Abstract
We have synthesized a small series of five 3-[4-arylmethoxy)phenyl]propanoic acids employing an easy and short synthetic pathway. The compounds were tested in vitro against a set of four protein targets identified as key elements in diabetes: G protein-coupled receptor 40 (GPR40), aldose reductase (AKR1B1), peroxisome proliferator-activated receptor gama (PPARγ) and solute carrier family 2 (facilitated glucose transporter), member 4 (GLUT-4). Compound 1 displayed an EC50 value of 0.075 μM against GPR40 and was an AKR1B1 inhibitor, showing IC50 = 7.4 μM. Compounds 2 and 3 act as slightly AKR1B1 inhibitors, potent GPR40 agonists and showed an increase of 2 to 4-times in the mRNA expression of PPARγ, as well as the GLUT-4 levels. Docking studies were conducted in order to explain the polypharmacological mode of action and the interaction binding mode of the most active molecules on these targets, showing several coincidences with co-crystal ligands. Compounds 1–3 were tested in vivo at an explorative 100 mg/kg dose, being 2 and 3 orally actives, reducing glucose levels in a non-insulin-dependent diabetes mice model. Compounds 2 and 3 displayed robust in vitro potency and in vivo efficacy, and could be considered as promising multitarget antidiabetic candidates. This is the first report of a single molecule with these four polypharmacological target action.
1. Introduction
Type 2 Diabetes Mellitus (T2DM) is a multifactorial metabolic disease that occurs with fasting blood glucose concentrations above 120 mg/dL (>7 mM), due to the abnormal pancreatic β-cells and/or insulin resistance [1]. Currently, experimental drug discovery for T2DM is focused on developing drugs for insulin sensitizing and/or releasing effect by several mechanisms. One of them is mediated by the G protein-coupled receptor 40 (GPR40 or FFA1), which is primarily expressed in pancreatic β-cells and enteroendocrine cells of the gut [2,3]. The GPR40 activation by medium to long chain fatty acids, stimulates insulin secretion only in the presence of high glucose concentration but does not disturb insulin exocytosis at low glucose levels [4,5]. This intriguing mechanism was suggestive in treating T2DM by small molecules agonists for GPR40, thus playing as novel insulin secretagogues with little or no risk of hypoglycemia [6]. An alternative mechanism for the control of glucose levels refers to the peroxisome proliferator-activated receptors (PPARs) action. PPARs, members of the nuclear hormone receptor family, occur in three subtypes: PPARα, PPARγ, and PPARβ/δ [7]. Each subtype regulates tissue-specific target genes acting as lipid sensors and regulators of glucose homeostasis, such as the solute carrier family 2 (facilitated glucose transporter), member 4 (GLUT-4) [8]. Glitazones function as insulin-sensitizing drugs, via activation of PPARγ [9,10], leading to enforced expression of target genes involved in glucose-sensing ability of pancreatic β-cells in diabetic subjects [11]. On the other hand, extended hyperglycemia plays a significant key in the development of diabetic complications: atherosclerosis, blindness, neuropathy and renal failure. The NADPH-dependent reduction of elevated levels of d-glucose to sorbitol catalyzed by aldose reductase (AKR1B1) is considered as one of the phenomena leading to the onset of long term diabetic complications [12]. Stressful and damaging cell conditions are produced by several factors such as the osmotic imbalance due to sorbitol accumulation, the loss of antioxidant power related with NADPH oxidation and the induction of additional advantageous conditions for protein glycation [13,14,15]. Inhibition of AKR1B1 represents a potential target of antidiabetic drug action. The purpose of the present work was to design, synthesize and screen in vitro, in vivo and in silico (in combo screens) a small series of five 3-[4-arylmethoxy)phenyl]propanoic acids to accomplish the treatment of diabetes with a single molecule with multitarget action: activation of PPARγ, GLUT-4 and GPR40 and inhibition of AKR1B1 (Figure 1). Multitarget of polypharmacological concept can be defined as the modulation