
Natural Alkaloids and Heterocycles as G-Quadruplex Ligands and Potential Anticancer Agents


Abstract
1. Introduction
2. G-Quadruplex Structures
3. Existence and Distribution of G-Quadruplexes
4. G-Quadruplexes as Promising Targets for Anticancer Drug Design
4.1. Telomeric G-Quadruplexes
4.2. Oncogene Promoter Region G-Quadruplexes
4.3. RNA G-Quadruplexes
5. Natural Alkaloids and Their Derivatives as G-Quadruplex Ligands
5.1. Methods to Discover G-Quadruplex Ligands
5.2. Quindoline Derivatives
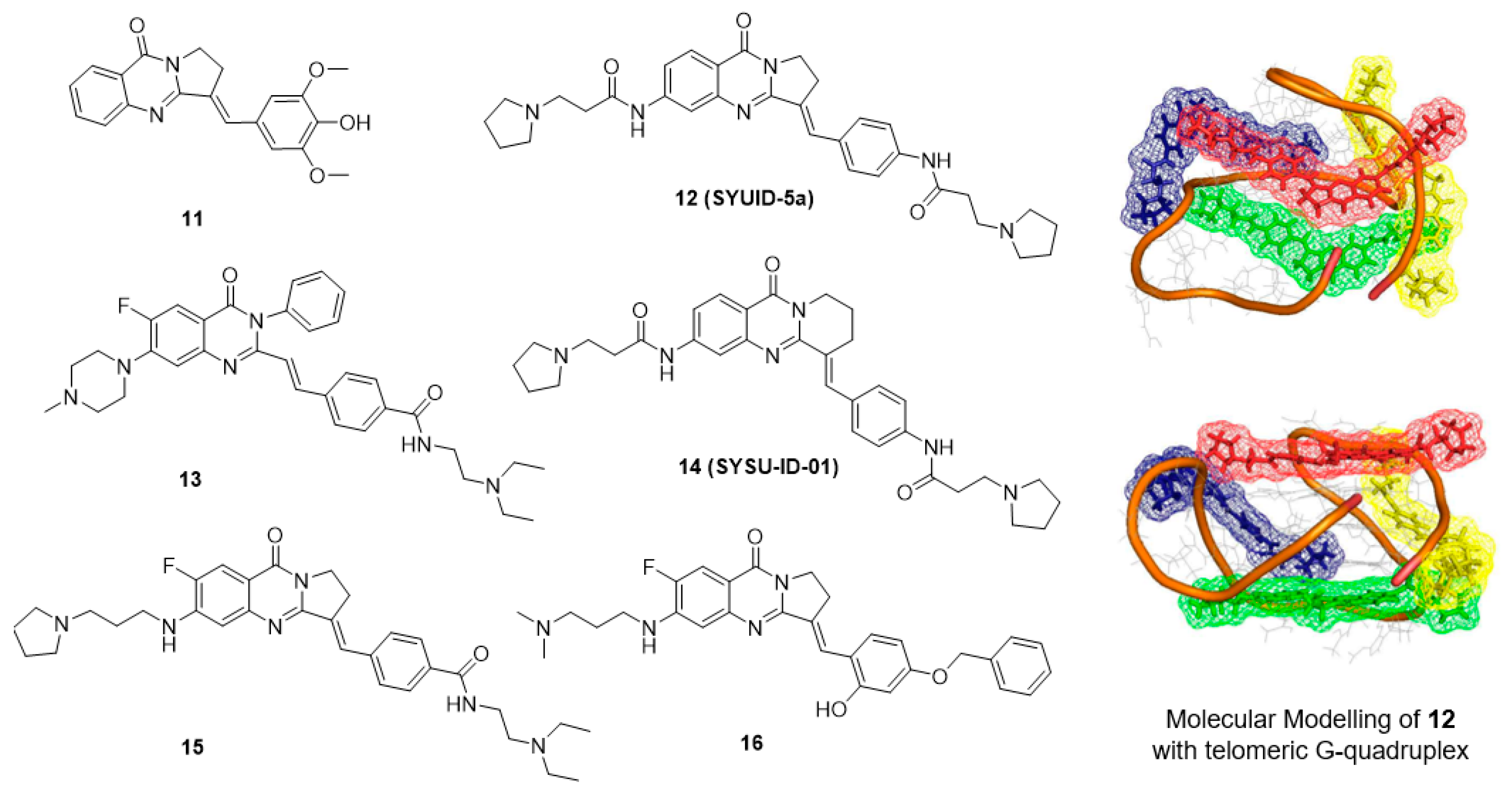
5.3. Isaindigotone Derivatives
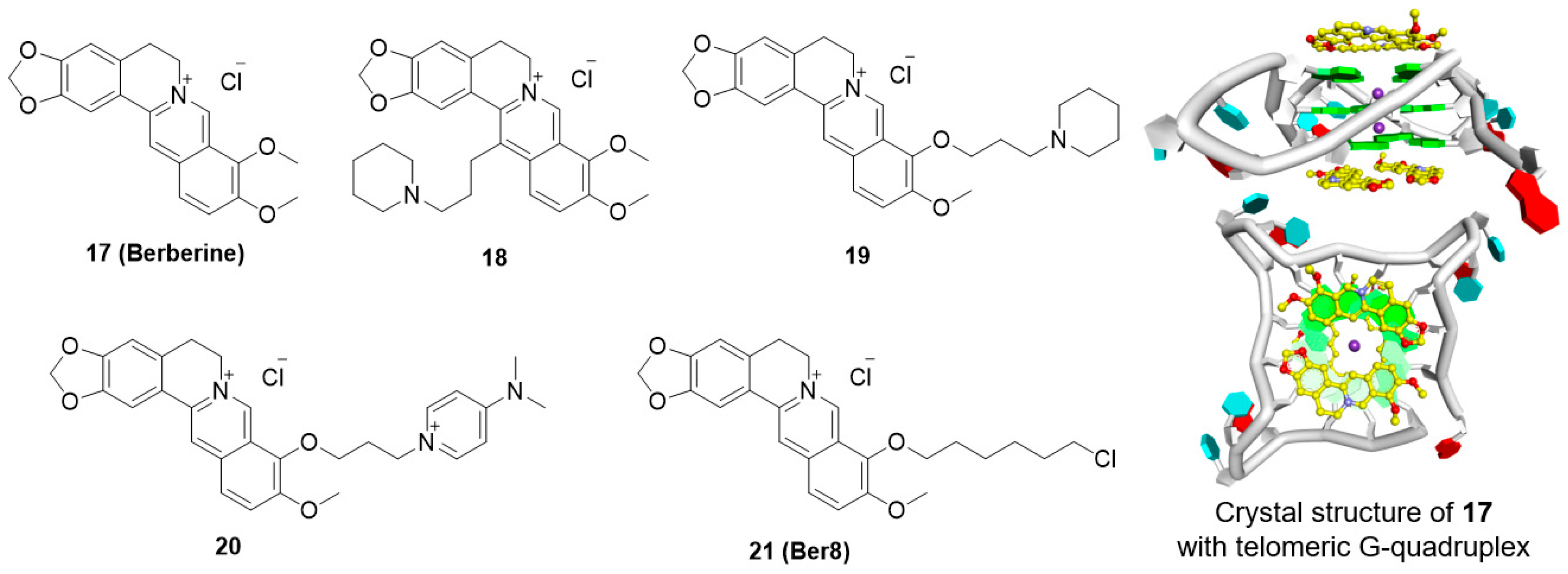
5.4. Berberine Derivatives
5.5. Other Alkaloid Derivatives
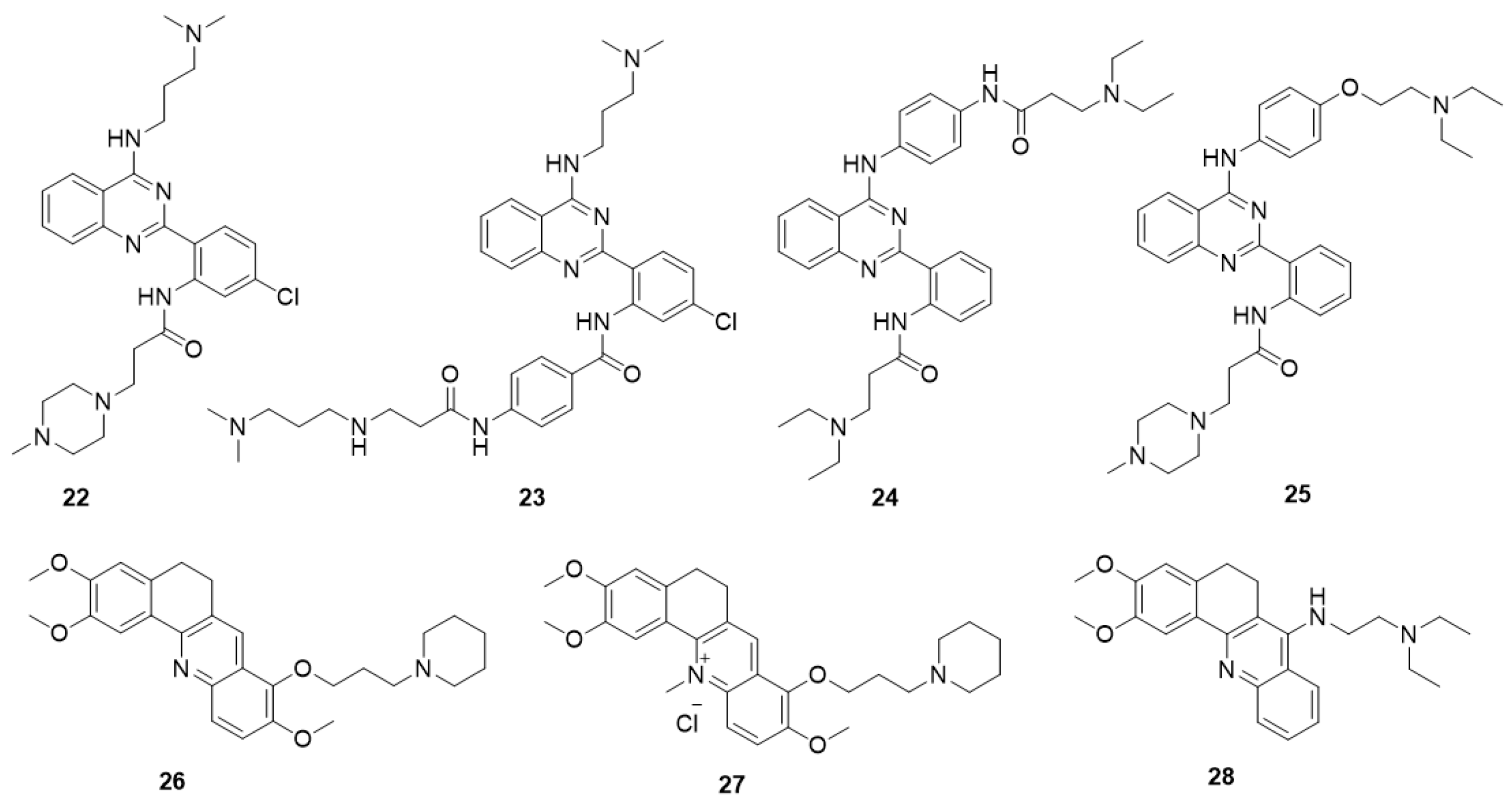
5.5.1. Quinazoline Derivatives
5.5.2. Acridine Derivatives
5.5.3. Multiaryl-Substituted Imidazole Derivatives
6. Summary and Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Smith, F.W.; Feigon, J. Quadruplex structure of Oxytricha telomeric DNA oligonucleotides. Nature 1992, 356, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Huppert, J.L.; Balasubramanian, S. Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 2005, 33, 2908–2916. [Google Scholar] [CrossRef] [PubMed]
- Eddy, J.; Maizels, N. Gene function correlates with potential for G4 DNA formation in the human genome. Nucleic Acids Res. 2006, 34, 3887–3896. [Google Scholar] [CrossRef] [PubMed]
- Chambers, V.S.; Marsico, G.; Boutell, J.M.; Di Antonio, M.; Smith, G.P.; Balasubramanian, S. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat. Biotechnol. 2015, 33, 877–881. [Google Scholar] [CrossRef] [PubMed]
- Hansel-Hertsch, R.; Beraldi, D.; Lensing, S.V.; Marsico, G.; Zyner, K.; Parry, A.; Di Antonio, M.; Pike, J.; Kimura, H.; Narita, M.; et al. G-quadruplex structures mark human regulatory chromatin. Nat. Genet. 2016, 48, 1267–1272. [Google Scholar] [CrossRef] [PubMed]
- Fedeles, B.I. G-quadruplex-forming promoter sequences enable transcriptional activation in response to oxidative stress. Proc. Natl. Acad. Sci. USA 2017, 114, 2788–2790. [Google Scholar] [CrossRef] [PubMed]
- Kwok, C.K.; Marsico, G.; Sahakyan, A.B.; Chambers, V.S.; Balasubramanian, S. rG4-seq reveals widespread formation of G-quadruplex structures in the human transcriptome. Nat. Methods 2016, 13, 841–844. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, A.L.; Singh, K.; Zhong, Y.; Drewe, P.; Rajasekhar, V.K.; Sanghvi, V.R.; Mavrakis, K.J.; Jiang, M.; Roderick, J.E.; Van der Meulen, J.; et al. Rna G-quadruplexes cause eif4a-dependent oncogene translation in cancer. Nature 2014, 513, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Hurley, L.H.; Neidle, S. Targeting G-quadruplexes in gene promoters: A novel anticancer strategy? Nat. Rev. Drug Discov. 2011, 10, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Phan, A.T.; Kuryavyi, V.; Gaw, H.Y.; Patel, D.J. Small-molecule interaction with a five-guanine-tract G-quadruplex structure from the human MYC promoter. Nat. Chem. Biol. 2005, 1, 234. [Google Scholar] [CrossRef]
- Kim, M.Y.; Vankayalapati, H.; Shin-Ya, K.; Wierzba, K.; Hurley, L.H. Telomestatin, a potent telomerase inhibitor that interacts quite specifically with the human telomeric intramolecular G-quadruplex. J. Am. Chem. Soc. 2002, 124, 2098–2099. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.J.; Heddi, B.; Tera, M.; Iida, K.; Nagasawa, K.; Phan, A.T. Solution structure of an intramolecular (3 + 1) human telomeric G-quadruplex bound to a telomestatin derivative. J. Am. Chem. Soc. 2013, 135, 13495–13501. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.; Miller, K.M.; Forment, J.V.; Bradshaw, C.R.; Nikan, M.; Britton, S.; Oelschlaegel, T.; Xhemalce, B.; Balasubramanian, S.; Jackson, S.P. Small-molecule-induced DNA damage identifies alternative DNA structures in human genes. Nat. Chem. Biol. 2012, 8, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Drygin, D.; Siddiqui-Jain, A.; O’Brien, S.; Schwaebe, M.; Lin, A.; Bliesath, J.; Ho, C.B.; Proffitt, C.; Trent, K.; Whitten, J.P.; et al. Anticancer activity of CX-3543: A direct inhibitor of rRNA biogenesis. Cancer Res. 2009, 69, 7653–7661. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.S.S.; Zhou, X.; Tan, Z. Biological function of G-quadruplex nucleic acids and potential application in medicinal chemistry. Curr. Top. Med. Chem. 2015, 15, 1939. [Google Scholar] [CrossRef] [PubMed]
