Suramin-Induced Neurotoxicity: Preclinical Models and Neuroprotective Strategies

 ,
,
Abstract
:1. Introduction
2. Results
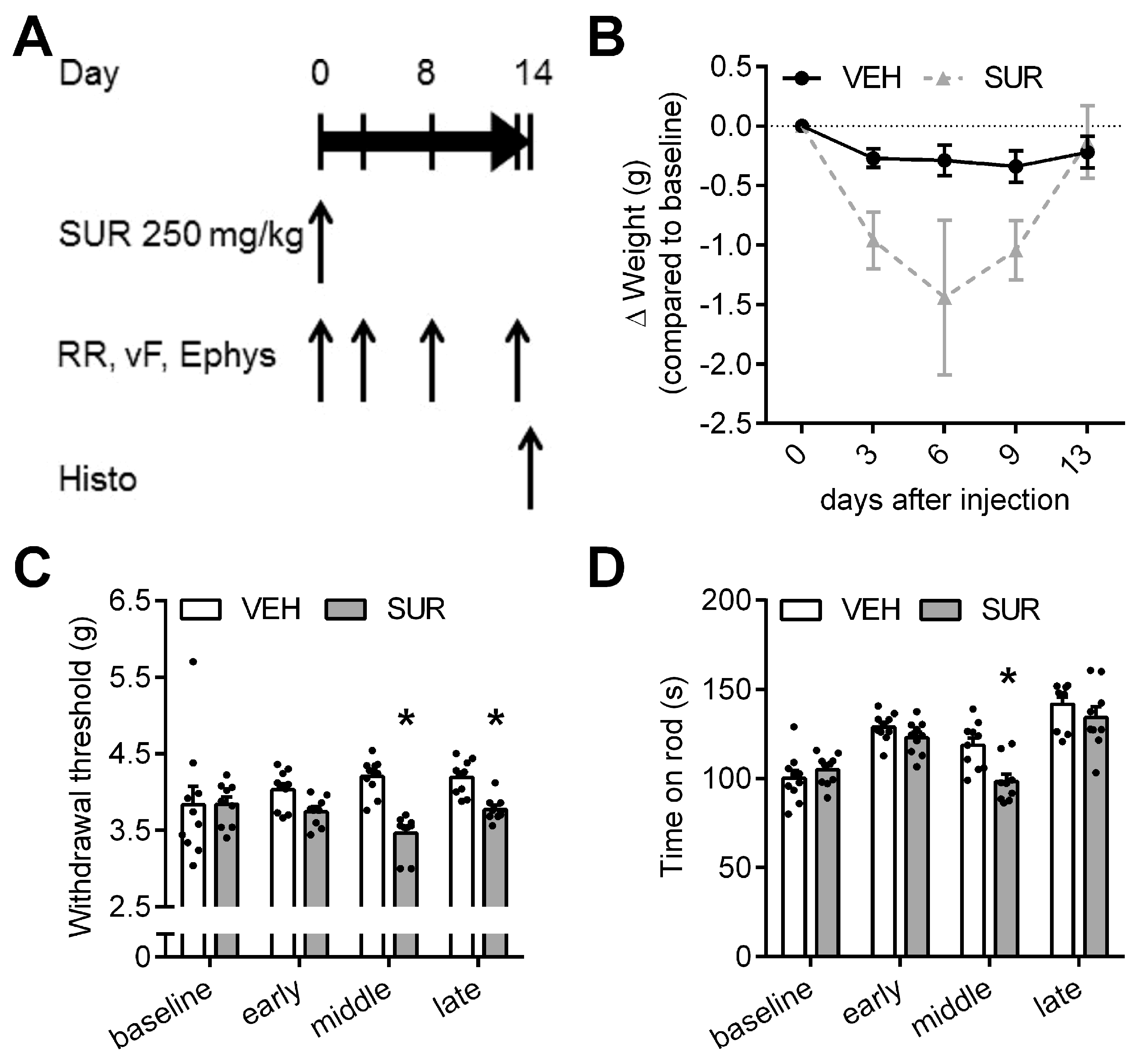
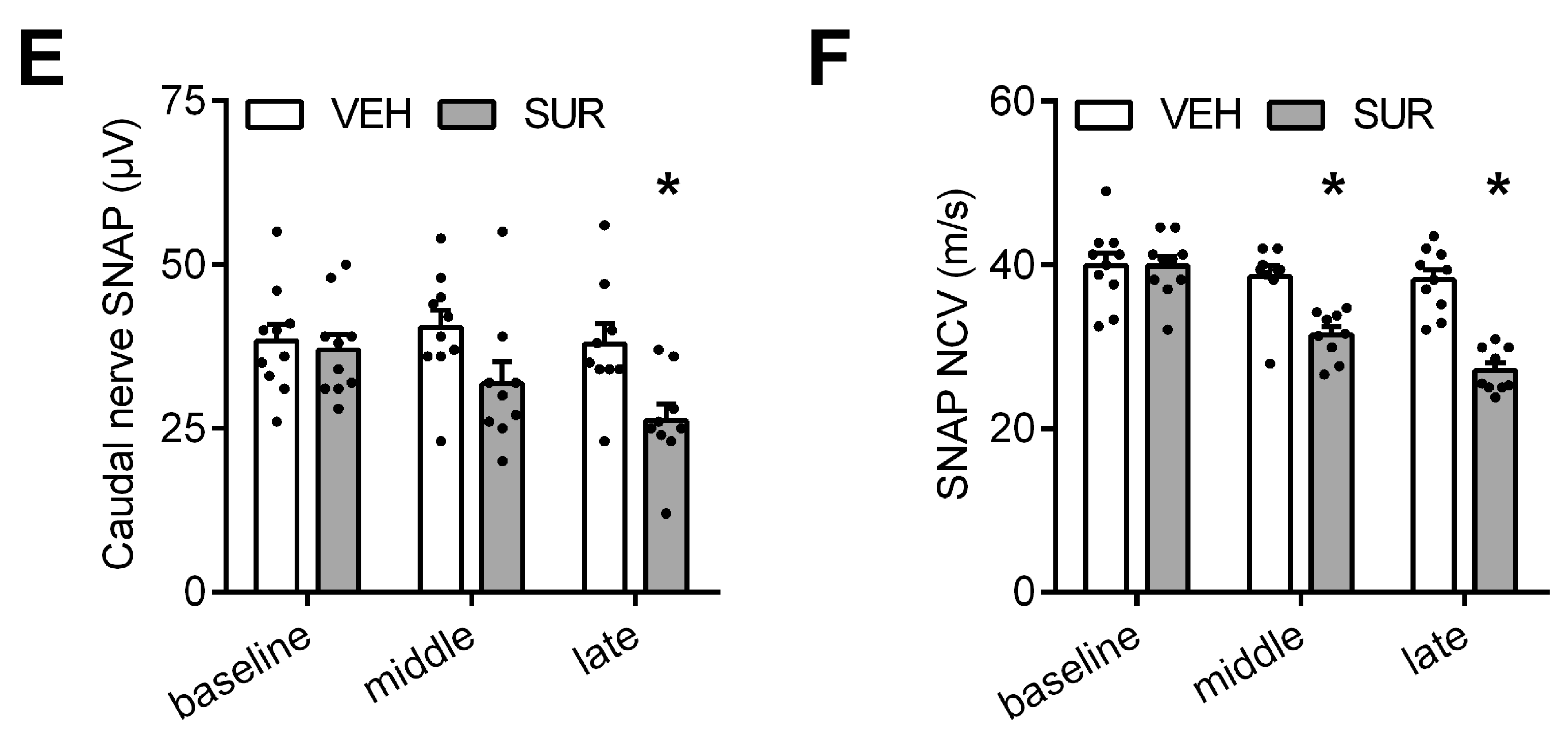
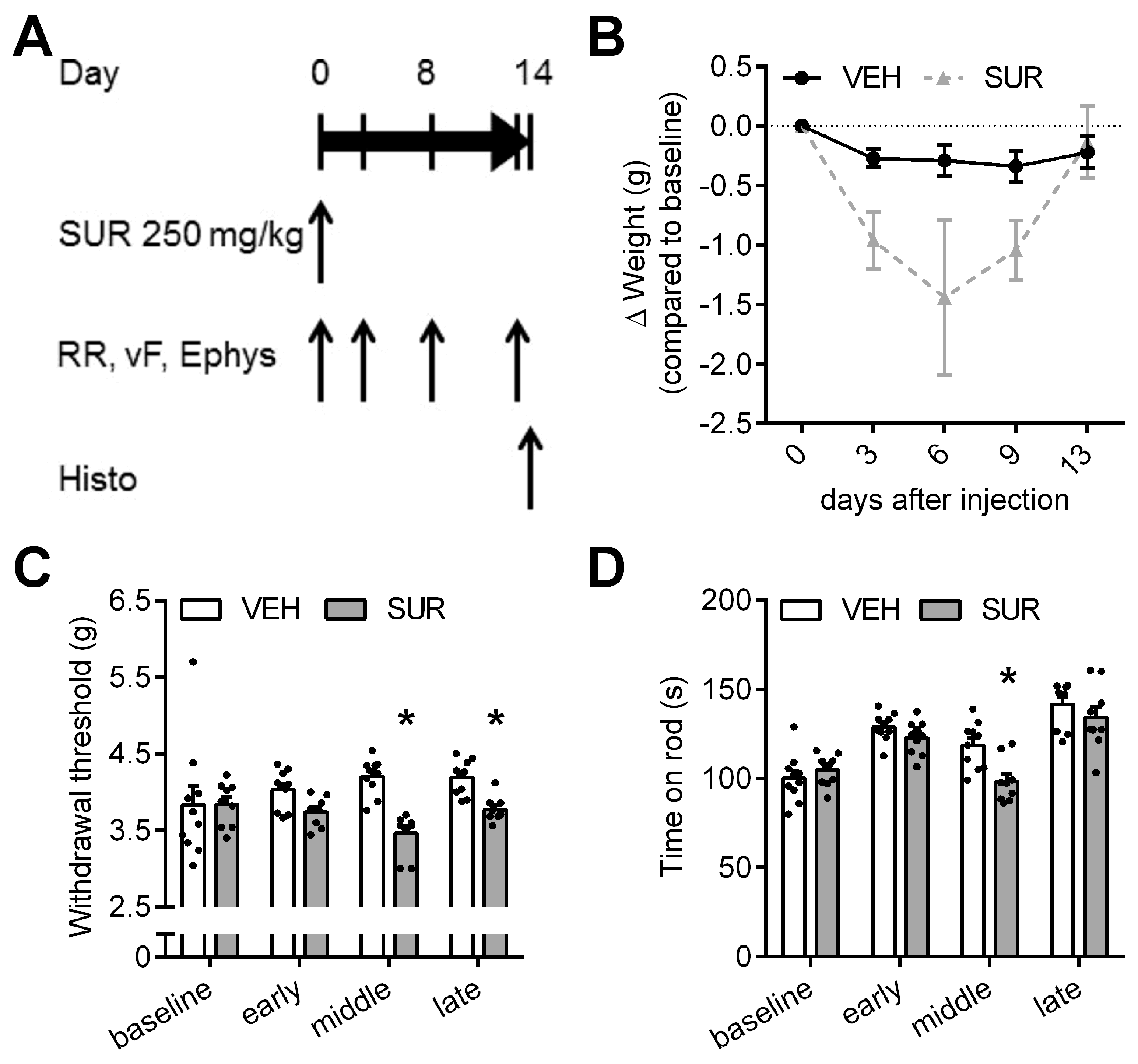
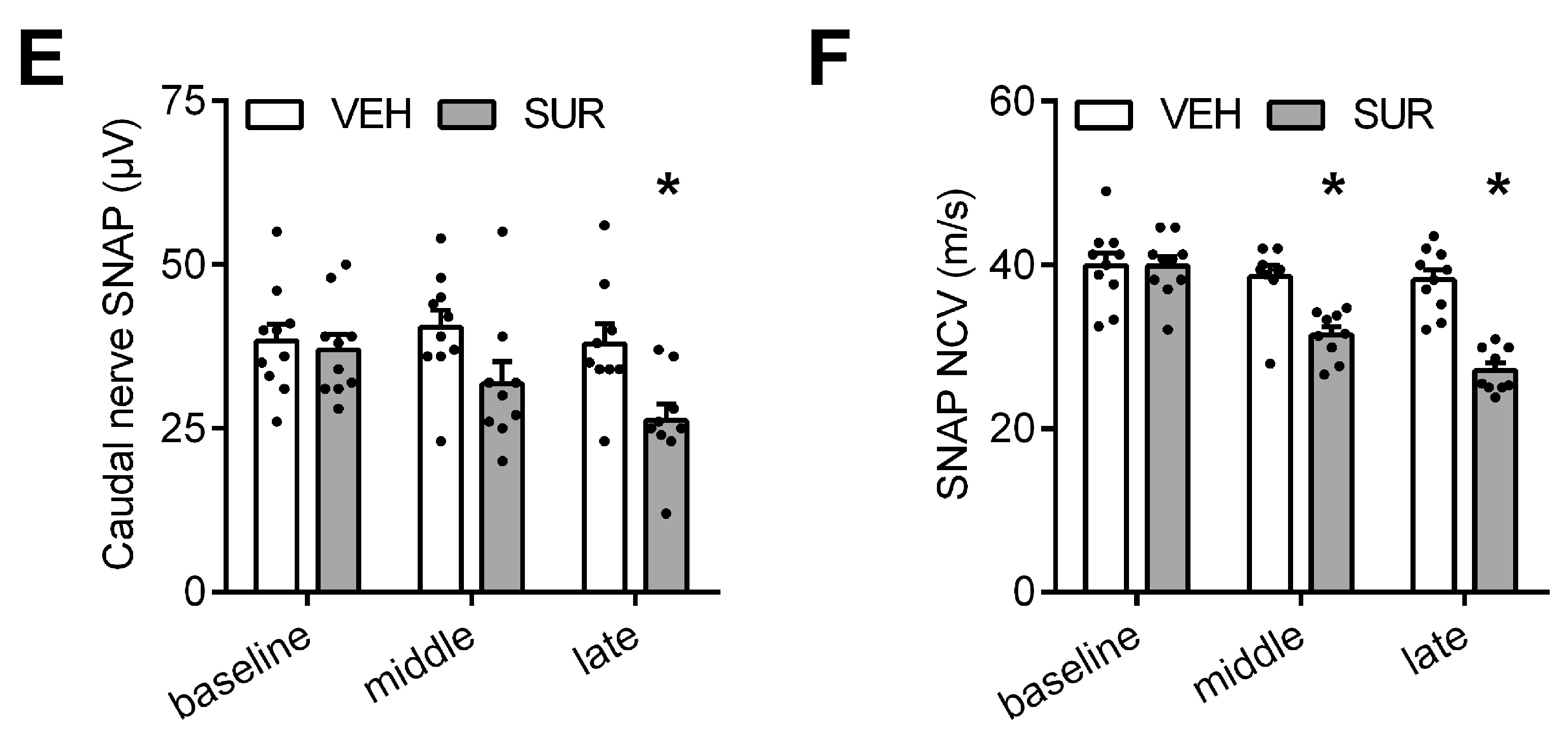
2.1. Suramin Induces a Sensory Axonal-Demyelinating Polyneuropathy in C57Bl/6 Mice
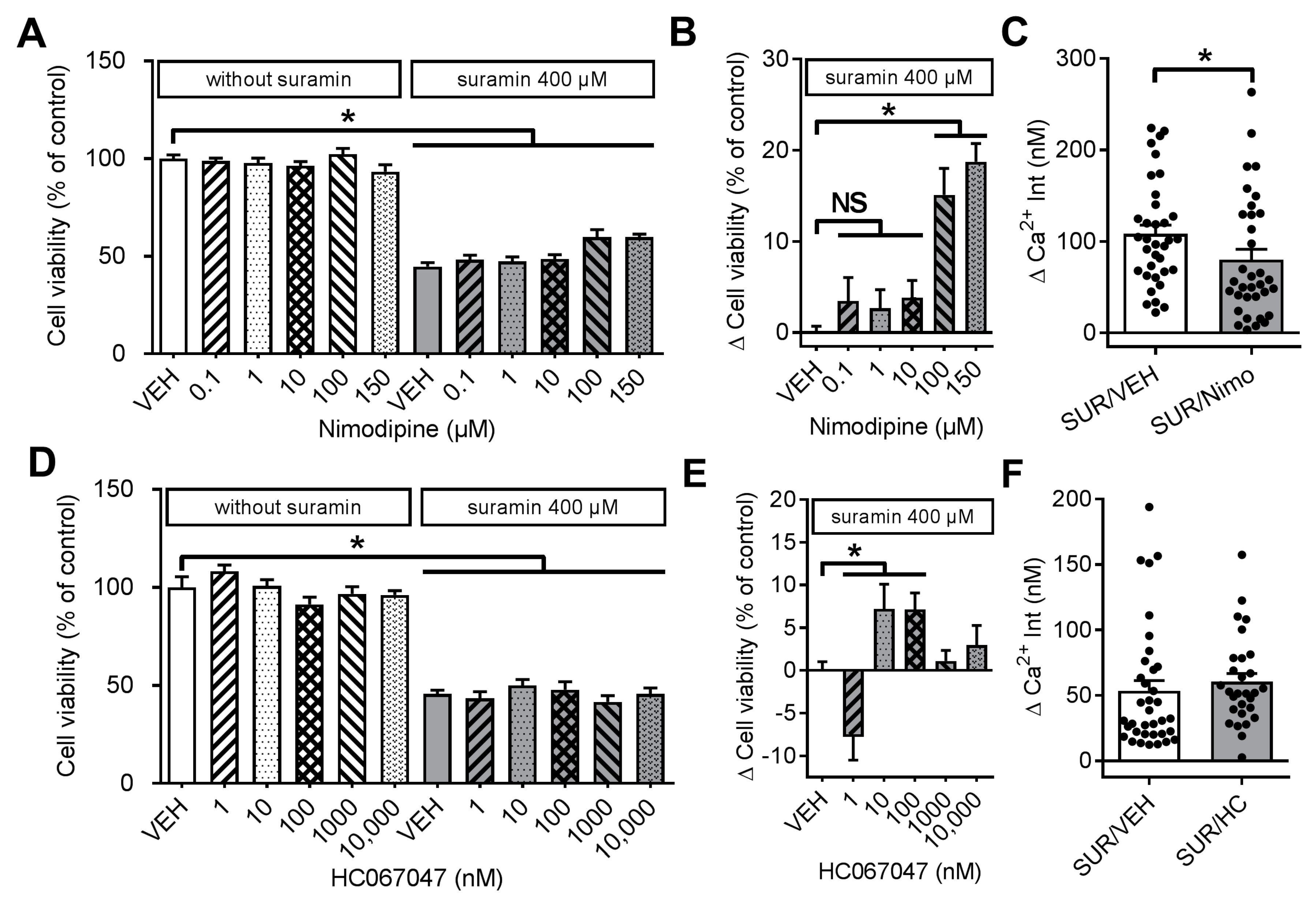
2.2. Effects of Suramin on Cell Viability and Calcium Homeostasis in Dorsal Root Ganglia Neurons
2.3. Effects of Various Inhibitors of Plasmamembrane Channels on Suramin-Induced Neurotoxicity
2.4. Downstream Apoptotic Pathways and Experimental Limitations
3. Discussion
4. Materials and Methods
4.1. In Vivo
4.1.1. Animal Housing, Sample Sizes and Methods of Randomization and Blinding
4.1.2. Drug Preparation and Injection
4.1.3. Behavior Analysis
4.1.4. Nerve Conduction Studies
4.2. In Vitro
4.2.1. DRGN Cell Culture
4.2.2. Calcium Imaging
4.2.3. MTT-Assay
4.2.4. Caspase Assay
4.3. Statistical Analysis and Exclusion Criteria
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Ca2+ | calcium |
| DRGN | dorsal root ganglia neurons |
| VGCC | voltage-gated calcium channels |
| TRP | transient-receptor-potential |
References
- McGeary, R.P.; Bennett, A.J.; Tran, Q.B.; Cosgrove, K.L.; Ross, B.P. Suramin: Clinical uses and structure-activity relationships. Mini Rev. Med. Chem. 2008, 8, 1384–1394. [Google Scholar] [CrossRef] [PubMed]
- Mitsuya, H.; Popovic, M.; Yarchoan, R.; Matsushita, S.; Gallo, R.C.; Broder, S. Suramin protection of T cells in vitro against infectivity and cytopathic effect of HTLV-III. Science 1984, 226, 172–174. [Google Scholar] [CrossRef] [PubMed]
- Cheson, B.D.; Levine, A.M.; Mildvan, D.; Kaplan, L.D.; Wolfe, P.; Rios, A.; Groopman, J.E.; Gill, P.; Volberding, P.A.; Poiesz, B.J. Suramin therapy in AIDS and related disorders: Report of the US Suramin Working Group. JAMA 1987, 258, 1347–1351. [Google Scholar] [CrossRef] [PubMed]