of several drug targets to achieve a desired therapeutic effect. This multitarget therapy may be an attractive option for the therapeutic treatment of diabetes that will not only control the glucose concentrations but also decrease complications linked to hyperglycemic conditions. Compounds 1–5 were designed based on the compounds GW9508 [16] and SHF-1 [17], maintaining the typical 4-point pharmacophore of synthetic GPR40 agonists [6], that mimic the fatty acid structure of endogenous agonists, such as docosahexaenoic acid (DHA, Figure 1): (I) an acidic group (carboxylate or surrogates); (II) a central p-substituted benzene; (III) a second lipophilic region and (IV) a flexible connector that joins regions (I) and (IV), and tolerating the structure to assume diverse conformations. The insulin-sensitizer targets PPARγ and GPR40 which are activated by long chain fatty acids and thiazolidine-2,4-diones are described to function as an integrated two-receptor signal transduction pathway [18] being sensitive to closely related pharmacophores, as represented by GW9508 and SHF-1 [17]. Then, it is reasonable to assume that also compounds 1–5 may act as agonists on these targets in a similar fashion. On the other hand, carboxylic acid derivatives comprise the majority of reported AKR1B1 inhibitors: epalrestat, tolrestat and zopolrestat. The other two chemical groups that function as inhibitors are: spirohydantoins (Sorbinil) and flavonoids such as quercetin [19]. Based on the relevance of the phenylpropanoic scaffold (Figure 1) in determining the inhibitory ability of zopolrestat towards AKR1B1, we expected that also compounds 1–5 may act as inhibitors of the enzyme. As a result of the multifactorial complexity of chronic diseases, the current therapeutic arsenal and the old-fashioned ‘‘one-molecule, one-target” model seem not so effective [20]. The design of new chemical entities (NCE) with two or more balancing bioactivities for the treatment of complex diseases would be very advantageous. Based on the above considerations, our work was focused on the design, preparation and evaluation of these novel multitarget molecules to be active against Diabetes. Although compound 1 was previously synthesized [21,22], in the current work the effectiveness as GPR40 agonist of this compound and of the accompanying molecules is extended and explored in vivo. A parallel investigation on the potential antidiabetic action of these compounds: the activation of PPARγ and GLUT-4 and the inhibition of AKR1B1, is also reported for first time. To our knowledge, this is the first report describing the potential of the simultaneous targeting of these four relevant processes linked to the diabetic pathology by a single molecule.
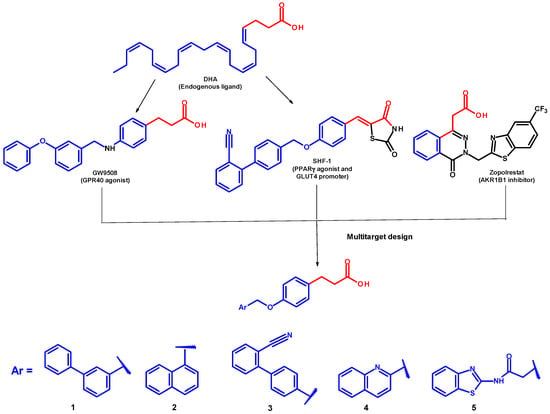
Figure 1.
Drug design of multitarget compounds 1–5, which were designed from pharmacophore extraction and reorganization from known modulators of four targets: GPR40, PPARγ, GLUT-4, and AKR1B1.
2. Results and Discussion
2.1. Chemical Synthesis
Compounds 1–5 were prepared starting from 4-hydroxybenzaldehyde (6), which was condensed with malonic acid (7), following by microwave Knoevenagel reaction and decarboxylation conditions to give compound 8. This compound was hydrogenated under catalytic reduction with 10% Pd/C giving 3-(4-hydroxyphenyl)propanoic acid (9), which was reacted via esterification with thionyl chloride and ethanol as solvent, obtaining ethyl 3-(4-hydroxyphenyl)propanoate (10). Subsequently, 10 was reacted with the appropriately substituted methylaryl halide in polar aprotic solvents and potassium carbonate to give compounds 11–14. All were treated separately by basic hydrolysis with potassium hydroxide, to give 1–4 (Scheme 1). Compound 5 was obtained by direct SN2 reaction between acid 9 and N-1,3-benzothiazol-2-yl-2-chloroacetamide (15). Compounds were recovered with modest yields and were purified by column chromatography or recrystallization (see experimental part). Their chemical structures were confirmed by spectroscopic (1D and 2D NMR, see supporting information) and spectrometric data, and their purity determined by microanalysis.