- Rocca, R.; Moraca, F.; Costa, G.; Alcaro, S.; Distinto, S.; Maccioni, E.; Ortuso, F.; Artese, A.; Parrotta, L. Structure-based virtual screening of novel natural alkaloid derivatives as potential binders of h-telo and c-myc DNA G-quadruplex conformations. Molecules 2014, 20, 206–223. [Google Scholar] [CrossRef] [PubMed]
- Ou, T.-M.; Lu, Y.-J.; Tan, J.-H.; Huang, Z.-S.; Wong, K.-Y.; Gu, L.-Q. G-quadruplexes: Targets in anticancer drug design. ChemMedChem 2008, 3, 690–713. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.J.; Cuesta, J.; Chessari, G.; Read, M.A.; Basra, S.K.; Reszka, A.P.; Morrell, J.; Gowan, S.M.; Incles, C.M.; Tanious, F.A.; et al. Trisubstituted acridine derivatives as potent and selective telomerase inhibitors. J. Med. Chem. 2003, 46, 4463–4476. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Schultes, C.M.; Cuesta, J.; Cuenca, F.; Gunaratnam, M.; Tanious, F.A.; Wilson, W.D.; Neidle, S. Trisubstituted acridines as G-quadruplex telomere targeting agents. Effects of extensions of the 3,6- and 9-side chains on quadruplex binding, telomerase activity, and cell proliferation. J. Med. Chem. 2006, 49, 582–599. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.; Sanders, D.A.; Di Antonio, M.; Matsis, S.; Riou, J.F.; Rodriguez, R.; Balasubramanian, S. Pyridostatin analogues promote telomere dysfunction and long-term growth inhibition in human cancer cells. Org. Biomol. Chem. 2012, 10, 6537–6546. [Google Scholar] [CrossRef] [PubMed]
- Gellert, M.; Lipsett, M.N.; Davies, D.R. Helix formation by guanylic acid. Proc. Natl. Acad. Sci. USA 1962, 48, 2013–2018. [Google Scholar] [CrossRef] [PubMed]
- Phan, A.T.; Modi, Y.S.; Patel, D.J. Propeller-type parallel-stranded G-quadruplexes in the human c-myc promoter. J. Am. Chem. Soc. 2004, 126, 8710–8716. [Google Scholar] [CrossRef] [PubMed]
- Hazel, P.; Parkinson, G.N.; Neidle, S. Topology variation and loop structural homology in crystal and simulated structures of a bimolecular DNA quadruplex. J. Am. Chem. Soc. 2006, 128, 5480–5487. [Google Scholar] [CrossRef] [PubMed]
- Campbell, N.; Collie, G.W.; Neidle, S. Crystallography of DNA and RNA G-quadruplex nucleic acids and their ligand complexes. Curr. Protoc. Nucleic Acid Chem. 2012. [Google Scholar] [CrossRef]
- Matsugami, A.; Okuizumi, T.; Uesugi, S.; Katahira, M. Intramolecular higher order packing of parallel quadruplexes comprising a G:G:G:G tetrad and a G(:A):G(:A):G(:A):G heptad of GGA triplet repeat DNA. J. Biol. Chem. 2003, 278, 28147–28153. [Google Scholar] [CrossRef] [PubMed]
- Neidle, S. The structures of quadruplex nucleic acids and their drug complexes. Curr. Opin. Struct. Biol. 2009, 19, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Collie, G.W.; Promontorio, R.; Hampel, S.M.; Micco, M.; Neidle, S.; Parkinson, G.N. Structural basis for telomeric G-quadruplex targeting by naphthalene diimide ligands. J. Am. Chem. Soc. 2012, 134, 2723–2731. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.J.; Heddi, B.; Hamon, F.; Teulade-Fichou, M.P.; Phan, A.T. Solution structure of a G-quadruplex bound to the bisquinolinium compound PHEN-DC(3). Angew. Chem. 2014, 53, 999–1002. [Google Scholar] [CrossRef] [PubMed]
- Collie, G.W.; Sparapani, S.; Parkinson, G.N.; Neidle, S. Structural basis of telomeric RNA quadruplex--acridine ligand recognition. J. Am. Chem. Soc. 2011, 133, 2721–2728. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.-L.L.; Wang, M.; Lin, S.; Han, Q.-B.B.; Leung, C.-H.H. Recent Development of G-Quadruplex Probes for Cellular Imaging. Curr. Top. Med. Chem. 2015, 15, 1957–1963. [Google Scholar] [CrossRef] [PubMed]
- Schaffitzel, C.; Berger, I.; Postberg, J.; Hanes, J.; Lipps, H.J.; Pluckthun, A. In vitro generated antibodies specific for telomeric guanine-quadruplex DNA react with Stylonychia lemnae macronuclei. Proc. Natl. Acad. Sci. USA 2001, 98, 8572–8577. [Google Scholar] [CrossRef] [PubMed]