- Woll, P.J.; Ranson, M.; Margison, J.; Thomson, Y.; van der Water, L.; George, N.; Howell, A. Suramin for breast and prostate cancer: A pilot study of intermittent short infusions without adaptive control. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 1994, 5, 597–600. [Google Scholar] [CrossRef]
- Small, E.J.; Meyer, M.; Marshall, M.E.; Reyno, L.M.; Meyers, F.J.; Natale, R.B.; Lenehan, P.F.; Chen, L.; Slichenmyer, W.J.; Eisenberger, M. Suramin therapy for patients with symptomatic hormone-refractory prostate cancer: Results of a randomized phase III trial comparing suramin plus hydrocortisone to placebo plus hydrocortisone. J. Clin. Oncol. 2000, 18, 1440–1450. [Google Scholar] [CrossRef] [PubMed]
- Eisenberger, M.A.; Reyno, L.M.; Jodrell, D.I.; Sinibaldi, V.J.; Tkaczuk, K.H.; Sridhara, R.; Zuhowski, E.G.; Lowitt, M.H.; Jacobs, S.C.; Egorin, M.J. Suramin, an active drug for prostate cancer: Interim observations in a phase I trial. J. Natl. Cancer Inst. 1993, 85, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Dawson, N.A.; Figg, W.D.; Cooper, M.R.; Sartor, O.; Bergan, R.C.; Senderowicz, A.M.; Steinberg, S.M.; Tompkins, A.; Weinberger, B.; Sausville, E.A.; et al. Phase II trial of suramin, leuprolide, and flutamide in previously untreated metastatic prostate cancer. J. Clin. Oncol. 1997, 15, 1470–1477. [Google Scholar] [CrossRef] [PubMed]
- Myers, C.; Cooper, M.; Stein, C.; LaRocca, R.; Walther, M.M.; Weiss, G.; Choyke, P.; Dawson, N.; Steinberg, S.; Uhrich, M.M.; et al. Suramin: A novel growth factor antagonist with activity in hormone-refractory metastatic prostate cancer. J. Clin. Oncol. 1992, 10, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, V.; Eisenberger, M.A.; Sinibaldi, V.J.; Sheikh, K.; Griffin, J.W.; Cornblath, D.R. A prospective study of suramin-induced peripheral neuropathy. Brain J. Neurol. 1996, 119, 2039–2052. [Google Scholar] [CrossRef]