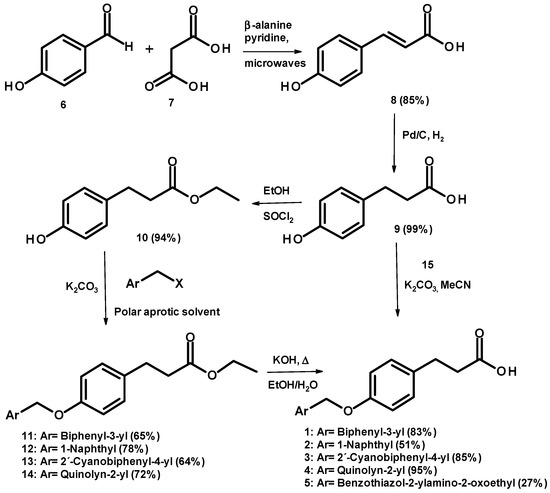
Scheme 1.
Synthesis of compounds 1–5.
2.2. In Vitro GPR40 Activity
Compounds 1–5 were in vitro tested as GPR40 agonist. This activity was measured with calcium flux assay in GPR40-transfected HEK293 cells [2,23]. The preliminary screening results are summarized in Table 1. Linoleic acid (LA), one of the endogenous ligands for GPR40, was designated as positive control. Compounds were tested in quadruplicates at 100 μM. After preliminary screening, compounds 1–5 were further tested in a concentration-dependent manner (Table 1).

Table 1.
Preliminary screening (Efficacy, maximal response) and GPR40 agonistic activities (Potency, EC50) of compounds 1–5 in calcium assay for hGPR40/Gα16/HEK293.
As we expected, the results indicated that compound 1 was the most potent agonist (EC50 = 0.075 μM), in accordance with the literature (EC50 = 0.260 μM) [21,22], followed by compounds 2 and 3, with naphthyl and biphenylcarbonitrile substituents, respectively. All of them showed potencies in the submicromolar order. Compound 4, with a quinolynyl substituent, provided a noteworthy reduction in agonistic action. Conversely, benzothiazole derivative 5 was inactive. Thus, compounds 2–5 were less active than previously reported 1. On the other hand, it is noteworthy that the potency of compounds 1–3 is orders of magnitude greater than that obtained by endogenous fatty acid ligand LA.
2.3. In Vitro Aldose Reductase (AKR1B1) Inhibition
The in vitro AKR1B1 inhibitory activity of compounds 1–5 (Table 2) was evaluated by using a highly purified human recombinant enzyme preparation [24,25]. Sorbinil was employed as reference drug.
Table 2.
Inhibition of human recombinant AKR1B1 by compounds 1–5.
2.4. Relative Expression of PPARγ and GLUT-4
Compounds tested were shown interesting AKR1B1 inhibitors in the low micromolar IC50 values. Once again, compound 1, previously identified as GPR40 agonist, was the most active inhibitor of the series, finding this new activity for this known compound. Compounds 2, 3 and 5 were almost equally active. Compound 4 was the least active. None of them were more active than sorbinil, a well-known AKR1B1 inhibitor. As we described previously, AKR1B1 plays a key role in the progress of chronic diabetic complications, such as retinopathy, nephropathy, neuropathy and also cardiovascular risk.
3T3-L1 fibroblasts were differentiated to the adipocyte phenotype in order to determine the action of compounds on PPARγ and GLUT-4 expression. Fibroblasts were treated during 24 h with compounds 1–5 and pioglitazone (10 μM) as reference drug and the mRNA expression level evaluated. Figure 2 shows that only compounds 2 and 3 enhance significantly the levels of relative expression for PPARγ (around three to four-times) and GLUT-4 mRNA's in the same way as pioglitazone. The activation of PPARγ could reduce the glucose levels in diabetic patients due to insulin resistance reduction. Our data also suggest that compounds 2 and 3 induces GLUT-4 expression in the same way than pioglitazone does. Several evidences indicate that increased levels of GLUT-4 expression in skeletal muscle are critical for the regulation of glucose homeostasis [7,17,26].
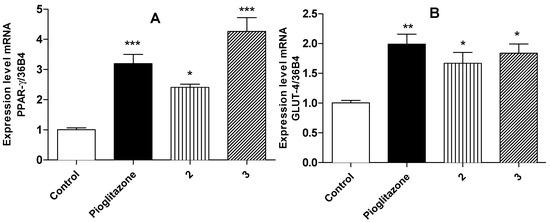
Figure 2.
