Electrochemical Aptamer-Based Sensors for Rapid Point-of-Use Monitoring of the Mycotoxin Ochratoxin A Directly in a Food Stream


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
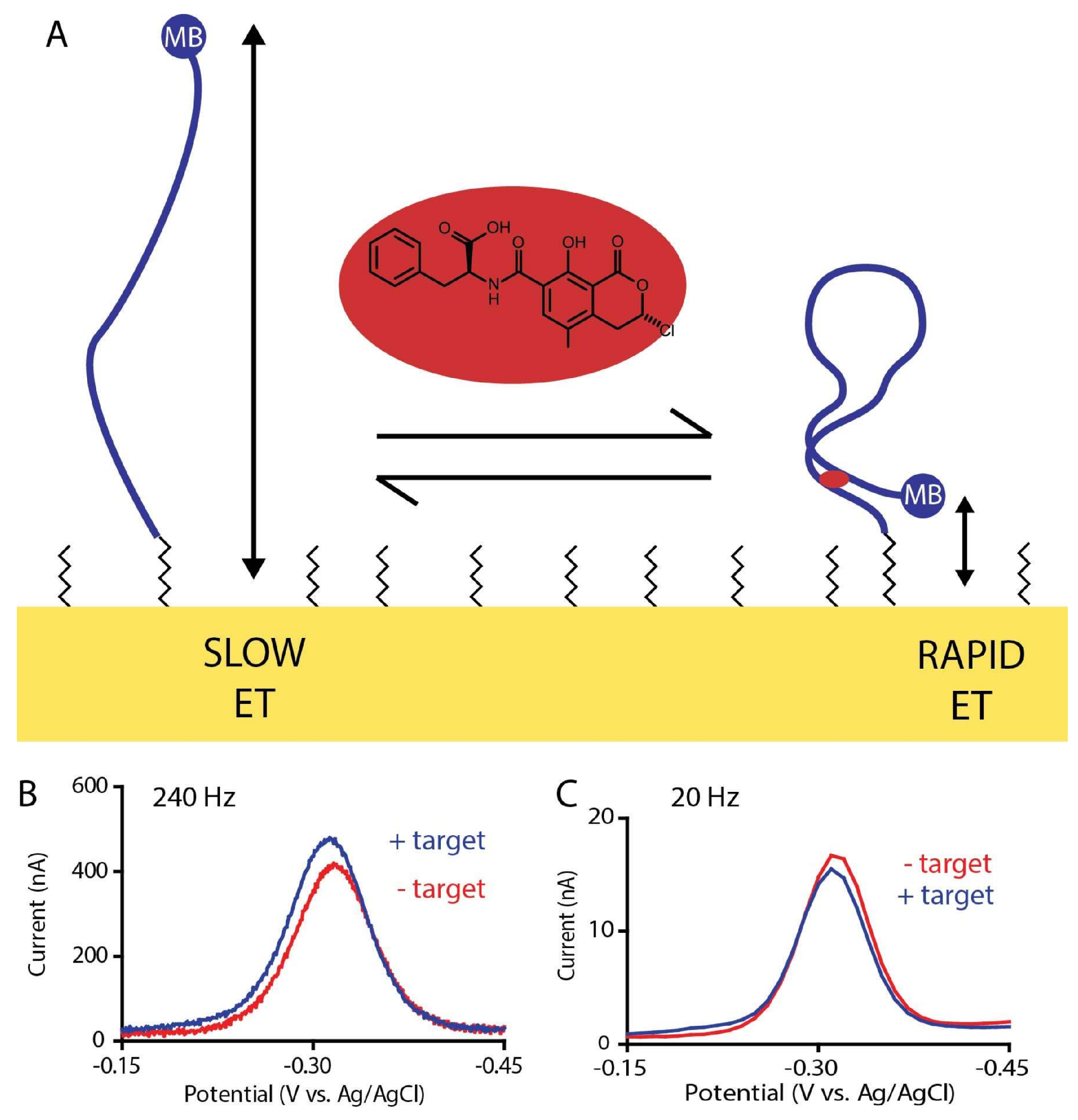
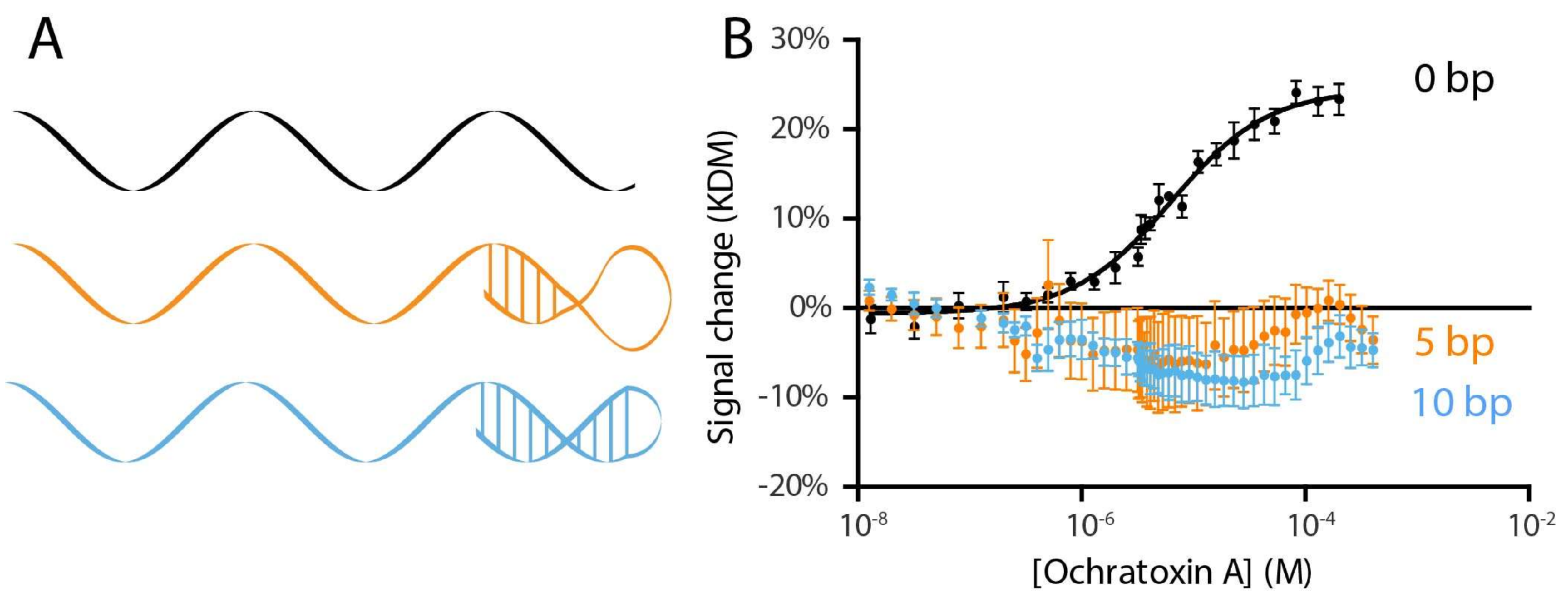
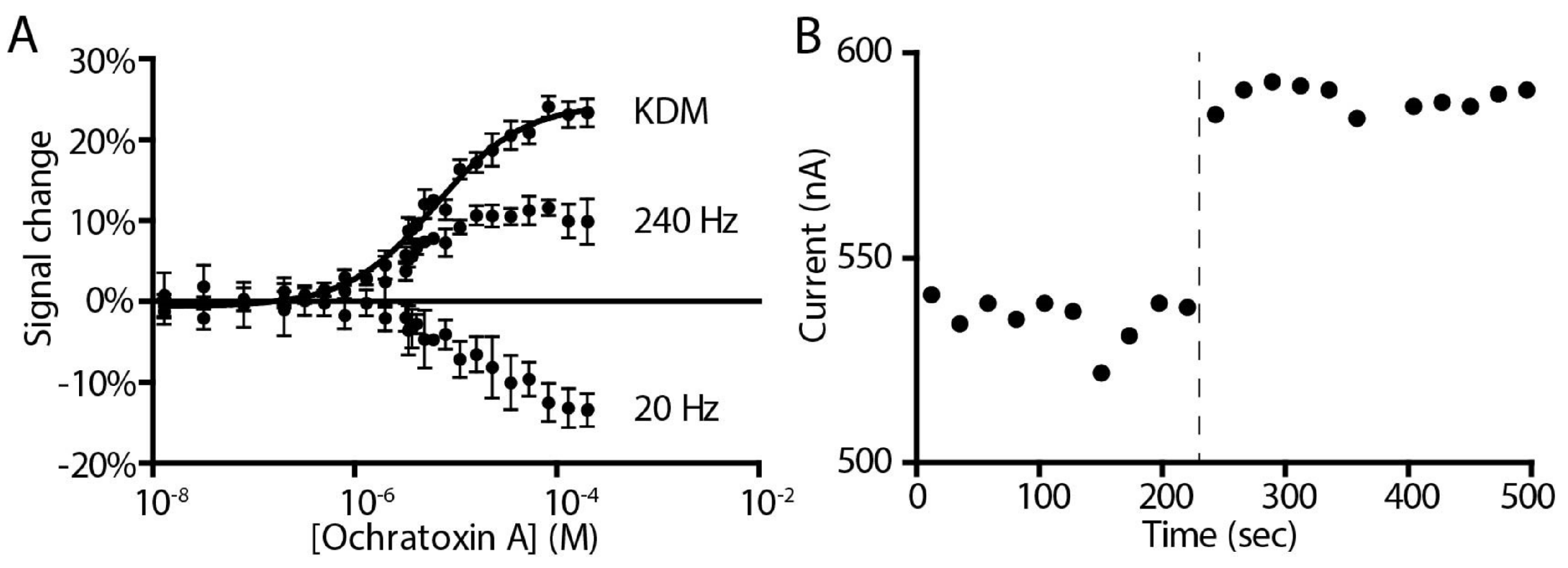
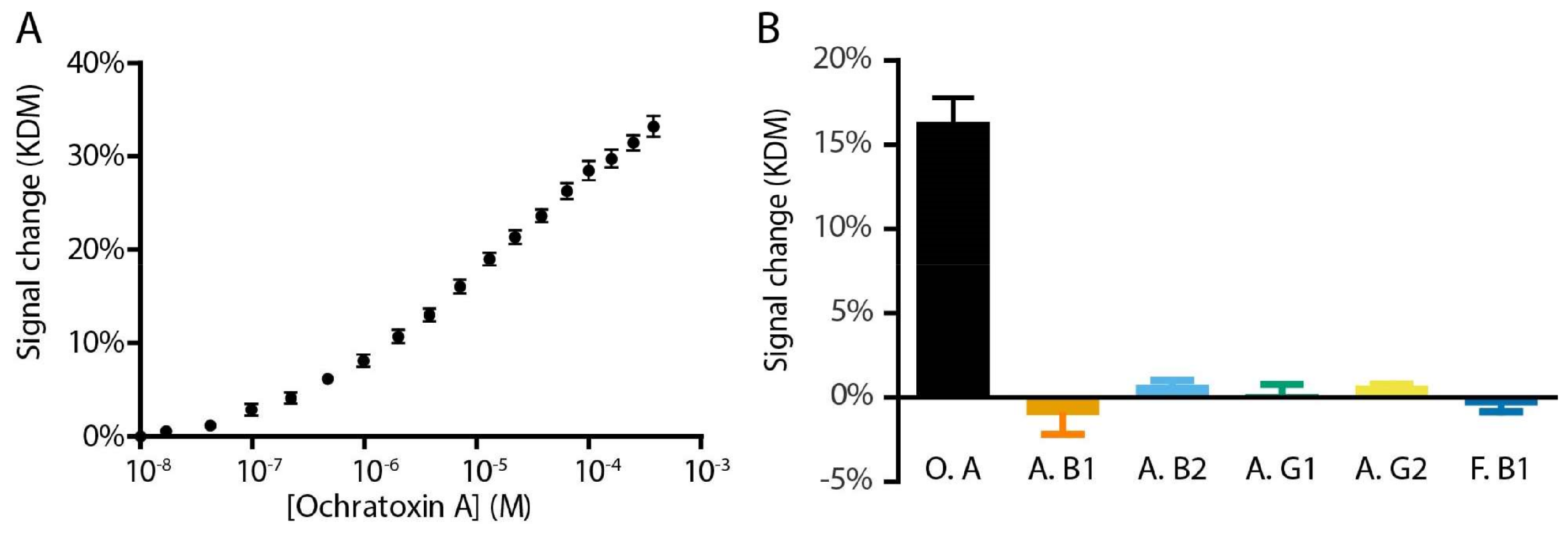
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Ochratoxin A Aptamer Sequences
3.3. Sensor Preparation
3.4. Electrochemical Measurements
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nyikal, J.; Misore, A.; Nzioka, C.; Njuguna, C.; Muchiri, E.; Njau, J.; Maingi, S.; Njoroge, J.; Mutiso, J.; Onteri, J.; et al. Outbreak of Aflatoxin Poisoning—Eastern and Central Provinces, Kenya, January–July 2004. Morb. Mortal. Wkly. Rep. 2004, 53, 790–793. [Google Scholar]
- Bennett, J.W.; Klich, M.; Mycotoxins, M. Mycotoxins. Clin. Microbiol. Rev. 2003, 16, 497–516. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wu, F. The need to revisit ochratoxin A risk in light of diabetes, obesity, and chronic kidney disease prevalence. Food Chem. Toxicol. 2017, 103, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Bui-Klimke, T.R.; Wu, F. Ochratoxin A and Human Health Risk: A Review of the Evidence. Crit. Rev. Food Sci. Nutr. 2015, 55, 1860–1869. [Google Scholar] [CrossRef] [PubMed]
