Targeting Receptor-Type Protein Tyrosine Phosphatases with Biotherapeutics: Is Outside-in Better than Inside-Out?

Abstract
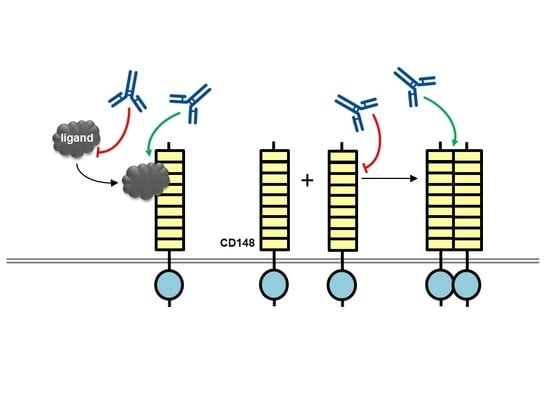
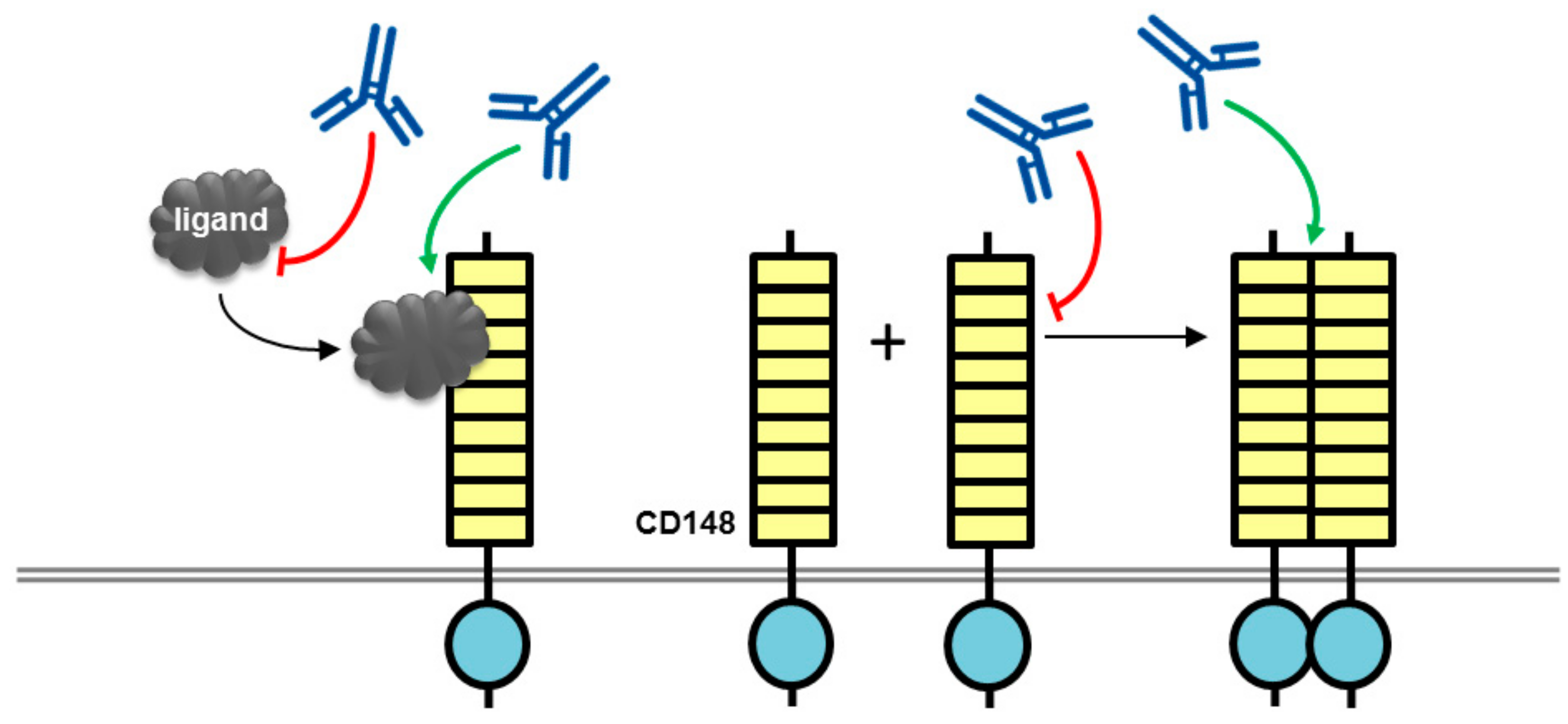
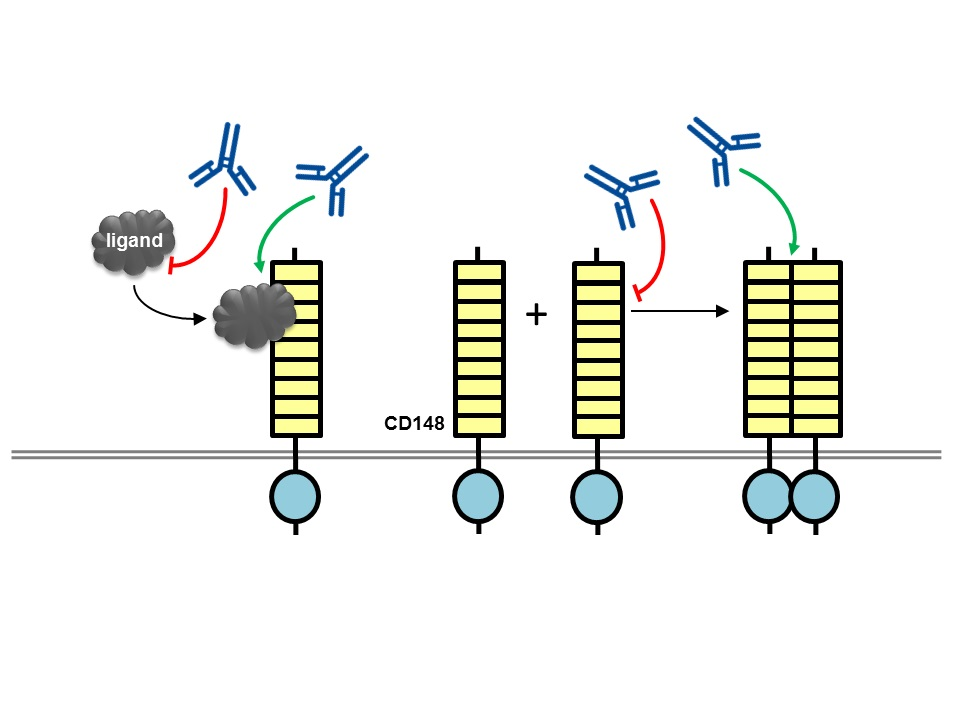{kind=link}
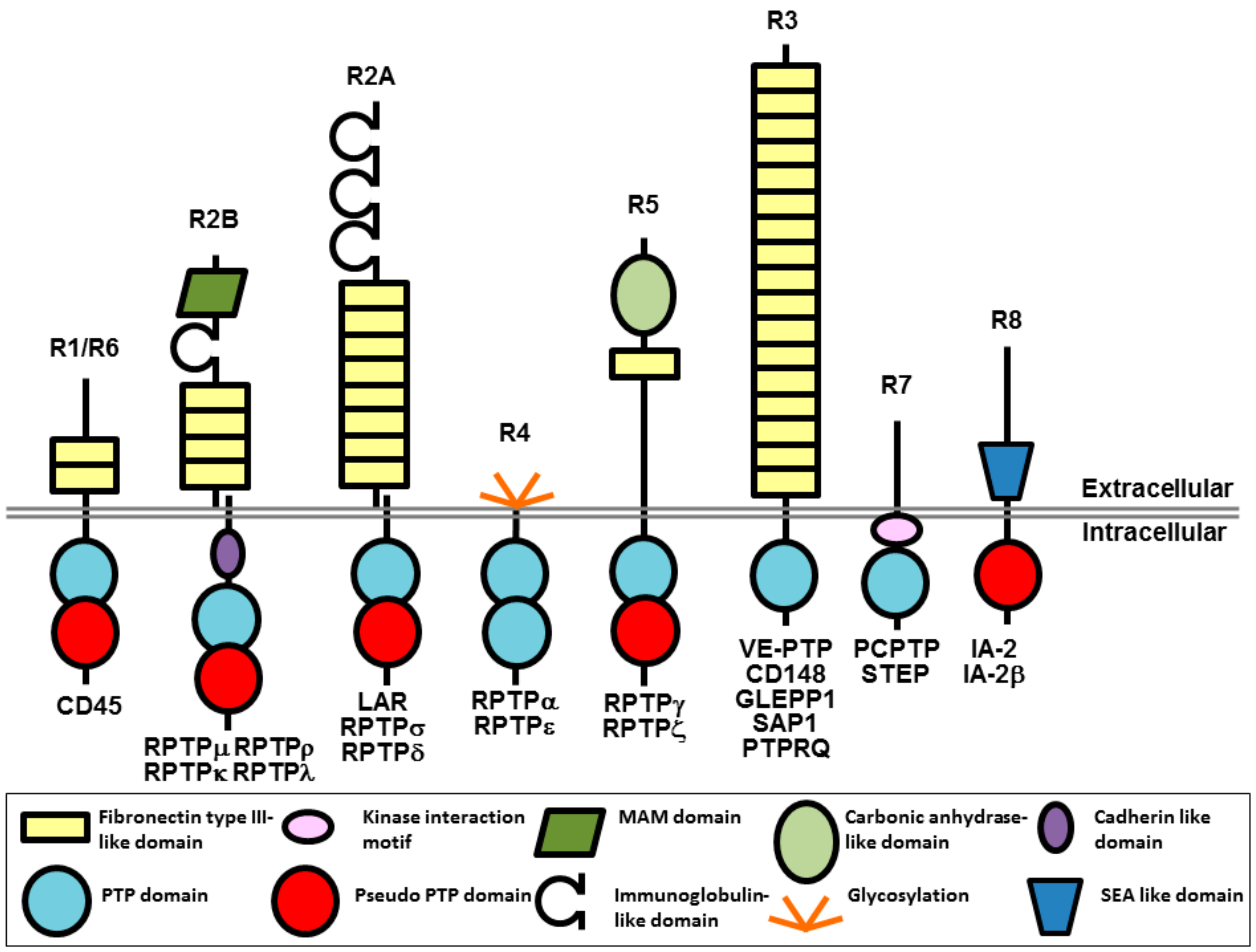{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Background on Currently Used Biotherapeutics
3. Therapeutic Potential of Biotherapeutics Targeting CD148 (PTPRJ)
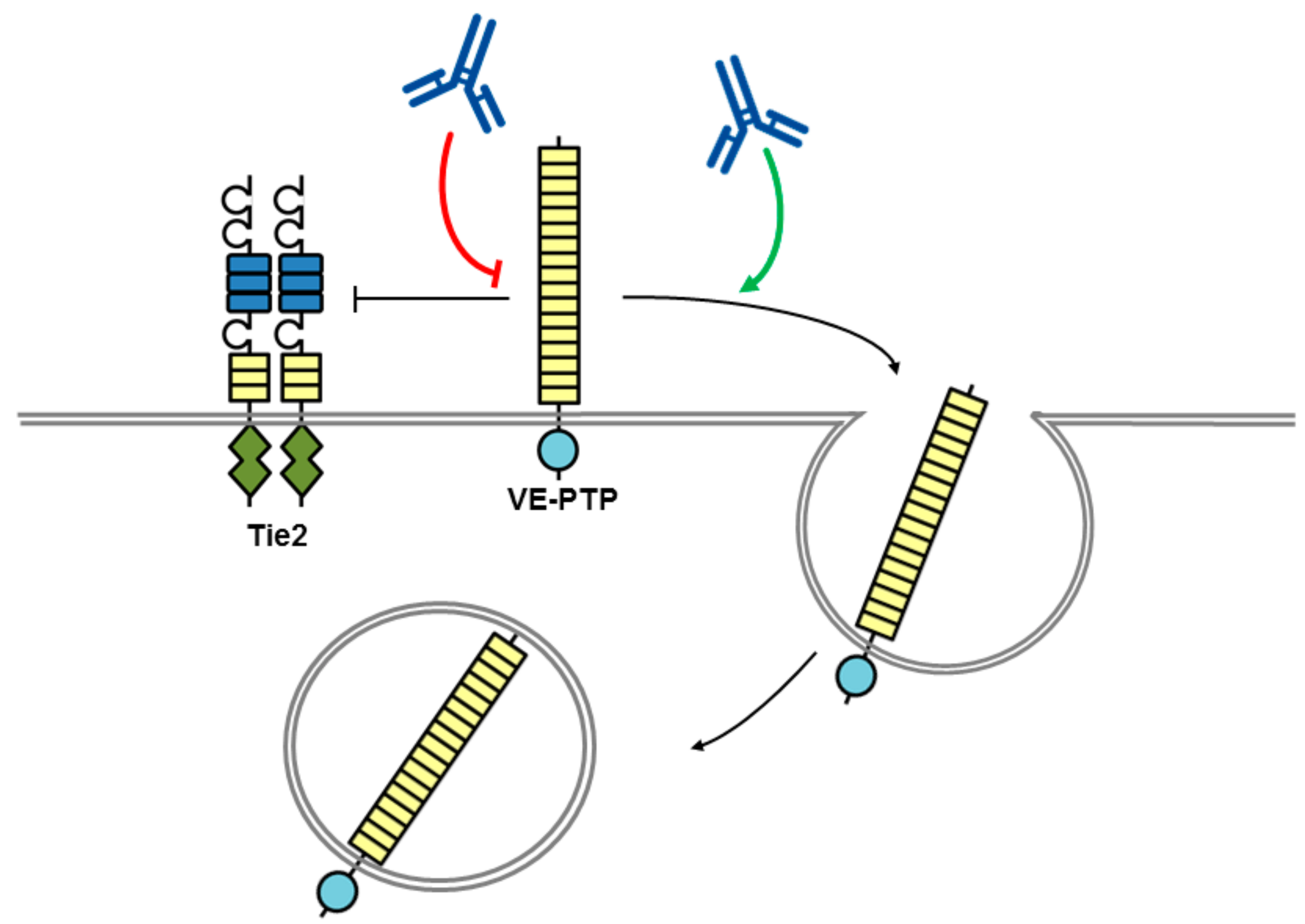
4. Therapeutic Potential of Biotherapeutics Targeting VE-PTP (PTPRB)
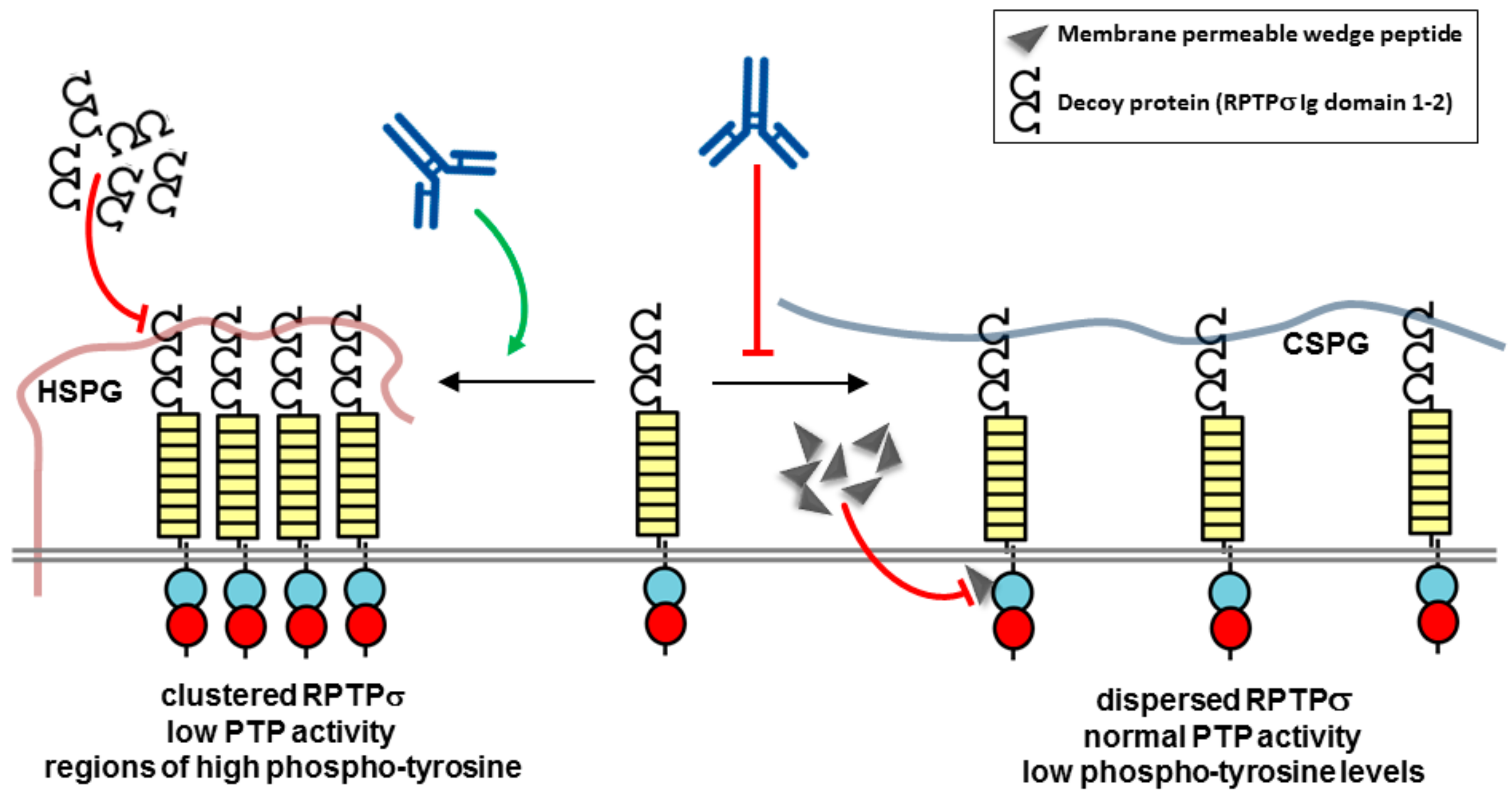
5. Therapeutic Potential of Biotherapeutics Targeting RPTPσ (PTPRS)
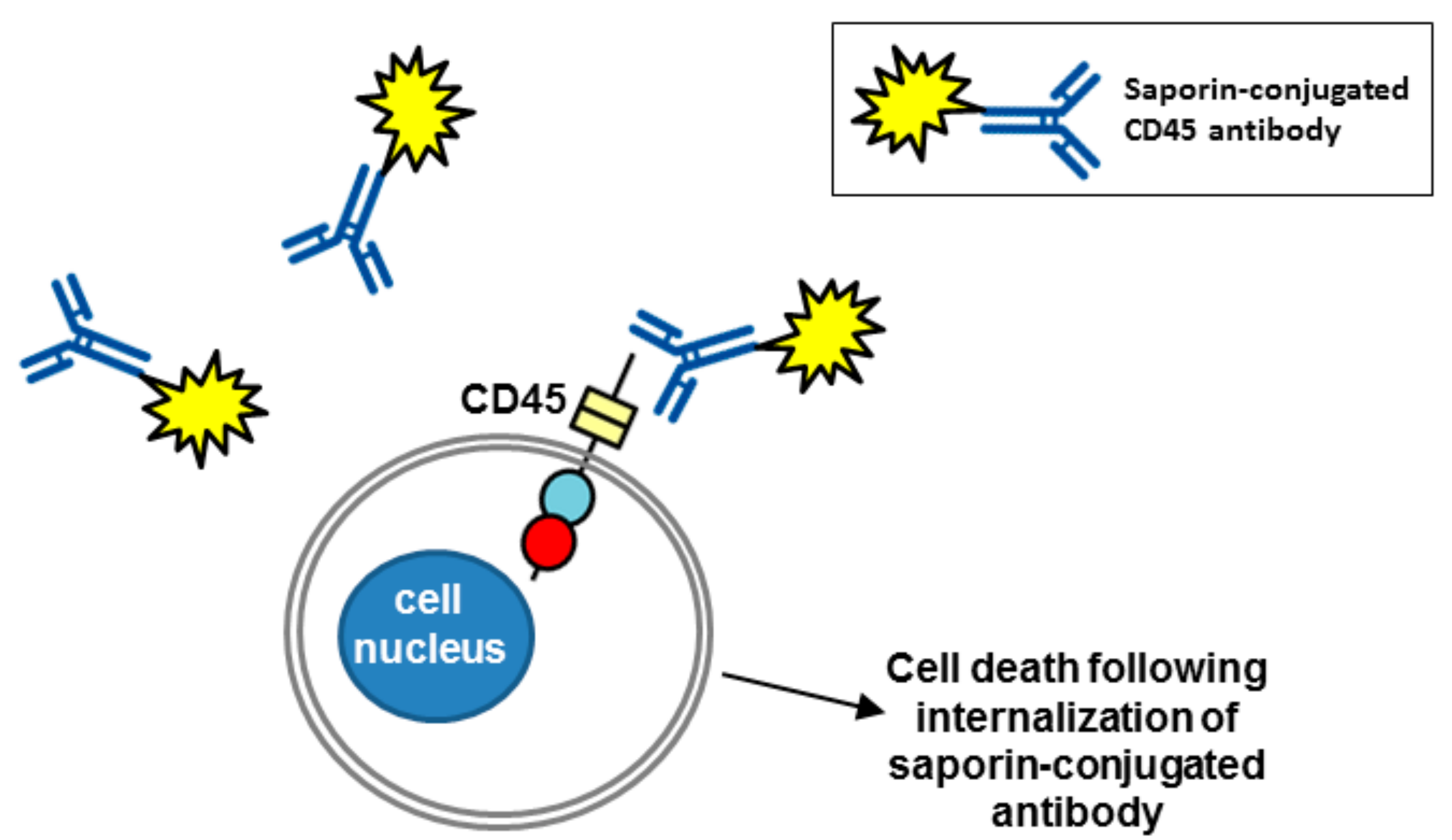
6. Therapeutic Potential of Biotherapeutics Targeting CD45 (PTPRC)
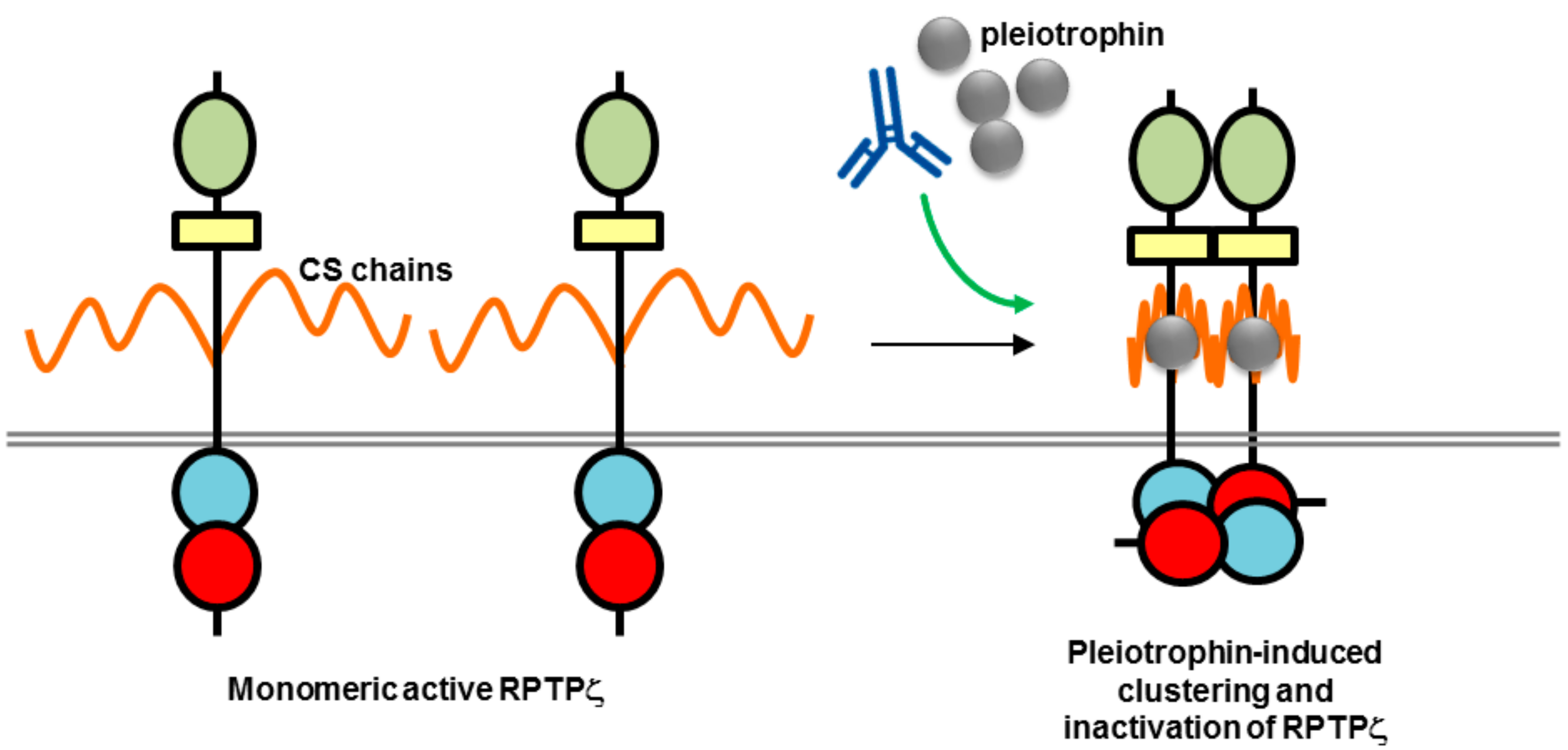
7. Therapeutic Potential of Biotherapeutics Targeting RPTPγ and RPTPζ (PTPRG and PTPRZ1)
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Alonso, A.; Sasin, J.; Bottini, N.; Friedberg, I.; Friedberg, I.; Osterman, A.; Godzik, A.; Hunter, T.; Dixon, J.; Mustelin, T. Protein tyrosine phosphatases in the human genome. Cell 2004, 117, 699–711. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.N.; Mortensen, O.H.; Peters, G.H.; Drake, P.G.; Iversen, L.F.; Olsen, O.H.; Jansen, P.G.; Andersen, H.S.; Tonks, N.K.; Moller, N.P. Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol. Cell. Biol. 2001, 21, 7117–7136. [Google Scholar] [CrossRef] [PubMed]
- Tonks, N.K. Protein tyrosine phosphatases: From genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 2006, 7, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Streuli, M.; Krueger, N.X.; Ariniello, P.D.; Tang, M.; Munro, J.M.; Blattler, W.A.; Adler, D.A.; Disteche, C.M.; Saito, H. Expression of the receptor-linked protein tyrosine phosphatase LAR: Proteolytic cleavage and shedding of the CAM-like extracellular region. EMBO J. 1992, 11, 897–907. [Google Scholar] [PubMed]
- Mohebiany, A.N.; Nikolaienko, R.M.; Bouyain, S.; Harroch, S. Receptor-type tyrosine phosphatase ligands: Looking for the needle in the haystack. FEBS J. 2013, 280, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Den Hertog, J.; Ostman, A.; Bohmer, F.D. Protein tyrosine phosphatases: Regulatory mechanisms. FEBS J. 2008, 275, 831–847. [Google Scholar] [CrossRef] [PubMed]
- Bilwes, A.M.; den Hertog, J.; Hunter, T.; Noel, J.P. Structural basis for inhibition of receptor protein-tyrosine phosphatase-alpha by dimerization. Nature 1996, 382, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Majeti, R.; Bilwes, A.M.; Noel, J.P.; Hunter, T.; Weiss, A. Dimerization-induced inhibition of receptor protein tyrosine phosphatase function through an inhibitory wedge. Science 1998, 279, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Nam, H.J.; Poy, F.; Saito, H.; Frederick, C.A. Structural basis for the function and regulation of the receptor protein tyrosine phosphatase CD45. J. Exp. Med. 2005, 201, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Barr, A.J.; Ugochukwu, E.; Lee, W.H.; King, O.N.; Filippakopoulos, P.; Alfano, I.; Savitsky, P.; Burgess-Brown, N.A.; Muller, S.; Knapp, S. Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell 2009, 136, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, W.J.; Pulido, R. Protein tyrosine phosphatase variants in human hereditary disorders and disease susceptibilities. Biochim. Biophys. Acta 2013, 1832, 1673–1696. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, W.J.; Elson, A.; Harroch, S.; Stoker, A.W. Protein tyrosine phosphatases: Functional inferences from mouse models and human diseases. FEBS J. 2008, 275, 816–830. [Google Scholar] [CrossRef] [PubMed]
- Ostman, A.; Hellberg, C.; Bohmer, F.D. Protein-tyrosine phosphatases and cancer. Nat. Rev. Cancer 2006, 6, 307–320. [Google Scholar] [CrossRef] [PubMed]
- He, R.J.; Yu, Z.H.; Zhang, R.Y.; Zhang, Z.Y. Protein tyrosine phosphatases as potential therapeutic targets. Acta Pharmacol. Sin. 2014, 35, 1227–1246. [Google Scholar] [CrossRef] [PubMed]
- Stanford, S.M.; Bottini, N. Targeting Tyrosine Phosphatases: Time to End the Stigma. Trends Pharmacol. Sci. 2017, 38, 524–540. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.N.; LaMarche, M.J.; Chan, H.M.; Fekkes, P.; Garcia-Fortanet, J.; Acker, M.G.; Antonakos, B.; Chen, C.H.; Chen, Z.; Cooke, V.G.; et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016, 535, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Stanford, S.M.; Aleshin, A.E.; Zhang, V.; Ardecky, R.J.; Hedrick, M.P.; Zou, J.; Ganji, S.R.; Bliss, M.R.; Yamamoto, F.; Bobkov, A.A.; et al. Diabetes reversal by inhibition of the low-molecular-weight tyrosine phosphatase. Nat. Chem. Biol. 2017, 13, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Ecker, D.M.; Jones, S.D.; Levine, H.L. The therapeutic monoclonal antibody market. mAbs 2015, 7, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Kato, C.; Kato, A. Therapeutic antibodies: Their mechanisms of action and the pathological findings they induce in toxicity studies. J. Toxicol. Pathol. 2015, 28, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Leader, B.; Baca, Q.J.; Golan, D.E. Protein therapeutics: A summary and pharmacological classification. Nat. Rev. Drug Discov. 2008, 7, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Ryman, J.T.; Meibohm, B. Pharmacokinetics of Monoclonal Antibodies. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Reichert, J.M.; Rosensweig, C.J.; Faden, L.B.; Dewitz, M.C. Monoclonal antibody successes in the clinic. Nat. Biotechnol. 2005, 23, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Ostman, A.; Yang, Q.; Tonks, N.K. Expression of DEP-1, a receptor-like protein-tyrosine-phosphatase, is enhanced with increasing cell density. Proc. Natl. Acad. Sci. USA 1994, 91, 9680–9684. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Takahashi, K.; Mernaugh, R.L.; Tsuboi, N.; Liu, H.; Daniel, T.O. A monoclonal antibody against CD148, a receptor-like tyrosine phosphatase, inhibits endothelial-cell growth and angiogenesis. Blood 2006, 108, 1234–1242. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Takahashi, K.; Mernaugh, R.L.; Friedman, D.B.; Weller, R.; Tsuboi, N.; Yamashita, H.; Quaranta, V.; Takahashi, T. Thrombospondin-1 acts as a ligand for CD148 tyrosine phosphatase. Proc. Natl. Acad. Sci. USA 2012, 109, 1985–1990. [Google Scholar] [CrossRef] [PubMed]
- Whiteford, J.R.; Xian, X.; Chaussade, C.; Vanhaesebroeck, B.; Nourshargh, S.; Couchman, J.R. Syndecan-2 is a novel ligand for the protein tyrosine phosphatase receptor CD148. Mol. Biol. Cell 2011, 22, 3609–3624. [Google Scholar] [CrossRef] [PubMed]
- Tarcic, G.; Boguslavsky, S.K.; Wakim, J.; Kiuchi, T.; Liu, A.; Reinitz, F.; Nathanson, D.; Takahashi, T.; Mischel, P.S.; Ng, T.; et al. An unbiased screen identifies DEP-1 tumor suppressor as a phosphatase controlling EGFR endocytosis. Curr. Biol. 2009, 19, 1788–1798. [Google Scholar] [CrossRef] [PubMed]
- Brunner, P.M.; Heier, P.C.; Mihaly-Bison, J.; Priglinger, U.; Binder, B.R.; Prager, G.W. Density enhanced phosphatase-1 down-regulates urokinase receptor surface expression in confluent endothelial cells. Blood 2011, 117, 4154–4161. [Google Scholar] [CrossRef] [PubMed]
- Ruivenkamp, C.A.; van Wezel, T.; Zanon, C.; Stassen, A.P.; Vlcek, C.; Csikos, T.; Klous, A.M.; Tripodis, N.; Perrakis, A.; Boerrigter, L.; et al. Ptprj is a candidate for the mouse colon-cancer susceptibility locus Scc1 and is frequently deleted in human cancers. Nat. Genet. 2002, 31, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Rollin, J.; Pouplard, C.; Gratacap, M.P.; Leroux, D.; May, M.A.; Aupart, M.; Gouilleux-Gruart, V.; Payrastre, B.; Gruel, Y. Polymorphisms of protein tyrosine phosphatase CD148 influence FcgammaRIIA-dependent platelet activation and the risk of heparin-induced thrombocytopenia. Blood 2012, 120, 1309–1316. [Google Scholar] [CrossRef] [PubMed]
- Walchli, S.; Espanel, X.; Hooft van Huijsduijnen, R. Sap-1/PTPRH activity is regulated by reversible dimerization. Biochem. Biophys. Res. Commun. 2005, 331, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Hower, A.E.; Beltran, P.J.; Bixby, J.L. Dimerization of tyrosine phosphatase PTPRO decreases its activity and ability to inactivate TrkC. J. Neurochem. 2009, 110, 1635–1647. [Google Scholar] [CrossRef] [PubMed]
- Tangye, S.G.; Phillips, J.H.; Lanier, L.L.; de Vries, J.E.; Aversa, G. CD148: A receptor-type protein tyrosine phosphatase involved in the regulation of human T cell activation. J. Immunol. 1998, 161, 3249–3255. [Google Scholar] [PubMed]
- Dave, R.K.; Naylor, A.J.; Young, S.P.; Bayley, R.; Hardie, D.L.; Haworth, O.; Rider, D.A.; Cook, A.D.; Buckley, C.D.; Kellie, S. Differential expression of CD148 on leukocyte subsets in inflammatory arthritis. Arthritis Res. Ther. 2013, 15, R108. [Google Scholar] [CrossRef] [PubMed]