(A) Effect of compounds 2 and 3 on expression level of PPARγ; (B) Effect of compounds on expression level of GLUT-4. Results are mean ± SEM (n = 6)/*** p < 0.001; ** p < 0.01; * p < 0.05 compared with control group.
2.5. Molecular Docking Studies
Based on the in vitro biological assays and the preliminary enzyme inhibition evaluations, the most active compounds were selected to explain the experimental activities on these relevant targets. A preliminary molecular docking simulation was performed to assess the presumed binding mode of 1–5 into the receptors GPR40, PPARγ and the enzyme AKR1B1. A pilot in silico calculation was done using DIA-DB [27], a web server for the prediction of antidiabetic drugs via inverse virtual screening of the input molecules 1–5 against a set of 18 protein targets identified as key elements in diabetes, within which are included PPARγ, GPR40 and AKR1B1, among others [28]. Subsequently, a more specific and refined analysis was carried out for the most active compounds (1–3).
Refined molecular docking reveals that compounds 2 and 3 internalize into the ligand binding site of PPARγ and interact by electrostatic and hydrogen bonds with Ser-289, His-323, His-449 and Tyr-473, all of them essential for the activation of this receptor. However, compound 3 (the most active in vitro) showed an additional interaction with Ser-342, characteristic of PPARγ partial agonists (Figure 3). Analyses of a huge number of crystallographic structures of the PPARχ ligand-binding site bound to an agonist have revealed that this isotype has two binding modes in a single pocket. These two binding modes correspond to full and partial agonists [29]. Side effects of glitazones, including weight gain, edema, congestive heart failure, and the recently reported increased risk of bone fracture are major undesired effects associated with the use of PPARγ full agonists [30]. On the other hand, partial agonists interact mainly through a hydrogen bond with Ser342. This interaction corresponds to several carboxylic ligands present in the majority of the PPARγ partial agonists that forms a hydrogen bond with the Ser342, such as showed by compound 3.
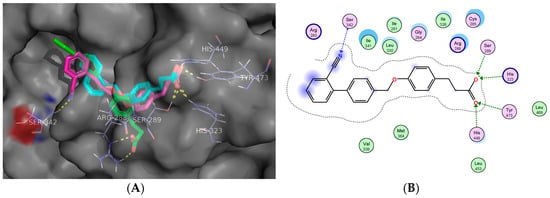
Figure 3.
(A) 3D binding model of compounds 1–3 into the ligand binding site of PPARγ. Compounds are presented as stick models: 1 (green), 2 (cyan) and 3 (magenta), and aminoacids as lines. Dashed line signifies polar interactions; (B) 2D interaction map of the most active compound 3 and PPARγ.
For GPR40, binding poses depicted in Figure 4 suggest that the in vitro active compounds 1, 2 and 3 interact through electrostatic bonds with residues of Arg-183 and Arg-2258, and by hydrogen bonds with Tyr-91, Asn-2244, Tyr-2240, all of them showed by well-known GPR40 allosteric agonists (such as TAK-875). On the other hand, the disposition of the biphenyl ring in 1, which was the most potent in the in vitro screening, fits into the GPR40 ligand-binding-pocket better than the other compounds generating π-π interactions with Phe-142 (Figure 4B). The docking score for compound 1 was the highest (∆G = −10.63 kcal/mol), in comparison with compounds 2 and 3 (∆G = −10.31 and −9.96 kcal/mol, respectively).
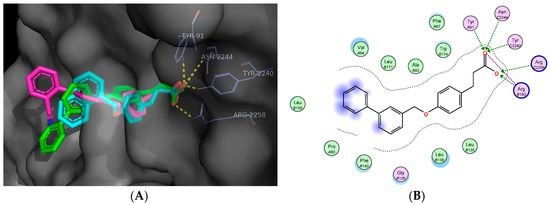
Figure 4.
(A) 3D binding model of compounds 1–3 into the allosteric ligand binding site of GPR40. Compounds are presented as stick models: 1 (green), 2 (cyan) and 3 (magenta). (B) 2D interaction map of the most active compound 1 and GPR40.
In the case of AKR1B1, solutions of molecular docking into the catalytic site of this enzyme show that acid moieties of compounds 1, 2 and 3 interact with Tyr-48, His-110 and Trp-111 showed in several currently inhibitors of this enzyme, such as zopolrestat and tolrestat. Also, the naphthyl ring of 2 conserves an interaction with Trp-111 through π-π stacking (Figure 5). All compounds showed moderate in vitro inhibition of this enzyme.