- Bayman, P.; Baker, J.L. Ochratoxins: A global perspective. Mycopathologia 2006, 162, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Studer-Rohr, I.; Dietrich, D.R.; Schlatter, J.; Schlatter, C. The occurrence of ochratoxin A in coffee. Food Chem. Toxicol. 1995, 33, 341–355. [Google Scholar] [CrossRef]
- Rai, M.; Jogee, P.S.; Ingle, A.P. Emerging nanotechnology for detection of mycotoxins in food and feed. Int. J. Food Sci. Nutr. 2015, 66, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Hyle, E.P.; Jani, I.V.; Lehe, J.; Su, A.E.; Wood, R.; Quevedo, J.; Losina, E.; Bassett, I.V.; Pei, P.P.; Paltiel, A.D.; et al. The Clinical and Economic Impact of Point-of-Care CD4 Testing in Mozambique and Other Resource-Limited Settings: A Cost-Effectiveness Analysis. PLoS Med. 2014, 11, e1001725. [Google Scholar] [CrossRef] [PubMed]
- Rowe, A.A.; Bonham, A.J.; White, R.J.; Zimmer, M.P.; Yadgar, R.J.; Hobza, T.M.; Honea, J.W.; Ben-Yaacov, I.; Plaxco, K.W. CheapStat: An Open-Source, “Do-It-Yourself” Potentiostat for Analytical and Educational Applications. PLoS ONE 2011, 6, e23783. [Google Scholar] [CrossRef] [PubMed]