- Granotier, C.; Pennarun, G.; Riou, L.; Hoffschir, F.; Gauthier, L.R.; De Cian, A.; Gomez, D.; Mandine, E.; Riou, J.F.; Mergny, J.L.; et al. Preferential binding of a G-quadruplex ligand to human chromosome ends. Nucleic Acids Res. 2005, 33, 4182–4190. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Kuo, I.C.; Ling, I.F.; Chen, C.T.; Chen, H.C.; Lou, P.J.; Lin, J.J.; Chang, T.C. Detection of quadruplex DNA structures in human telomeres by a fluorescent carbazole derivative. Anal. Chem. 2004, 76, 4490–4494. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Tannahill, D.; McCafferty, J.; Balasubramanian, S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 2013, 5, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chu, J.F.; Kao, F.J.; Chiu, Y.C.; Lou, P.J.; Chen, H.C.; Chang, T.C. Verification of antiparallel G-quadruplex structure in human telomeres by using two-photon excitation fluorescence lifetime imaging microscopy of the 3,6-Bis(1-methyl-4-vinylpyridinium)carbazole diiodide molecule. Anal. Chem. 2006, 78, 2810–2815. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.W.; Chen, S.B.; Liu, H.Y.; Ye, W.J.; Ou, T.M.; Tan, J.H.; Li, D.; Gu, L.Q.; Huang, Z.S. Development of a new colorimetric and red-emitting fluorescent dual probe for G-quadruplex nucleic acids. Chem. Commun. 2014, 50, 6927–6930. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yan, S.; Yuan, L.; Zhou, Y.; Song, Y.; Xiao, H.; Weng, X.; Zhou, X. Nonlinear optical dye TSQ1 as an efficiently selective fluorescent probe for G-quadruplex DNA. Org. Chem. Front. 2014, 1, 267. [Google Scholar] [CrossRef]
- Vummidi, B.R.; Alzeer, J.; Luedtke, N.W. Fluorescent probes for G-quadruplex structures. Chem. Biol. Chem. 2013, 14, 540–558. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.J.; Yan, S.C.; Chan, F.Y.; Zou, L.; Chung, W.H.; Wong, W.L.; Qiu, B.; Sun, N.; Chan, P.H.; Huang, Z.S.; et al. Benzothiazole-substituted benzofuroquinolinium dye: A selective switch-on fluorescent probe for G-quadruplex. Chem. Commun. 2011, 47, 4971–4973. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Di Antonio, M.; Tannahill, D.; Balasubramanian, S. Visualization and selective chemical targeting of RNA G-quadruplex structures in the cytoplasm of human cells. Nat. Chem. 2014, 6, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Laguerre, A.; Hukezalie, K.; Winckler, P.; Katranji, F.; Chanteloup, G.; Pirrotta, M.; Perrier-Cornet, J.M.; Wong, J.M.; Monchaud, D. Visualization of RNA-quadruplexes in live cells. J. Am. Chem. Soc. 2015, 137, 8521–8525. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-B.; Hu, M.-H.; Liu, G.-C.; Wang, J.; Ou, T.-M.; Gu, L.-Q.; Huang, Z.-S.; Tan, J.-H. Visualization of NRAS RNA G-quadruplex structures in cells with an engineered fluorogenic hybridization probe. J. Am. Chem. Soc. 2016, 138, 10382–10385. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E.H. Structure and function of telomeres. Nature 1991, 350, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Chadeneau, C.; Hay, K.; Hirte, H.W.; Gallinger, S.; Bacchetti, S. Telomerase activity associated with acquisition of malignancy in human colorectal cancer. Cancer Res. 1995, 55, 2533–2536. [Google Scholar] [PubMed]
- Phatak, P.; Cookson, J.C.; Dai, F.; Smith, V.; Gartenhaus, R.B.; Stevens, M.F.; Burger, A.M. Telomere uncapping by the G-quadruplex ligand RHPS4 inhibits clonogenic tumour cell growth in vitro and in vivo consistent with a cancer stem cell targeting mechanism. Br. J. Cancer 2007, 96, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, G.N.; Lee, M.P.; Neidle, S. Crystal structure of parallel quadruplexes from human telomeric DNA. Nature 2002, 417, 876–880. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Carver, M.; Punchihewa, C.; Jones, R.A.; Yang, D. Structure of the Hybrid-2 type intramolecular human telomeric G-quadruplex in K+ solution: Insights into structure polymorphism of the human telomeric sequence. Nucleic Acids Res. 2007, 35, 4927–4940. [Google Scholar] [CrossRef] [PubMed]