- Dave, R.K.; Hume, D.A.; Elsegood, C.; Kellie, S. CD148/DEP-1 association with areas of cytoskeletal organisation in macrophages. Exp. Cell Res. 2009, 315, 1734–1744. [Google Scholar] [CrossRef] [PubMed]
- Paduano, F.; Ortuso, F.; Campiglia, P.; Raso, C.; Iaccino, E.; Gaspari, M.; Gaudio, E.; Mangone, G.; Carotenuto, A.; Bilotta, A.; et al. Isolation and functional characterization of peptide agonists of PTPRJ, a tyrosine phosphatase receptor endowed with tumor suppressor activity. ACS Chem. Biol. 2012, 7, 1666–1676. [Google Scholar] [CrossRef] [PubMed]
- Katsumoto, T.R.; Kudo, M.; Chen, C.; Sundaram, A.; Callahan, E.C.; Zhu, J.W.; Lin, J.; Rosen, C.E.; Manz, B.N.; Lee, J.W.; et al. The phosphatase CD148 promotes airway hyperresponsiveness through SRC family kinases. J. Clin. Investig. 2013, 123, 2037–2048. [Google Scholar] [CrossRef] [PubMed]
- Shintani, T.; Higashi, S.; Takeuchi, Y.; Gaudio, E.; Trapasso, F.; Fusco, A.; Noda, M. The R3 receptor-like protein tyrosine phosphatase subfamily inhibits insulin signaling by dephosphorylating the insulin receptor at specific sites. J. Biochem. 2015, 158, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Kruger, J.; Brachs, S.; Trappiel, M.; Kintscher, U.; Meyborg, H.; Wellnhofer, E.; Thone-Reineke, C.; Stawowy, P.; Ostman, A.; Birkenfeld, A.L.; et al. Enhanced insulin signaling in density-enhanced phosphatase-1 (DEP-1) knockout mice. Mol. Metab. 2015, 4, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Shintani, T.; Higashi, S.; Suzuki, R.; Takeuchi, Y.; Ikaga, R.; Yamazaki, T.; Kobayashi, K.; Noda, M. PTPRJ Inhibits Leptin Signaling, and Induction of PTPRJ in the Hypothalamus Is a Cause of the Development of Leptin Resistance. Sci. Rep. 2017, 7, 11627. [Google Scholar] [CrossRef] [PubMed]
- Senis, Y.A.; Tomlinson, M.G.; Ellison, S.; Mazharian, A.; Lim, J.; Zhao, Y.; Kornerup, K.N.; Auger, J.M.; Thomas, S.G.; Dhanjal, T.; et al. The tyrosine phosphatase CD148 is an essential positive regulator of platelet activation and thrombosis. Blood 2009, 113, 4942–4954. [Google Scholar] [CrossRef] [PubMed]
- Mori, J.; Nagy, Z.; Di Nunzio, G.; Smith, C.W.; Geer, M.J.; Al Ghaithi, R.; van Geffen, J.P.; Heising, S.; Boothman, L.; Tullemans, B.M.E.; et al. Maintenance of murine platelet homeostasis by the kinase Csk and the phosphatase CD148. Blood 2018. [Google Scholar] [CrossRef] [PubMed]
- Winderlich, M.; Keller, L.; Cagna, G.; Broermann, A.; Kamenyeva, O.; Kiefer, F.; Deutsch, U.; Nottebaum, A.F.; Vestweber, D. VE-PTP controls blood vessel development by balancing Tie-2 activity. J. Cell Biol. 2009, 185, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Frye, M.; Dierkes, M.; Kuppers, V.; Vockel, M.; Tomm, J.; Zeuschner, D.; Rossaint, J.; Zarbock, A.; Koh, G.Y.; Peters, K.; et al. Interfering with VE-PTP stabilizes endothelial junctions in vivo via Tie-2 in the absence of VE-cadherin. J. Exp. Med. 2015, 212, 2267–2287. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Frye, M.; Lee, B.L.; Reinardy, J.L.; McClung, J.M.; Ding, K.; Kojima, M.; Xia, H.; Seidel, C.; Lima e Silva, R.; et al. Targeting VE-PTP activates TIE2 and stabilizes the ocular vasculature. J. Clin. Investig. 2014, 124, 4564–4576. [Google Scholar] [CrossRef] [PubMed]