- White, R.J.; Plaxco, K.W. Exploiting binding-induced changes in probe flexibility for the optimization of electrochemical biosensors. Anal. Chem. 2010, 82, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Aguado, J.A.; Penner, G. Determination of Ochratoxin A with a DNA Aptamer. J. Agric. Food Chem. 2008, 56, 10456–10461. [Google Scholar] [CrossRef] [PubMed]
- Ha, T.H. Recent advances for the detection of ochratoxin A. Toxins 2015, 7, 5276–5300. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Fang, Z.; Liu, J.; Zeng, L. A simple and rapid biosensor for ochratoxin A based on a structure-switching signaling aptamer. Food Control 2012, 25, 555–560. [Google Scholar] [CrossRef]
- Tan, Z.; Feagin, T.A.; Heemstra, J.M. Temporal Control of Aptamer Biosensors Using Covalent Self-Caging to Shift Equilibrium. J. Am. Chem. Soc. 2016, 138, 6328–6331. [Google Scholar] [CrossRef] [PubMed]
- Arroyo-Currás, N.; Somerson, J.; Vieira, P.A.; Ploense, K.L.; Kippin, T.E.; Plaxco, K.W. Real-time measurement of small molecules directly in awake, ambulatory animals. Proc. Natl. Acad. Sci. USA 2017, 114, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Cash, K.J.; Ricci, F.; Plaxco, K.W. An electrochemical sensor for the detection of protein-small molecule interactions directly in serum and other complex matrices. J. Am. Chem. Soc. 2009, 131, 6955–6957. [Google Scholar] [CrossRef] [PubMed]