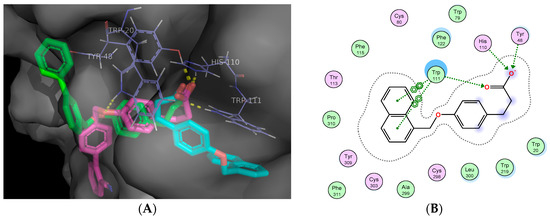
Figure 5.
(A) 3D binding model of compounds 1–3 into the active site of Aldose reductase (AKR1B1). Compounds are presented as stick models: 1 (green), 2 (cyan) and 3 (magenta); (B) 2D interaction map of the second most active compound 2 and AKR1B1.
2.6. In Vivo Antidiabetic Effect of Compounds 1–3
Compounds 1–3 were the most active against some of targets identified as key elements in diabetes in this work, and they were chosen in order to assess their in vivo activity over Streptozotocin (STZ) and Nicotinamide (NA) diabetic induced mice, a well-known non-insulin-dependent diabetes murine model [7,17]. Glibenclamide was used as a positive control, in order to ensure that the damage over β-cells was partial and mice pancreas still produces insulin, that responds to a secretagogue such as sulfonylurea drug used in this work. The antidiabetic activity of compounds 1–3 were determined via intragastric route of a single dose of 100 mg/kg (Figure 6).
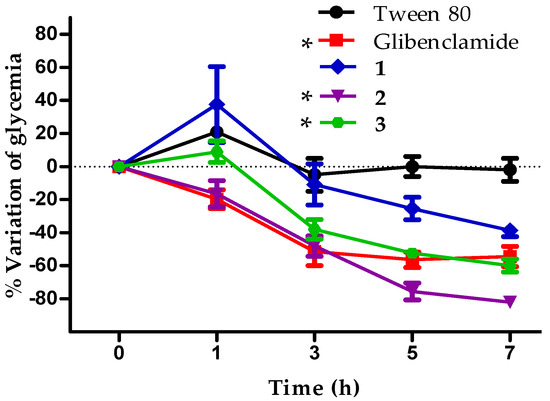
Figure 6.
Effect of a single dose (100 mg/kg) of compounds 1–3 and glibenclamide (5 mg/kg) in STZ-NA induced diabetes mice model (intragastric, n = 6). * p < 0.05 versus Tween 80 (10%) group.
In the in vivo assay, compound 1 exhibited potential differences in bioavailability that could explain the observed lack of in vivo activity for this compound, in comparison with the demonstrated activity of 2 and 3. In particular, compounds 2 and 3 showed the best in vivo activity, compared with control group (Tween 80). Also, compound 2 producing a sustained decrease of blood glucose levels, close to 80% less, 7 h after compound administration (Figure 6).
This compound showed a statistically significant hypoglycemic effect like glibenclamide during the time that the experiment lasted. The glucose lowering effect of compound 3 was increasing after the first hour of administration (−20% at 3 h, −50% at 5 h, and −60% at 7 h). Besides, statistically significant difference is detected between 3 and tween 80 group, and the maximum effect was detected in the last hours of post-intragastric administration (Figure 6).
2.7. Off-Target Toxicity Predictions
With the aim of anticipating potential off-targets affinity and toxicity issues of the most active compounds 1–3, a virtual prediction of safety profiles was accomplished. The toxicity parameters of 1–3, pioglitazone, sorbinil and glibenclamide (in vitro and in vivo antidiabetic reference drugs) were calculated through the ACD/ToxSuite software, v. 2.95 (Table 3).
Table 3.
Toxicity profiles predicted for compounds 1–3, pioglitazone, sorbinil and glibenclamide.
The in silico calculation of inhibition for the three main isoforms of CYP450 for compounds 1–3 were comparable to pioglitazone, sorbinil and glibenclamide at relevant clinical concentrations (<10 μM), showing low probabilities of drug-drug interactions and undesirable adverse effects [31]. Besides, several lipophilic compounds are associated with cardiovascular risks due to human ether-a-go-go related gene (hERG) channel blockade [7,32]. Compounds 1–3, contemplated as polypharmacological molecules, showed low prediction of hERG channel blockage at clinically relevant concentrations (Ki < 10 μM), being considered as potentially non-cardiotoxic compounds. In the calculation of acute toxicity, compounds 1–3 demonstrated similar or even high predicted LD50 than pioglitazone, sorbinil and glibenclamide by two different administration routes, being predicted less toxic than these common antidiabetic drugs used as reference in this work.