- Voogd, T.E.; Vansterkenburg, E.L.; Wilting, J.; Janssen, L.H. Recent research on the biological activity of suramin. Pharmacol. Rev. 1993, 45, 177–203. [Google Scholar] [PubMed]
- Peltier, A.C.; Russell, J.W. Recent advances in drug-induced neuropathies. Curr. Opin. Neurol. 2002, 15, 633–638. [Google Scholar] [CrossRef] [PubMed]
- La Rocca, R.V.; Meer, J.; Gilliatt, R.W.; Stein, C.A.; Cassidy, J.; Myers, C.E.; Dalakas, M.C. Suramin-induced polyneuropathy. Neurology 1990, 40, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Bitton, R.J.; Figg, W.D.; Venzon, D.J.; Dalakas, M.C.; Bowden, C.; Headlee, D.; Reed, E.; Myers, C.E.; Cooper, M.R. Pharmacologic variables associated with the development of neurologic toxicity in patients treated with suramin. J. Clin. Oncol. 1995, 13, 2223–2229. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.S.; Hobday, K.L.; Windebank, A.J. Mechanism of suramin toxicity in stable myelinating dorsal root ganglion cultures. Exp. Neurol. 1995, 133, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.W.; Gill, J.S.; Sorenson, E.J.; Schultz, D.A.; Windebank, A.J. Suramin-induced neuropathy in an animal model. J. Neurol. Sci. 2001, 192, 71–80. [Google Scholar] [CrossRef]
- Russell, J.W.; Windebank, A.J.; Podratz, J.L. Role of nerve growth factor in suramin neurotoxicity studied in vitro. Ann.Neurol. 1994, 36, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.S.; Connolly, D.C.; McManus, M.J.; Maihle, N.J.; Windebank, A.J. Suramin induces phosphorylation of the high-affinity nerve growth factor receptor in PC12 cells and dorsal root ganglion neurons. J. Neurochem. 1996, 66, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Windebank, A.J. Calcium in suramin-induced rat sensory neuron toxicity in vitro. Brain Res. 1996, 742, 149–156. [Google Scholar] [PubMed]
- Boehmerle, W.; Endres, M. Salinomycin induces calpain and cytochrome c-mediated neuronal cell death. Cell Death Dis. 2011, 2, e168. [Google Scholar] [CrossRef] [PubMed]