- Swensen, J.S.; Xiao, Y.; Ferguson, B.S.; Lubin, A.A.; Lai, R.Y.; Heeger, A.J.; Plaxco, K.W.; Soh, H.T. Continuous, real-time monitoring of cocaine in undiluted blood serum via a microfluidic, electrochemical aptamer-based sensor. J. Am. Chem. Soc. 2009, 131, 4262–4266. [Google Scholar] [CrossRef] [PubMed]
- Bonham, A.J.; Hsieh, K.; Ferguson, B.S.; Vallée-Bélisle, A.; Ricci, F.; Soh, H.T.; Plaxco, K.W. Quantification of transcription factor binding in cell extracts using an electrochemical, structure-switching biosensor. J. Am. Chem. Soc. 2012, 134, 3346–3348. [Google Scholar] [CrossRef] [PubMed]
- Porchetta, A.; Vallée-Bélisle, A.; Plaxco, K.W.; Ricci, F. Using Distal-Site Mutations and Allosteric Inhibition to Tune, Extend, and Narrow the Useful Dynamic Range of Aptamer-Based Sensors. J. Am. Chem. Soc. 2012, 134, 20601–20604. [Google Scholar] [CrossRef] [PubMed]
- Vallée-Bélisle, A.; Ricci, F.; Plaxco, K.W. Thermodynamic basis for the optimization of binding-induced biomolecular switches and structure-switching biosensors. Proc. Natl. Acad. Sci. USA 2009, 106, 13802–13807. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, B.S.; Hoggarth, D.A.; Maliniak, D.; Ploense, K.; White, R.J.; Woodward, N.; Hsieh, K.; Bonham, A.J.; Eisenstein, M.; Kippin, T.E.; et al. Real-time, aptamer-based tracking of circulating therapeutic agents in living animals. Sci. Transl. Med. 2013, 5, 213ra165. [Google Scholar] [CrossRef] [PubMed]