- Campochiaro, P.A.; Sophie, R.; Tolentino, M.; Miller, D.M.; Browning, D.; Boyer, D.S.; Heier, J.S.; Gambino, L.; Withers, B.; Brigell, M.; et al. Treatment of diabetic macular edema with an inhibitor of vascular endothelial-protein tyrosine phosphatase that activates Tie2. Ophthalmology 2015, 122, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Campochiaro, P.A.; Khanani, A.; Singer, M.; Patel, S.; Boyer, D.; Dugel, P.; Kherani, S.; Withers, B.; Gambino, L.; Peters, K.; et al. Enhanced Benefit in Diabetic Macular Edema from AKB-9778 Tie2 Activation Combined with Vascular Endothelial Growth Factor Suppression. Ophthalmology 2016, 123, 1722–1730. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; Gupta, N.; Walcott, B.P.; Snuderl, M.; Kesler, C.T.; Kirkpatrick, N.D.; Heishi, T.; Huang, Y.; Martin, J.D.; Ager, E.; et al. Effects of vascular-endothelial protein tyrosine phosphatase inhibition on breast cancer vasculature and metastatic progression. J. Natl. Cancer Inst. 2013, 105, 1188–1201. [Google Scholar] [CrossRef] [PubMed]
- Gurnik, S.; Devraj, K.; Macas, J.; Yamaji, M.; Starke, J.; Scholz, A.; Sommer, K.; Di Tacchio, M.; Vutukuri, R.; Beck, H.; et al. Angiopoietin-2-induced blood-brain barrier compromise and increased stroke size are rescued by VE-PTP-dependent restoration of Tie2 signaling. Acta Neuropathol. 2016, 131, 753–773. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Tenney, A.P.; Busch, S.A.; Horn, K.P.; Cuascut, F.X.; Liu, K.; He, Z.; Silver, J.; Flanagan, J.G. PTPsigma is a receptor for chondroitin sulfate proteoglycan, an inhibitor of neural regeneration. Science 2009, 326, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Coles, C.H.; Shen, Y.; Tenney, A.P.; Siebold, C.; Sutton, G.C.; Lu, W.; Gallagher, J.T.; Jones, E.Y.; Flanagan, J.G.; Aricescu, A.R. Proteoglycan-specific molecular switch for RPTPsigma clustering and neuronal extension. Science 2011, 332, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Giger, R.J. A new role for RPTPsigma in spinal cord injury: Signaling chondroitin sulfate proteoglycan inhibition. Sci. Signal. 2010, 3. [Google Scholar] [CrossRef] [PubMed]