Several examples of polypharmacology have been reported elsewhere [33,34]. Actually, some multikinase inhibitors used in cancer treatment and all marketed antipsychotics are polypharmacological drugs, even recently approved compounds. Multitargeting concept explains the efficacy of these drugs by their modulation of a network of disease relevant targets correcting a pathological imbalance [33].
3. Materials and Methods
3.1. Chemistry
All compounds were purchased from Sigma-Aldrich (Saint Louis, MO, USA). Melting points were recorded using an EZ-Melt MPA120 automated apparatus from Stanford Research Systems (Sunnyvale, CA, USA). Reactions were monitored by TLC on 0.2 mm precoated silica gel 60 F254 plates (Merck, Darmstadt, Germany). 1H-NMR spectra were determined on a Varian Oxford instrument (Palo Alto, CA, USA) operating at 600 MHz (1H-NMR) and 150 MHz (13C-NMR) instruments. Chemical shifts are given in ppm in DMSO-d6 and CDCl3 as deuterated solvents. Mass spectrometry were determined on a JMS-700 spectrometer (JEOL, Tokyo, Japan) operating in electronic impact mode.
3.2. General Procedure for the Synthesis of Compounds 1–4
Ethyl esters 11–14 (0.70 mmol) were treated with a mixture of EtOH/H2O (1:1, v/v) and KOH (2 equiv.). The mixture was stirred at reflux for 3.5–5 h. Subsequently, the ethanol was removed under vacuum and HCl solution (10%, v/v) was added until reach pH 5, to obtain a precipitate which was filtered and washed with cold water to give a solid. The crude products were recrystallized from ethanol or methanol affording title compounds.
3-[4-(Biphenyl-3-ylmethoxy)phenyl]propanoic acid (1). Yield 83%, pearly flakes m.p. 123.7–126 °C; 1H-NMR (DMSO-d6) δ: 2.43 (t, 2H, CH2), 2.74 (t, 2H, CH2), 5.13 (s, 2H, CH2), 6.94 (d, 2H, H-3A, H-5A, J = 7.74 Hz), 7.14 (d, 2H, H-2A, H-6A, J = 8.04 Hz), 7.4 (t, 1H, H-4C), 7.43 (d, 1H, H-4B, J = 7.2 Hz), 7.48–7.46 (m, 3H, H-6B, H-3C, H-5C), 7.61 (t, 1H, H-5B, J = 7.5 6Hz), 7.66 (d, 2H, H-2C, H-6C, J = 8.22 Hz), 7.72 (s, 1H, H-2C) ppm. 13C-NMR (DMSO-d6) δ: 30.4 (CH2), 36.9 (CH2), 69.6 (O-CH2), 115.1 (C-3A, C-5A), 126.4 (C-6B), 126.6 (C-4B), 127.1 (C-2C, C-6C), 128 (C-4C), 129.4 (C-3C, C-5C), 129.5 (C-5B), 129.7 (C-2A, C-6A), 134 (C-1A), 138.5 (C-1C), 140.4 (C-1B), 140.8 (C-3B), 157.0 (C-4A), 174.7 (C=O) ppm. MS/EI: m/z (% int. rel). 353 (M + Na), 178 (M + Na − 175) 100%. Anal. Calcd. for C22H20O3: C 79.50, H 6.06; Found C 79.54, H 6.04.
3-[4-(1-Naphthylmethoxy)phenyl]propanoic acid (2). Yield 51%, white powder m.p. 124.8–127.8 °C; 1H-NMR (DMSO-d6) δ: 2.49 (t, 2H, CH2), 2.76 (t, 2H, CH2), 5.50 (s, 2H, CH2), 7.0 (d, 2H, H-3A, H-5A, J = 8.46 Hz), 7.16 (d, 2H, H-2A, H-6A, J = 8.46 Hz), 7.51 (t, 1H, H-3′, J = 7.56 Hz), 7.58–7.55 (m, 2H, H-6′, H-7′), 7.66 (d, 1H, J = 6.9 Hz), 7.92 (d, 1H, J = 8.52 Hz), 7.97 (d, 1H, J = 7.86 Hz), 8.08 (d, 1H, J = 7.97 Hz). δ: 13C-NMR (DMSO-d6) δ: 30.0 (CH2), 36.1 (CH2), 68.2 (CH2), 115.1 (C-3A, C-5A), 124.3 (C-8′), 125.8 (C-3′), 126.4 (C-6′), 126.9 (C-7′), 127.03 (C-2′). 128.9 (C-5′), 129.0 (C-4′), 129.7 (C-2A, C-6A), 131.6 (C-8a), 133.1 (C-1A), 133.6 (C-4a), 133.7 (C-1′), 157.2 (C-4A, 174.3 (C=O) ppm. MS/EI: m/z (% int. rel.). 306 (M+), 141(M − 165)100%. Anal. Calcd. for C20H18O3: C 78.41, H 5.92; Found C 78.43, H 5.91.