- Ambrus, A.; Chen, D.; Dai, J.; Bialis, T.; Jones, R.A.; Yang, D. Human telomeric sequence forms a hybrid-type intramolecular G-quadruplex structure with mixed parallel/antiparallel strands in potassium solution. Nucleic Acids Res. 2006, 34, 2723–2735. [Google Scholar] [CrossRef] [PubMed]
- Abraham Punnoose, J.; Ma, Y.; Li, Y.; Sakuma, M.; Mandal, S.; Nagasawa, K.; Mao, H. Adaptive and specific recognition of telomeric G-quadruplexes via polyvalency induced unstacking of binding units. J. Am. Chem. Soc. 2017, 139, 7476–7484. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.-H.; Chen, S.-B.; Wang, B.; Ou, T.-M.; Gu, L.-Q.; Tan, J.-H.; Huang, Z.-S. Specific targeting of telomeric multimeric G-quadruplexes by a new triaryl-substituted imidazole. Nucleic Acids Res. 2016, 45, 1606–1618. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui-Jain, A.; Grand, C.L.; Bearss, D.J.; Hurley, L.H. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [Google Scholar] [CrossRef] [PubMed]
- Phan, A.T.; Kuryavyi, V.; Burge, S.; Neidle, S.; Patel, D.J. Structure of an unprecedented G-quadruplex scaffold in the human c-KIT promoter. J. Am. Chem. Soc. 2007, 129, 4386–4392. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Dexheimer, T.S.; Chen, D.; Carver, M.; Ambrus, A.; Jones, R.A.; Yang, D. An intramolecular G-quadruplex structure with mixed parallel/antiparallel g-strands formed in the human BCL-2 promoter region in solution. J. Am. Chem. Soc. 2006, 128, 1096–1098. [Google Scholar] [CrossRef] [PubMed]
- Cogoi, S.; Xodo, L.E. G-quadruplex formation within the promoter of the KRAS proto-oncogene and its effect on transcription. Nucleic Acids Res. 2006, 34, 2536–2549. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Guo, K.; Rusche, J.J.; Hurley, L.H. Facilitation of a structural transition in the polypurine/polypyrimidine tract within the proximal promoter region of the human VEGF gene by the presence of potassium and G-quadruplex-interactive agents. Nucleic Acids Res. 2005, 33, 6070–6080. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Sugiyama, H. Formation of the G-quadruplex and i-motif structures in retinoblastoma susceptibility genes (RB). Nucleic Acids Res. 2006, 34, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Rezler, E.M.; Gokhale, V.; Sun, D.; Hurley, L.H. Characterization of the G-quadruplexes in the duplex nuclease hypersensitive element of the PDGF-A promoter and modulation of PDGF-A promoter activity by TMPyP4. Nucleic Acids Res. 2007, 35, 7698–7713. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.; Lemarteleur, T.; Lacroix, L.; Mailliet, P.; Mergny, J.L.; Riou, J.F. Telomerase downregulation induced by the G-quadruplex ligand 12459 in A549 cells is mediated by hTERT RNA alternative splicing. Nucleic Acids Res. 2004, 32, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Soucek, L.; Whitfield, J.; Martins, C.P.; Finch, A.J.; Murphy, D.J.; Sodir, N.M.; Karnezis, A.N.; Swigart, L.B.; Nasi, S.; Evan, G.I. Modelling MYC inhibition as a cancer therapy. Nature 2008, 455, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Bartel, D.P. RNA G-quadruplexes are globally unfolded in eukaryotic cells and depleted in bacteria. Science 2016, 353, aaf5371. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.B.; Tan, J.H.; Ou, T.M.; Huang, S.L.; An, L.K.; Luo, H.B.; Li, D.; Gu, L.Q.; Huang, Z.S. Pharmacophore-based discovery of triaryl-substituted imidazole as new telomeric G-quadruplex ligand. Bioorg. Med. Chem. Lett. 2011, 21, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Cosconati, S.; Marinelli, L.; Trotta, R.; Virno, A.; Mayol, L.; Novellino, E.; Olson, A.J.; Randazzo, A. Tandem application of virtual screening and NMR experiments in the discovery of brand new DNA quadruplex groove binders. J. Am. Chem. Soc. 2009, 131, 16336–16337. [Google Scholar] [CrossRef] [PubMed]