- Boehmerle, W.; Splittgerber, U.; Lazarus, M.B.; McKenzie, K.M.; Johnston, D.G.; Austin, D.J.; Ehrlich, B.E. Paclitaxel induces calcium oscillations via an inositol 1,4,5-trisphosphate receptor and neuronal calcium sensor 1-dependent mechanism. Proc. Natl. Acad. Sci. USA 2006, 103, 18356–18361. [Google Scholar] [CrossRef] [PubMed]
- Boehmerle, W.; Huehnchen, P.; Peruzzaro, S.; Balkaya, M.; Endres, M. Electrophysiological, behavioral and histological characterization of paclitaxel, cisplatin, vincristine and bortezomib-induced neuropathy in C57Bl/6 mice. Sci. Rep. 2014, 4, 6370. [Google Scholar] [CrossRef] [PubMed]
- Balzarini, J.; Mitsuya, H.; De Clercq, E.; Broder, S. Aurintricarboxylic acid and evans blue represent two different classes of anionic compounds which selectively inhibit the cytopathogenicity of human T-cell lymphotropic virus type III/lymphadenopathy-associated virus. Biochem. Biophys. Res. Commun. 1986, 136, 64–71. [Google Scholar] [CrossRef]
- Boehmerle, W.; Zhang, K.; Sivula, M.; Heidrich, F.M.; Lee, Y.; Jordt, S.E.; Ehrlich, B.E. Chronic exposure to paclitaxel diminishes phosphoinositide signaling by calpain-mediated neuronal calcium sensor-1 degradation. Proc. Natl. Acad. Sci. USA 2007, 104, 11103–11108. [Google Scholar] [CrossRef] [PubMed]
- Boehmerle, W.; Muenzfeld, H.; Springer, A.; Huehnchen, P.; Endres, M. Specific targeting of neurotoxic side effects and pharmacological profile of the novel cancer stem cell drug salinomycin in mice. J. Mol. Med. 2014, 92, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Eichhorst, S.T.; Krueger, A.; Muerkoster, S.; Fas, S.C.; Golks, A.; Gruetzner, U.; Schubert, L.; Opelz, C.; Bilzer, M.; Gerbes, A.L.; et al. Suramin inhibits death receptor-induced apoptosis in vitro and fulminant apoptotic liver damage in mice. Nat. Med. 2004, 10, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Prigozhina, N.L.; Heisel, A.J.; Seldeen, J.R.; Cosford, N.D.; Price, J.H. Amphiphilic suramin dissolves Matrigel, causing an ‘inhibition’ artefact within in vitro angiogenesis assays. Int. J. Exp. Pathol. 