3-{4-[(2′-Cyanobiphenyl-4-yl)methoxy]phenyl} propanoic acid (3). Yield 85%, pearly flakes m.p. 174–176.9; 1H-NMR (CDCl3) δ: 2.66 (t, 2H, CH2), 2.91 (t, 2H, CH2), 5.11 (s, 2H, CH2), 6.93 (d, 2H, H-3A, H-5A, J = 8.52 Hz), 7.15 (d, 2H, H-2A, H-6A, J = 8.46 Hz), 7.44 (t, 1H, H-4C, J = 7.68 Hz), 7.51 (d, 1H, H-6C, J = 7.86 Hz), 7.55 (2H, H-3B, H-5B, J = 8.22 Hz), 7.58 (2H, H-2B, H-6B, J = 8.16 Hz), 7.63 (t, 1H, H-5C, J = 7.62 Hz), 7.76 (d, 1H, H-3C, J = 7.74 Hz), 12.10 (s, 1H, OH) ppm. 13C-NMR (CDCl3) δ: 29.8 (CH2), 35.9 (CH2), 69.6 (CH2), 111.3 (C-2C), 118.6 (CN), 114.9 (C-3A, C-5A), 127.6 (C-4C), 127.7 (C-3B, C-5B), 129 (C-2B, C-6B), 129.3 (C-2A, C-6A), 130.0 (C-6C), 132.8 (C-5C), 133.8 (C-3C), 137.7 (C-4B), 137.8 (C-1B), 145.1 (C-1C), 157.2 (C-4A), 178.7 (C=O) ppm. MS/EI: m/z (% int. rel). 355 (M+), 192 (M − 165) 100%. Anal. Calcd. for C23H19NO3: C 77.29, H 5.36, N 3.92; Found C 77.31, H 5.34, N 3.94.
3-[4-(Quinolin-2-ylmethoxy)phenyl]propanoic acid (4). Yield 95%, yellow crystals m.p. 125.8–128.4 °C; 1H-NMR (DMSO-d6) δ: 2.49 (t, 2H, CH2), 2.74 (t, 2H, CH2), 5.33 (s, 2H, CH2), 6.97 (d, 2H, H-3A, H-5A, J = 8.82 Hz), 7.15 (d, 2H, H-2A, H-6a, J = 8.34 Hz), 7.62 (t, 1H, H-6′, J = 7.8, J = 7.14 Hz), 7.66 (d, 1H, H-3′, J = 8.4 Hz), 7.79 (t, 1H, H-7′, J = 6.96, J = 8.34 Hz), 7.99, d, 1H, H-5′, J = 8.16 Hz), 8.02 (d, 1H, H-4′, J = 8.46 Hz), 8.41, d, 1H, H-8′, J = 8.52 Hz), 12.1 (s, 1H, OH) ppm. 13C-NMR (DMSO-d6) δ: 29.9 (CH2), 35.9 (CH2), 71.2 (CH2), 115.1 (C-3A, C-5A), 119.9 (C-3′), 127.01 (C-6′), 128.4 (C-4a), 129.0 (C-7′), 129.8 (C-2A, C-6A), 130.3 (C-5′), 133.8 (C-1A), 137.5 (C-4′), 147.4 (C-8a), 156.9 (C-4A), 158.2 (C-2′), 174.2 (C=O) ppm. MS/EI: m/z (% int. rel). 307 (M+) 100%, 142 (M − 165) 75%. Anal. Calcd. for C19H17NO3: C 74.25, H 5.58, N 4.56; Found C 74.20, H 5.52, N 4.56.