- Monchaud, D.; Allain, C.; Teulade-Fichou, M.P. Development of a fluorescent intercalator displacement assay (G4-FID) for establishing quadruplex-DNA affinity and selectivity of putative ligands. Bioorg. Med. Chem. Lett. 2006, 16, 4842–4845. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Li, L.; Xiang, J.; Sun, H.; Tang, Y. Fast screening and structural elucidation of G-quadruplex ligands from a mixture via G-quadruplex recognition and NMR methods. Biochimie 2009, 91, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Renciuk, D.; Zhou, J.; Beaurepaire, L.; Guedin, A.; Bourdoncle, A.; Mergny, J.L. A fret-based screening assay for nucleic acid ligands. Methods 2012, 57, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Redman, J.E. Surface plasmon resonance for probing quadruplex folding and interactions with proteins and small molecules. Methods 2007, 43, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, D.; Amato, J.; Zizza, P.; Platella, C.; Cosconati, S.; Cingolani, C.; Biroccio, A.; Novellino, E.; Randazzo, A.; Giancola, C.; et al. Tandem application of ligand-based virtual screening and G4-OAS assay to identify novel G-quadruplex-targeting chemotypes. Biochim. Biophys. Acta 2017, 1861, 1341–1352. [Google Scholar] [CrossRef] [PubMed]
- Dwuma-Badu, D.; Ayim, J.S.; Fiagbe, N.I.; Knapp, J.E.; Schiff, P.L., Jr.; Slatkin, D.J. Constituents of West African medicinal plants XX: Quindoline from cryptolepis sanguinolenta. J. Pharm. Sci. 1978, 67, 433–434. [Google Scholar] [CrossRef] [PubMed]
- Caprio, V.; Guyen, B.; Opoku-Boahen, Y.; Mann, J.; Gowan, S.M.; Kelland, L.M.; Read, M.A.; Neidle, S. A novel inhibitor of human telomerase derived from 10H-indolo[3,2-b]quinoline. Bioorg. Med. Chem. Lett. 2000, 10, 2063–2066. [Google Scholar] [CrossRef]
- Zhou, J.-L.; Lu, Y.-J.; Ou, T.-M.; Zhou, J.-M.; Huang, Z.-S.; Zhu, X.-F.; Du, C.-J.; Bu, X.-Z.; Ma, L.; Gu, L.-Q.; et al. Synthesis and evaluation of quindoline derivatives as G-quadruplex inducing and stabilizing ligands and potential inhibitors of telomerase. J. Med. Chem. 2005, 48, 7315–7321. [Google Scholar] [CrossRef] [PubMed]
- Ou, T.-M.; Lu, Y.-J.; Zhang, C.; Huang, Z.-S.; Wang, X.-D.; Tan, J.-H.; Chen, Y.; Ma, D.-L.; Wong, K.-Y.; Tang, J.-C.; et al. Stabilization of G-quadruplex DNA and down-regulation of oncogene c-myc by quindoline derivatives. J. Med. Chem. 2007, 50, 1465–1474. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.-M.; Zhu, X.-F.; Lu, Y.-J.; Deng, R.; Huang, Z.-S.; Mei, Y.-P.; Wang, Y.; Huang, W.-L.; Liu, Z.-C.; Gu, L.-Q.; et al. Senescence and telomere shortening induced by novel potent G-quadruplex interactive agents, quindoline derivatives, in human cancer cell lines. Oncogene 2006, 25, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-J.; Ou, T.-M.; Tan, J.-H.; Hou, J.-Q.; Shao, W.-Y.; Peng, D.; Sun, N.; Wang, X.-D.; Wu, W.-B.; Bu, X.-Z.; et al. 5-N-methylated quindoline derivatives as telomeric G-quadruplex stabilizing ligands: Effects of 5-N positive charge on quadruplex binding affinity and cell proliferation. J. Med. Chem. 2008, 51, 6381–6392. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Li, Z.; Tan, J.-H.; Ou, T.-M.; Li, D.; Gu, L.-Q.; Huang, Z.-S. Benzofuroquinoline derivatives had remarkable improvement of their selectivity for telomeric G-quadruplex DNA over duplex DNA upon introduction of peptidyl group. Bioconjug. Chem. 2012, 23, 1821–1831. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Carver, M.; Hurley, L.H.; Yang, D. Solution structure of a 2:1 quindoline-c-MYC G-quadruplex: Insights into G-quadruplex-interactive small molecule drug design. J. Am. Chem. Soc. 2011, 133, 17673–17680. [Google Scholar] [CrossRef] [PubMed]
- Zeng, D.-Y.; Kuang, G.-T.; Wang, S.-K.; Peng, W.; Lin, S.-L.; Zhang, Q.; Su, X.-X.; Hu, M.-H.; Wang, H.; Tan, J.-H.; et al. Discovery of novel 11-triazole substituted benzofuro[3,2-b]quinolone derivatives as c-myc G-quadruplex specific stabilizers via click chemistry. J. Med. Chem. 2017, 60, 5407–5423. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-Y.; Chen, A.-C.; Yin, Q.-K.; Li, Z.; Huang, S.-M.; Du, G.; He, J.-H.; Zan, L.-P.; Wang, S.-K.; Xu, Y.-H.; et al. New disubstituted quindoline derivatives inhibiting burkitt’s lymphoma cell proliferation by impeding c-MYC transcription. J. Med. Chem. 2017, 60, 5438–5454. [Google Scholar] [CrossRef] [PubMed]