2013, 94, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Zabrenetzky, V.S.; Kohn, E.C.; Roberts, D.D. Suramin inhibits laminin- and thrombospondin-mediated melanoma cell adhesion and migration and binding of these adhesive proteins to sulfatide. Cancer Res. 1990, 50, 5937–5942. [Google Scholar] [PubMed]
- Constantopoulos, G.; Rees, S.; Cragg, B.G.; Barranger, J.A.; Brady, R.O. Experimental animal model for mucopolysaccharidosis: Suramin-induced glycosaminoglycan and sphingolipid accumulation in the rat. Proc. Natl. Acad. Sci. USA 1980, 77, 3700–3704. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; DeChavigny, A.; Johnson, C.E.; Hamada, J.; Stein, C.A.; Nicolson, G.L. Suramin. A potent inhibitor of melanoma heparanase and invasion. J. Biol. Chem. 1991, 266, 9661–9666. [Google Scholar] [PubMed]
- Nakagawa, T.; Yuan, J. Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J. Cell Biol. 2000, 150, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Liljelund, P.; Netzeband, J.G.; Gruol, D.L. L-Type calcium channels mediate calcium oscillations in early postnatal Purkinje neurons. J. Neurosci. 2000, 20, 7394–7403. [Google Scholar] [PubMed]
- Galvan, E.J.; Calixto, E.; Barrionuevo, G. Bidirectional Hebbian plasticity at hippocampal mossy fiber synapses on CA3 interneurons. J. Neurosci. 2008, 28, 14042–14055. [Google Scholar] [CrossRef] [PubMed]
- Pascaud, C.; Garrigos, M.; Orlowski, S. Multidrug resistance transporter P-glycoprotein has distinct but interacting binding sites for cytotoxic drugs and reversing agents. Biochem. J. 1998, 333, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.L.; Gold, M.S. Dihydropyridine block of voltage-dependent K+ currents in rat dorsal root ganglion neurons. Neuroscience 2009, 161, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Alessandri-Haber, N.; Dina, O.A.; Yeh, J.J.; Parada, C.A.; Reichling, D.B.; Levine, J.D. Transient receptor potential vanilloid 4 is essential in chemotherapy-induced neuropathic pain in the rat. J. Neurosci. 2004, 24, 4444–4452. [Google Scholar] [CrossRef] [PubMed]
