell Growth. Molecules 2018, 23, 1093. https://doi.org/10.3390/molecules23051093
Neves AR, Correia-da-Silva M, Silva PMA, Ribeiro D, Sousa E, Bousbaa H, Pinto M. Synthesis of New Glycosylated Flavonoids with Inhibitory Activity on Cell Growth. Molecules. 2018; 23(5):1093. https://doi.org/10.3390/molecules23051093
Chicago/Turabian StyleNeves, Ana R., Marta Correia-da-Silva, Patrícia M. A. Silva, Diana Ribeiro, Emília Sousa, Hassan Bousbaa, and Madalena Pinto. 2018. "Synthesis of New Glycosylated Flavonoids with Inhibitory Activity on Cell Growth" Molecules 23, no. 5: 1093. https://doi.org/10.3390/molecules23051093
APA StyleNeves, A. R., Correia-da-Silva, M., Silva, P. M. A., Ribeiro, D., Sousa, E., Bousbaa, H., & Pinto, M. (2018). Synthesis of New Glycosylated Flavonoids with Inhibitory Activity on Cell Growth. Molecules, 23(5), 1093. https://doi.org/10.3390/molecules23051093