- Shan, C.; Lin, J.; Hou, J.-Q.; Liu, H.-Y.; Chen, S.-B.; Chen, A.-C.; Ou, T.-M.; Tan, J.-H.; Li, D.; Gu, L.-Q.; et al. Chemical intervention of the NM23-H2 transcriptional programme on c-MYC via a novel small molecule. Nucleic Acids Res. 2015, 43, 6677–6691. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Qin, G.; Cheung, K.K.; Cheng, K.F. New alkaloids from isatis indigotica. Tetrahedron 1997, 53, 13323–13328. [Google Scholar] [CrossRef]
- Tan, J.-H.; Ou, T.-M.; Hou, J.-Q.; Lu, Y.-J.; Huang, S.-L.; Luo, H.-B.; Wu, J.-Y.; Huang, Z.-S.; Wong, K.-Y.; Gu, L.-Q. Isaindigotone derivatives: A new class of highly selective ligands for telomeric G-quadruplex DNA. J. Med. Chem. 2009, 52, 2825–2835. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.-Q.; Tan, J.-H.; Wang, X.-X.; Chen, S.-B.; Huang, S.-Y.; Yan, J.-W.; Chen, S.-H.; Ou, T.-M.; Luo, H.-B.; Li, D.; et al. Impact of planarity of unfused aromatic molecules on G-quadruplex binding: Learning from isaindigotone derivatives. Org. Biomol. Chem. 2011, 9, 6422–6436. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhou, C.-X.; Yan, J.-W.; Hou, J.-Q.; Chen, S.-B.; Ou, T.-M.; Gu, L.-Q.; Huang, Z.-S.; Tan, J.-H. Synthesis and evaluation of quinazolone derivatives as a new class of c-KIT G-quadruplex binding ligands. ACS Med. Chem. Lett. 2013, 4, 909–914. [Google Scholar] [CrossRef] [PubMed]
- Vy Thi Le, T.; Han, S.; Chae, J.; Park, H.J. G-quadruplex binding ligands: From naturally occurring to rationally designed molecules. Curr. Pharm. Des. 2012, 18, 1948–1972. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.; Rodriguez, R. G-quadruplex interacting small molecules and drugs: From bench toward bedside. Expert Rev. Clin. Pharmacol. 2014, 7, 663–679. [Google Scholar] [CrossRef] [PubMed]
- Grande, V.; Doria, F.; Freccero, M.; Wurthner, F. An aggregating amphiphilic squaraine: A light-up probe that discriminates parallel G-quadruplexes. Angew. Chem. 2017, 56, 7520–7524. [Google Scholar] [CrossRef] [PubMed]
- Laguerre, A.; Stefan, L.; Larrouy, M.; Genest, D.; Novotna, J.; Pirrotta, M.; Monchaud, D. A twice-as-smart synthetic g-quartet: PyroTASQ is both a smart quadruplex ligand and a smart fluorescent probe. J. Am. Chem. Soc. 2014, 136, 12406–12414. [Google Scholar] [CrossRef] [PubMed]
- Thakur, R.K.; Kumar, P.; Halder, K.; Verma, A.; Kar, A.; Parent, J.L.; Basundra, R.; Kumar, A.; Chowdhury, S. Metastases suppressor NM23-H2 interaction with G-quadruplex DNA within c-myc promoter nuclease hypersensitive element induces c-MYC expression. Nucleic Acids Res. 2009, 37, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Shan, C.; Yan, J.-W.; Wang, Y.-Q.; Che, T.; Huang, Z.-L.; Chen, A.-C.; Yao, P.-F.; Tan, J.-H.; Li, D.; Ou, T.-M.; et al. Design, synthesis, and evaluation of isaindigotone derivatives to downregulate c-myc transcription via disrupting the interaction of NM23-H2 with G-quadruplex. J. Med. Chem. 2017, 60, 1292–1308. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-Q.; Huang, Z.-L.; Chen, S.-B.; Wang, C.-X.; Shan, C.; Yin, Q.-K.; Ou, T.-M.; Li, D.; Gu, L.-Q.; Tan, J.-H.; et al. Design, synthesis, and evaluation of new selective NM23-H2 binders as c-MYC transcription inhibitors via disruption of the NM23-H2/G-quadruplex interaction. J. Med. Chem. 2017, 60, 6924–6941. [Google Scholar] [CrossRef] [PubMed]
- Naasani, I.; Seimiya, H.; Yamori, T.; Tsuruo, T. FJ5002: A potent telomerase inhibitor identified by exploiting the disease-oriented screening program with COMPARE analysis. Cancer Res. 1999, 59, 4004–4011. [Google Scholar] [PubMed]