- Yusaf, S.P.; Goodman, J.; Pinnock, R.D.; Dixon, A.K.; Lee, K. Expression of voltage-gated calcium channel subunits in rat dorsal root ganglion neurons. Neurosci. Lett. 2001, 311, 137–141. [Google Scholar] [CrossRef]
- Scroggs, R.S.; Fox, A.P. Calcium current variation between acutely isolated adult rat dorsal root ganglion neurons of different size. J. Physiol. 1992, 445, 639–658. [Google Scholar] [CrossRef] [PubMed]
- Acosta, C.G.; Lopez, H.S. delta opioid receptor modulation of several voltage-dependent Ca2+ currents in rat sensory neurons. J. Neurosci. 1999, 19, 8337–8348. [Google Scholar] [PubMed]
- Vandewauw, I.; Owsianik, G.; Voets, T. Systematic and quantitative mRNA expression analysis of TRP channel genes at the single trigeminal and dorsal root ganglion level in mouse. BMC Neurosci. 2013, 14, 21. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.S.; Windebank, A.J. Suramin induced ceramide accumulation leads to apoptotic cell death in dorsal root ganglion neurons. Cell Death Differ. 1998, 5, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, C. P1- and P2-purinoceptor subtypes—An update. Arch. Int. Pharmacodyn. Ther. 1990, 303, 30–50. [Google Scholar] [PubMed]
- Rago, R.P.; Miles, J.M.; Sufit, R.L.; Spriggs, D.R.; Wilding, G. Suramin-induced weakness from hypophosphatemia and mitochondrial myopathy. Association of suramin with mitochondrial toxicity in humans. Cancer 1994, 73, 1954–1959. [Google Scholar] [CrossRef]