- Lang, B.T.; Cregg, J.M.; DePaul, M.A.; Tran, A.P.; Xu, K.; Dyck, S.M.; Madalena, K.M.; Brown, B.P.; Weng, Y.L.; Li, S.; et al. Modulation of the proteoglycan receptor PTPsigma promotes recovery after spinal cord injury. Nature 2015, 518, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.T.; Wang, L.; Lang, B.T.; Cregg, J.M.; Dunbar, C.L.; Woodward, W.R.; Silver, J.; Ripplinger, C.M.; Habecker, B.A. Targeting protein tyrosine phosphatase sigma after myocardial infarction restores cardiac sympathetic innervation and prevents arrhythmias. Nat. Commun. 2015, 6, 6235. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Massa, S.M.; Ensslen-Craig, S.E.; Major, D.L.; Yang, T.; Tisi, M.A.; Derevyanny, V.D.; Runge, W.O.; Mehta, B.P.; Moore, L.A.; et al. Protein-tyrosine phosphatase (PTP) wedge domain peptides: A novel approach for inhibition of PTP function and augmentation of protein-tyrosine kinase function. J. Biol. Chem. 2006, 281, 16482–16492. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.L.; Hardy, S.; Aubry, I.; Landry, M.; Haggarty, A.; Saragovi, H.U.; Tremblay, M.L. Identification of function-regulating antibodies targeting the receptor protein tyrosine phosphatase sigma ectodomain. PLoS ONE 2017, 12, e0178489. [Google Scholar] [CrossRef] [PubMed]
- Doody, K.M.; Stanford, S.M.; Sacchetti, C.; Svensson, M.N.; Coles, C.H.; Mitakidis, N.; Kiosses, W.B.; Bartok, B.; Fos, C.; Cory, E.; et al. Targeting phosphatase-dependent proteoglycan switch for rheumatoid arthritis therapy. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015, 42, 11–55. [Google Scholar] [CrossRef] [PubMed]
- Korb-Pap, A.; Stratis, A.; Muhlenberg, K.; Niederreiter, B.; Hayer, S.; Echtermeyer, F.; Stange, R.; Zwerina, J.; Pap, T.; Pavenstadt, H.; et al. Early structural changes in cartilage and bone are required for the attachment and invasion of inflamed synovial tissue during destructive inflammatory arthritis. Ann. Rheum. Dis. 2012, 71, 1004–1011. [Google Scholar] [CrossRef] [PubMed]
- Bunin, A.; Sisirak, V.; Ghosh, H.S.; Grajkowska, L.T.; Hou, Z.E.; Miron, M.; Yang, C.; Ceribelli, M.; Uetani, N.; Chaperot, L.; et al. Protein Tyrosine Phosphatase PTPRS Is an Inhibitory Receptor on Human and Murine Plasmacytoid Dendritic Cells. Immunity 2015, 43, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Muise, A.M.; Walters, T.; Wine, E.; Griffiths, A.M.; Turner, D.; Duerr, R.H.; Regueiro, M.D.; Ngan, B.Y.; Xu, W.; Sherman, P.M.; et al. Protein-tyrosine phosphatase sigma is associated with ulcerative colitis. Curr. Biol. 2007, 17, 1212–1218. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, Y.; Kong, W.; Hussain, R.; Horiuchi, M.; Tremblay, M.L.; Ganea, D.; Li, S. Protein tyrosine phosphatase sigma regulates autoimmune encephalomyelitis development. Brain Behav. Immun. 2017, 65, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Tchilian, E.Z.; Beverley, P.C. Altered CD45 expression and disease. Trends Immunol. 2006, 27, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Wulf, G.G.; Luo, K.L.; Goodell, M.A.; Brenner, M.K. Anti-CD45-mediated cytoreduction to facilitate allogeneic stem cell transplantation. Blood 2003, 101, 2434–2439. [Google Scholar] [CrossRef] [PubMed]