- Zhang, W.-J.; Ou, T.-M.; Lu, Y.-J.; Huang, Y.-Y.; Wu, W.-B.; Huang, Z.-S.; Zhou, J.-L.; Wong, K.-Y.; Gu, L.-Q. 9-substituted berberine derivatives as G-quadruplex stabilizing ligands in telomeric DNA. Bioorg. Med. Chem. 2007, 15, 5493–5501. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Ou, T.-M.; Hou, J.-Q.; Lu, Y.-J.; Tan, J.-H.; Gu, L.-Q.; Huang, Z.-S. 9-N-substituted berberine derivatives: Stabilization of G-quadruplex DNA and down-regulation of oncogene c-myc. Bioorg. Med. Chem. 2008, 16, 7582–7591. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Ou, T.-M.; Tan, J.-H.; Hou, J.-Q.; Huang, S.-L.; Gu, L.-Q.; Huang, Z.-S. Synthesis and evaluation of 9-O-substituted berberine derivatives containing aza-aromatic terminal group as highly selective telomeric G-quadruplex stabilizing ligands. Bioorg. Med. Chem. Lett. 2009, 19, 3414–3417. [Google Scholar] [CrossRef] [PubMed]
- Bazzicalupi, C.; Ferraroni, M.; Bilia, A.R.; Scheggi, F.; Gratteri, P. The crystal structure of human telomeric DNA complexed with berberine: An interesting case of stacked ligand to g-tetrad ratio higher than 1:1. Nucleic Acids Res. 2013, 41, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Xiao, N.; Chen, S.; Ma, Y.; Qiu, J.; Tan, J.-H.; Ou, T.-M.; Gu, L.-Q.; Huang, Z.-S.; Li, D. Interaction of berberine derivative with protein POT1 affect telomere function in cancer cells. Biochem. Biophys. Res. Commun. 2012, 419, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.-X.; Su, H.-F.; Lv, P.; Ma, Y.; Wang, S.-K.; Miao, H.; Liu, H.-Y.; Tan, J.-H.; Ou, T.-M.; Gu, L.-Q.; et al. A newly identified berberine derivative induces cancer cell senescence by stabilizing endogenous G-quadruplexes and sparking a DNA damage response at the telomere region. Oncotarget 2015, 6, 35625–35635. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Tan, J.-H.; He, J.-H.; Long, Y.; Ou, T.-M.; Li, D.; Gu, L.-Q.; Huang, Z.-S. Disubstituted quinazoline derivatives as a new type of highly selective ligands for telomeric G-quadruplex DNA. Eur. J. Med. Chem. 2012, 47, 299–311. [Google Scholar] [CrossRef] [PubMed]
- He, J.-H.; Liu, H.-Y.; Li, Z.; Tan, J.-H.; Ou, T.-M.; Huang, S.-L.; An, L.-K.; Li, D.; Gu, L.-Q.; Huang, Z.-S. New quinazoline derivatives for telomeric G-quadruplex DNA: Effects of an added phenyl group on quadruplex binding ability. Eur. J. Med. Chem. 2013, 63, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Chen, A.C.; Kuang, G.T.; Wang, S.K.; Ou, T.M.; Tan, J.H.; Li, D.; Huang, Z.S. Design, synthesis and biological evaluation of 4-anilinoquinazoline derivatives as new c-myc G-quadruplex ligands. Eur. J. Med. Chem. 2016, 122, 264–279. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-K.; Wu, Y.; Wang, X.-Q.; Kuang, G.-T.; Zhang, Q.; Lin, S.-L.; Liu, H.-Y.; Tan, J.-H.; Huang, Z.-S.; Ou, T.-M. Discovery of small molecules for repressing cap-independent translation of human vascular endothelial growth factor (hVEGF) as novel antitumor agents. J. Med. Chem. 2017, 60, 5306–5319. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.-R.; Zhou, C.-X.; Wu, W.-B.; Ou, T.-M.; Tan, J.-H.; Li, D.; Gu, L.-Q.; Huang, Z.-S. 12-N-Methylated 5,6-dihydrobenzo[c]acridine derivatives: A new class of highly selective ligands for c-myc G-quadruplex DNA. Eur. J. Med. Chem. 2012, 53, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.L.; Su, H.F.; Wang, N.; Liao, S.R.; Lu, Y.T.; Ou, T.M.; Tan, J.H.; Li, D.; Huang, Z.S. Synthesis and evaluation of 7-substituted-5,6-dihydrobenzo[c]acridine derivatives as new c-KIT promoter G-quadruplex binding ligands. Eur. J. Med. Chem. 2017, 130, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-B.; Wu, W.-B.; Hu, M.-H.; Ou, T.-M.; Gu, L.-Q.; Tan, J.-H.; Huang, Z.-S. Discovery of a new fluorescent light-up probe specific to parallel G-quadruplexes. Chem. Commun. 2014, 50, 12173–12176. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.-H.; Chen, S.-B.; Guo, R.-J.; Ou, T.-M.; Huang, Z.-S.; Tan, J.-H. Development of a highly sensitive fluorescent light-up probe for G-quadruplexes. Analyst 2015, 140, 4616–4625. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.-H.; Chen, X.; Chen, S.-B.; Ou, T.-M.; Yao, M.; Gu, L.-Q.; Huang, Z.-S.; Tan, J.-H. A new application of click chemistry in situ: Development of fluorescent probe for specific G-quadruplex topology. Sci. Rep. 2015, 5, 17202. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.-H.; Chen, S.-B.; Wang, Y.-Q.; Zeng, Y.-M.; Ou, T.-M.; Li, D.; Gu, L.-Q.; Huang, Z.-S.; Tan, J.-H. Accurate high-throughput identification of parallel G-quadruplex topology by a new tetraaryl-substituted imidazole. Biosens. Bioelectron. 2016, 83, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Shin-ya, K.; Wierzba, K.; Matsuo, K.; Ohtani, T.; Yamada, Y.; Furihata, K.; Hayakawa, Y.; Seto, H. Telomestatin, a novel telomerase inhibitor from Streptomyces anulatus. J. Am. Chem. Soc. 2001, 123, 1262–1263. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | Screening Index | Ref. |