- Light, A.R.; Wu, Y.; Hughen, R.W.; Guthrie, P.B. Purinergic receptors activating rapid intracellular Ca increases in microglia. Neuron Glia Biol. 2006, 2, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Faul, F.; Erdfelder, E.; Buchner, A.; Lang, A.G. Statistical power analyses using G*Power 3.1: Tests for correlation and regression analyses. Behav. Res. Methods 2009, 41, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Huehnchen, P.; Boehmerle, W.; Endres, M. Assessment of paclitaxel induced sensory polyneuropathy with "Catwalk" automated gait analysis in mice. PLoS ONE 2013, 8, e76772. [Google Scholar] [CrossRef] [PubMed]
- Huehnchen, P.; Boehmerle, W.; Springer, A.; Freyer, D.; Endres, M. A novel preventive therapy for paclitaxel-induced cognitive deficits: Preclinical evidence from C57BL/6 mice. Transl. Psychiatry 2017, 7, e1185. [Google Scholar] [CrossRef] [PubMed]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. Osteoarthr. Cartil. 2012, 20, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Dardis, C. Peirce’s criterion for the rejection of non-normal outliers: Defining the range of applicability. J. Stat. Softw. 2004, 10, 1–8. [Google Scholar]
- Ross, S.M. Peirce’s criterion for the elimination of suspect experimental data. J. Eng. Technol. 2003, 20, 38–41. [Google Scholar]
Sample Availability: Samples of the compounds are available from commercial sources. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substance | Nimodipine (L-Type VGCC Inhibitor) | A 967079 (TRPA1-Inhibitor) | ||||||||
| SUR 400 µM + | 0.1 µM | 1 µM | 10 µM | 100 µM | 150 µM | 1 nM | 10 nM | 100 nM | 1 µM | 10 µM |
| Change attributable to intervention (Δ% of SUR/VEH) | +3.5 ±2.6 | +2.7 ±2.0 | +3.9 ±1.9 | +15.1 * ±2.9 | +18.7 * ±2.1 | +0.2 ±2.3 | +7.1 ±3.5 | +3.0 ±2.4 | +2.6 ±1.2 | +2.8 ±4.8 |
| Substance | Efonidipine (L-Type VGCC Inhibitor) | HC 067047 (TRPV4 Inhibitor) | ||||||||
| SUR 400 µM + | 0.1 µM | 1 µM | 10 µM | 50 µM | 1 nM | 10 nM | 100 nM | 1 µM | 10 µM | |
| Change attributable to intervention (Δ% of SUR/VEH) | +3.5 ±2.7 | +0.5 ±2.6 | +0.6 ±2.7 | −1.6 ±3.2 | −7.5 * ±3.0 | +7.2 * ±2.9 | +7.1 * ±2.0 | +1.1 ±1.2 | +2.9 ±2.3 | |