- Brenner, M.K.; Wulf, G.G.; Rill, D.R.; Luo, K.L.; Goodell, M.A.; Mei, Z.; Kuehnle, I.; Brown, M.P.; Pule, M.; Heslop, H.E.; et al. Complement-fixing CD45 monoclonal antibodies to facilitate stem cell transplantation in mouse and man. Ann. N. Y. Acad. Sci. 2003, 996, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.C.; Appelbaum, F.R.; Eary, J.F.; Fisher, D.R.; Durack, L.D.; Hui, T.E.; Martin, P.J.; Mitchell, D.; Press, O.W.; Storb, R.; et al. Phase I study of (131)I-anti-CD45 antibody plus cyclophosphamide and total body irradiation for advanced acute leukemia and myelodysplastic syndrome. Blood 1999, 94, 1237–1247. [Google Scholar] [PubMed]
- Pagel, J.M.; Appelbaum, F.R.; Eary, J.F.; Rajendran, J.; Fisher, D.R.; Gooley, T.; Ruffner, K.; Nemecek, E.; Sickle, E.; Durack, L.; et al. 131I-anti-CD45 antibody plus busulfan and cyclophosphamide before allogeneic hematopoietic cell transplantation for treatment of acute myeloid leukemia in first remission. Blood 2006, 107, 2184–2191. [Google Scholar] [CrossRef] [PubMed]
- Frost, S.H.; Miller, B.W.; Back, T.A.; Santos, E.B.; Hamlin, D.K.; Knoblaugh, S.E.; Frayo, S.L.; Kenoyer, A.L.; Storb, R.; Press, O.W.; et al. alpha-Imaging Confirmed Efficient Targeting of CD45-Positive Cells After 211At-Radioimmunotherapy for Hematopoietic Cell Transplantation. J. Nucl. Med. 2015, 56, 1766–1773. [Google Scholar] [CrossRef] [PubMed]
- Palchaudhuri, R.; Saez, B.; Hoggatt, J.; Schajnovitz, A.; Sykes, D.B.; Tate, T.A.; Czechowicz, A.; Kfoury, Y.; Ruchika, F.; Rossi, D.J.; et al. Non-genotoxic conditioning for hematopoietic stem cell transplantation using a hematopoietic-cell-specific internalizing immunotoxin. Nat. Biotechnol. 2016, 34, 738–745. [Google Scholar] [CrossRef] [PubMed]
- Bagga, S.; Seth, D.; Batra, J.K. The cytotoxic activity of ribosome-inactivating protein saporin-6 is attributed to its rRNA N-glycosidase and internucleosomal DNA fragmentation activities. J. Biol. Chem. 2003, 278, 4813–4820. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, G.; Perfetti, V.; Tonon, L.; Novella, A.; Lucotti, C.; Danova, M.; Glennie, M.J.; Merlini, G.; Cazzola, M. Saporin, a ribosome-inactivating protein used to prepare immunotoxins, induces cell death via apoptosis. Br. J. Haematol. 1996, 93, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Shintani, T.; Maeda, N.; Nishiwaki, T.; Noda, M. Characterization of rat receptor-like protein tyrosine phosphatase gamma isoforms. Biochem. Biophys. Res. Commun. 1997, 230, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Lissandrini, D.; Vermi, W.; Vezzalini, M.; Sozzani, S.; Facchetti, F.; Bellone, G.; Mafficini, A.; Gentili, F.; Ennas, M.G.; Tecchio, C.; et al. Receptor-type protein tyrosine phosphatase gamma (PTPgamma), a new identifier for myeloid dendritic cells and specialized macrophages. Blood 2006, 108, 4223–4231. [Google Scholar] [CrossRef] [PubMed]
- Krueger, N.X.; Saito, H. A human transmembrane protein-tyrosine-phosphatase, PTP zeta, is expressed in brain and has an N-terminal receptor domain homologous to carbonic anhydrases. Proc. Natl. Acad. Sci. USA 1992, 89, 7417–7421. [Google Scholar] [CrossRef] [PubMed]