- Rowe, A.A.; White, R.J.; Bonham, A.J.; Plaxco, K.W. Fabrication of Electrochemical-DNA Biosensors for the Reagentless Detection of Nucleic Acids, Proteins and Small Molecules. J. Vis. Exp. 2011, 2922. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Somerson, J.; Plaxco, K.W. Electrochemical Aptamer-Based Sensors for Rapid Point-of-Use Monitoring of the Mycotoxin Ochratoxin A Directly in a Food Stream. Molecules 2018, 23, 912. https://doi.org/10.3390/molecules23040912
Somerson J, Plaxco KW. Electrochemical Aptamer-Based Sensors for Rapid Point-of-Use Monitoring of the Mycotoxin Ochratoxin A Directly in a Food Stream. Molecules. 2018; 23(4):912. https://doi.org/10.3390/molecules23040912
Chicago/Turabian StyleSomerson, Jacob, and Kevin W. Plaxco. 2018. "Electrochemical Aptamer-Based Sensors for Rapid Point-of-Use Monitoring of the Mycotoxin Ochratoxin A Directly in a Food Stream" Molecules 23, no. 4: 912. https://doi.org/10.3390/molecules23040912
APA StyleSomerson, J., & Plaxco, K. W. (2018). Electrochemical Aptamer-Based Sensors for Rapid Point-of-Use Monitoring of the Mycotoxin Ochratoxin A Directly in a Food Stream. Molecules, 23(4), 912. https://doi.org/10.3390/molecules23040912