| Substance | Ruthenium Red (unselective including VGCC and TRP Inhibition) | Pyr 3 (TRPC3 Inhibitor) | ||||||||
| SUR 400 µM + | 10 nM | 100 nM | 1 µM | 10 µM | 1 nM | 10 nM | 100 nM | 1 µM | 10 µM | |
| Change attributable to intervention (Δ% of SUR/VEH) | +8.1 ±4.2 | +4.4 ±1.6 | +7.6 ±3.9 | −1.5 ±2.8 | +0.2 ±2.5 | −3.4 ±4.2 | −1.3 ±3.8 | −2.8 ±3.0 | −2.6 ±2.5 | |
| Substance | SNX 482 (R-Type VGCC Inhibitor) | Ononetin (TRPM3 Inhibitor) | ||||||||
| SUR 400 µM + | 2 nM | 20 nM | 200 nM | 3 nM | 30 nM | 300 nM | 3 µM | 30 µM | ||
| Change attributable to intervention (Δ% of SUR/VEH) | −3.2 ±1.8 | −2.2 ±3.0 | +2.1 ±2.3 | −5.9 ±2.6 | +2.5 ±2.1 | −2.2 ±2.4 | −2.7 ±3.3 | −1.9 ±2.7 | ||
| Substance | Ω-Conotoxin MVIIC (N-, P-, Q-Type VGCC Inh) | |||||||||
| SUR 400 µM + | 1 nM | 10 nM | 100 nM | 1 µM | ||||||
| Change attributable to intervention (Δ% of SUR/VEH) | −2.2 ±1.8 | −2.0 ±2.6 | +1.9 ±1.6 | +1.9 ±3.3 | ||||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Von der Ahe, D.; Huehnchen, P.; Balkaya, M.; Peruzzaro, S.; Endres, M.; Boehmerle, W. Suramin-Induced Neurotoxicity: Preclinical Models and Neuroprotective Strategies. Molecules 2018, 23, 346. https://doi.org/10.3390/molecules23020346
Von der Ahe D, Huehnchen P, Balkaya M, Peruzzaro S, Endres M, Boehmerle W. Suramin-Induced Neurotoxicity: Preclinical Models and Neuroprotective Strategies. Molecules. 2018; 23(2):346. https://doi.org/10.3390/molecules23020346
Chicago/Turabian StyleVon der Ahe, David, Petra Huehnchen, Mustafa Balkaya, Sarah Peruzzaro, Matthias Endres, and Wolfgang Boehmerle. 2018. "Suramin-Induced Neurotoxicity: Preclinical Models and Neuroprotective Strategies" Molecules 23, no. 2: 346. https://doi.org/10.3390/molecules23020346
APA StyleVon der Ahe, D., Huehnchen, P., Balkaya, M., Peruzzaro, S., Endres, M., & Boehmerle, W. (2018). Suramin-Induced Neurotoxicity: Preclinical Models and Neuroprotective Strategies. Molecules, 23(2), 346. https://doi.org/10.3390/molecules23020346