- LaForgia, S.; Morse, B.; Levy, J.; Barnea, G.; Cannizzaro, L.A.; Li, F.; Nowell, P.C.; Boghosian-Sell, L.; Glick, J.; Weston, A.; et al. Receptor protein-tyrosine phosphatase gamma is a candidate tumor suppressor gene at human chromosome region 3p21. Proc. Natl. Acad. Sci. USA 1991, 88, 5036–5040. [Google Scholar] [CrossRef] [PubMed]
- Vezzalini, M.; Mafficini, A.; Tomasello, L.; Lorenzetto, E.; Moratti, E.; Fiorini, Z.; Holyoake, T.L.; Pellicano, F.; Krampera, M.; Tecchio, C.; et al. A new monoclonal antibody detects downregulation of protein tyrosine phosphatase receptor type gamma in chronic myeloid leukemia patients. J. Hematol. Oncol. 2017, 10, 129. [Google Scholar] [CrossRef] [PubMed]
- Lamprianou, S.; Vacaresse, N.; Suzuki, Y.; Meziane, H.; Buxbaum, J.D.; Schlessinger, J.; Harroch, S. Receptor protein tyrosine phosphatase gamma is a marker for pyramidal cells and sensory neurons in the nervous system and is not necessary for normal development. Mol. Cell. Biol. 2006, 26, 5106–5119. [Google Scholar] [CrossRef] [PubMed]
- Shintani, T.; Watanabe, E.; Maeda, N.; Noda, M. Neurons as well as astrocytes express proteoglycan-type protein tyrosine phosphatase zeta/RPTPbeta: Analysis of mice in which the PTPzeta/RPTPbeta gene was replaced with the LacZ gene. Neurosci. Lett. 1998, 247, 135–138. [Google Scholar] [CrossRef]
- Kuboyama, K.; Fujikawa, A.; Suzuki, R.; Tanga, N.; Noda, M. Role of Chondroitin Sulfate (CS) Modification in the Regulation of Protein-tyrosine Phosphatase Receptor Type Z (PTPRZ) Activity: Pleiotrophin-Ptprz-a signaling is involved in oligodendrocyte differentiation. J. Biol. Chem. 2016, 291, 18117–18128. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.W.; Keough, M.B.; Haylock-Jacobs, S.; Cua, R.; Doring, A.; Sloka, S.; Stirling, D.P.; Rivest, S.; Yong, V.W. Chondroitin sulfate proteoglycans in demyelinated lesions impair remyelination. Ann. Neurol. 2012, 72, 419–432. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Senis, Y.A.; Barr, A.J. Targeting Receptor-Type Protein Tyrosine Phosphatases with Biotherapeutics: Is Outside-in Better than Inside-Out? Molecules 2018, 23, 569. https://doi.org/10.3390/molecules23030569
Senis YA, Barr AJ. Targeting Receptor-Type Protein Tyrosine Phosphatases with Biotherapeutics: Is Outside-in Better than Inside-Out? Molecules. 2018; 23(3):569. https://doi.org/10.3390/molecules23030569
Chicago/Turabian StyleSenis, Yotis A., and Alastair J. Barr. 2018. "Targeting Receptor-Type Protein Tyrosine Phosphatases with Biotherapeutics: Is Outside-in Better than Inside-Out?" Molecules 23, no. 3: 569. https://doi.org/10.3390/molecules23030569
APA StyleSenis, Y. A., & Barr, A. J. (2018). Targeting Receptor-Type Protein Tyrosine Phosphatases with Biotherapeutics: Is Outside-in Better than Inside-Out? Molecules, 23(3), 569. https://doi.org/10.3390/molecules23030569