|---|---|---|
| 3D-QSAR Pharmacophore | Chemical feature of active compounds | [62] |
| Molecular docking | Estimated binding affinity to G-quadruplex | [63] |
| Fluorescence displacement (FID) | Competed binding towards G-quadruplex | [64] |
| NMR | Binding site with G-quadruplex | [65] |
| Fluorescence resonance energy transfer (FRET) | Stability effect of compounds on G-quadruplex | [66] |
| Surface plasmon resonance (SPR) | Binding affinity to G-quadruplex | [67] |
| G-quadruplex on Oligo Affinity Support (G4-OAS) | Binding affinity to G-quadruplex | [68] |
| Derivatives | Xenograft Model | Dosage (mg·kg−1) | Tumor Growth Inhibition | Days to Complete Response | Ref. |
|---|---|---|---|---|---|
| 9 | A549 human lung cancer | 6.25; i.p. | 38.1% | 12 | [77] |
| 10 | RAJI human Burkitt’s lymphoma | 30; i.p. | 27.4% | 14 | [78] |
| 15 | SiHa human cervical squamous cancer | 20; i.p. | 35.2% | 20 | [79] |
| 16 | 5; i.p. | 64.8% | 21 | [79] | |
| 21 | 1; i.p. | 49.3% | 20 | [79] | |
| 24 | MCF-7 human breast cancer | 7.5; i.p. | 60.1% | 20 | [79] |
| Compounds | Target G4 | Biochemical Activity | Cytotoxicity (IC50) | Ref. | |||
|---|---|---|---|---|---|---|---|
| ΔTm | KD | telIC50 | |||||
| Quindoline derivatives | 3 | telomere | n.d. | n.d. | 16 μM b | 6.3 μM (SKOV-6) | [69] |
| 4 | telomere | 13.0 °C | n.d. | 6.4 μM b | n.d. | [70] | |
| 5 | telomere | 21.0 °C | n.d. | 0.44 μM b | 1.68 μM (SW620) | [73] | |
| 6 | telomere | 14.0 °C | n.d. | 0.31 μM b | 1.21 μM (HL-60) | [74] | |
| 7 | telomere | 24.3 °C | 0.19 μM (SPR) | 5.5 μM c | 45 μM (HL-60) | [75] | |
| 9 | c-MYC | 13.9 °C | 1.1 μM (MST) | n.d. | 0.19 μM (RAJI) | [77] | |
| 10 | c-MYC | 27.0 °C | 1.3 μM (MST) | n.d. | 4.7 μM (RAJI) | [78] | |
| Isaindigotone derivatives | 12 | telomere | 21.9 °C | 0.1 μM (FIT) | 7.8 μM c | 29 μM (CA46) | [81,82,83] |
| 13 | c-KIT | 13.6 °C | 0.6 μM (SPR) | n.d. | 6.8 μM (HGC-27) | [81,82,83] | |
| 14 | c-MYC a | n.d. | 5.29 μM (SPR) | n.d. | 13.1 μM (Hela) | [79] | |
| 15 | c-MYC a | 12.1 °C | 17.0 μM (MST) | n.d. | 16.0 μM (SiHA) | [79] | |
| 16 | c-MYC a | 0.5 °C | 2.0 μM (MST) | n.d. | 1.78 μM (SiHA) | [79] | |
| Berberine derivatives | 18 | telomere | 23.4 °C | n.d. | n.d. | n.d. | [95] |
| 19 | telomere | 28.2 °C | 0.3 μM (FIT) | 16 μM b | n.d. | [95] | |
| 20 | telomere | 25.0 °C | 0.1 μM (FIT) | 14 μM | n.d. | [95] | |
| 21 | telomere | 9.0 °C | 14.8 μM | n.d. | 1.7 μM (HL-60) | [95] | |
| BRACO-19 | telomere | 27.5 °C | 0.032 μM (SPR) | 0.11 μM c | 8 μM (21NT) | [19] | |
| Telomestatin | telomere | n.d. | n.d. | 0.005 μM b | 0.8 μM (LAN1) | [108] | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Che, T.; Wang, Y.-Q.; Huang, Z.-L.; Tan, J.-H.; Huang, Z.-S.; Chen, S.-B. Natural Alkaloids and Heterocycles as G-Quadruplex Ligands and Potential Anticancer Agents. Molecules 2018, 23, 493. https://doi.org/10.3390/molecules23020493
Che T, Wang Y-Q, Huang Z-L, Tan J-H, Huang Z-S, Chen S-B. Natural Alkaloids and Heterocycles as G-Quadruplex Ligands and Potential Anticancer Agents. Molecules. 2018; 23(2):493. https://doi.org/10.3390/molecules23020493
Chicago/Turabian StyleChe, Tong, Yu-Qing Wang, Zhou-Li Huang, Jia-Heng Tan, Zhi-Shu Huang, and Shuo-Bin Chen. 2018. "Natural Alkaloids and Heterocycles as G-Quadruplex Ligands and Potential Anticancer Agents" Molecules 23, no. 2: 493. https://doi.org/10.3390/molecules23020493
APA StyleChe, T., Wang, Y.-Q., Huang, Z.-L., Tan, J.-H., Huang, Z.-S., & Chen, S.-B. (2018). Natural Alkaloids and Heterocycles as G-Quadruplex Ligands and Potential Anticancer Agents. Molecules, 23(2), 493. https://doi.org/10.3390/molecules23020493