3.3. Synthesis of 3-{4-[2-(1,3-Benzothiazol-2-ylamino)-2 oxoethoxy]phenyl}propanoic Acid (5)
A stirred mixture of N-1,3-benzothiazol-2-yl-2-chloroacetamide 15 (0.0006 mol), 3-(4-hydroxyphenyl)propanoic acid 9 (1 equiv.), potassium carbonate (3 equiv.), potassium iodide (10% mol) in 3 mL of acetonitrile was refluxed. The reaction solvent was dried and the mixture was basified at pH = 3 with HCl 10% (v/v). The solid was filtered and dried to give compound 5. Yield 27%, low melting point solid; 1H-NMR (DMSO-d6) δ: 2.41 (t, CH2, H-2), 2.70 (t, CH2, H-3), 4.78 (s, CH2, H-4), 7.00 (t, H-5A, J = 7.74 Hz), 7.20 (t, H-6A, J = 7.68 Hz), 7.32 (d, H-2, H-6B, J = 8.64 Hz), 7.45 (s, H-3B, H-5B), 7.64 (d, H-4A, H-7A, J = 7.80 Hz). 13C-NMR (DMSO-d6) δ: 19.24 (C-3), 35.09 (C-2), 65.00 (C-4), 119.47 (C-4B), 120.16 (C-1B), 122.86 (C-4A, C-7A), 125.98 (C-3B, C-5B), 125.99 (C-2B, C-6B), 130.55 (C-5A, C-6A), 134.17 (C-7A), 134.29 (C-3A), 136.06 (C-2 A), 157.95 (C-5), 171.56 (C-1); MS-ESI: m/z 357 (M + H+). Anal. Calcd. for C18H16N2O4S: C 60.66, H 4.53, N 7.86; Found C 60.71, H 4.54, N 7.84.
3.4. Synthesis of (2E)-3-(4-Hydroxyphenyl)acrylic Acid (8)
4-Hydroxybenzaldehyde 6 (0.25 g, 2.04 mmol), β-alanine (0.1 equiv, 0.20 mmol) and malonic acid 7 (0.21g, 1.1 equiv, 2.06 mmol) were suspended in pyridine (5 mL), following Knoevenagel reaction with subsequent decarboxylation. The flask was shaken well and heated under microwave irradiation system (Discovery Microwave System apparatus, 2450 MHz, 300 W, CEM, Matthews, NC, USA) for 10 min at 70 °C. After irradiation, the mixture was verted in cold water and acidified until pH 5. The precipitate was collected by filtration, washed with water, dried and recrystallized from ethanol. Yield 80%, yellow crystals m.p. 214 °C (dec); 1H-NMR (DMSO-d6) δ: 6.35 (d, 1H, =CH trans, J = 16 Hz), 6.86 (d, 2H, H-3′, H-5′, J = 8.4 Hz), 7.55 (d, 2H, H-2′, H-6′, J = 8.4 Hz), 7.58 (d, 1H, =CH trans, J = 16 Hz), 8.87 (bs, 2H, OH, COOH) ppm.13C-NMR (DMSO-d6) δ: 115.4 (C-2), 116.2 (C-3′, C-5′), 126.1 (C-1′), 130.4 (C-2′, C-6′), 145.3 (C-3), 158.3 (C-4′), 168.6 (C=O) ppm. MS/EI: m/z (% int. rel). 164 (M+).
3.5. Synthesis of 3-(4-Hydroxyphenyl)propanoic Acid (9)
A mixture of acid 8 (0.2 g, 1.20 mmol) and Pd/C 10% (0.02 g) in ethanol (40 mL) was hidrogenated at 60 lb/in2 in a hydrogenation apparatus for 30 min. After filtration and evaporation of the solvent uder reduce pressure a yellow oil was obtained. After cooling, a solid compound was obtained which purificated by recrystallyzation in ethanol. Yield 99%, yellow crystals m.p. 129–131 °C; 1H-NMR (DMSO-d6) δ:2.45 (t, 2H, CH2), 2.72 (t, 2H, CH2), 6.65 (d, 2H, H-3′, H-5′, J = 8.01 Hz), 7.05 (d, 2H, H-2′, H-6′, J = 8.02 Hz), 9.54 (bs, 2H, OH, COOH) ppm.13C-NMR (DMSO-d6) δ: 30.1 (CH2), 36.9 (CH2), 115.5 (C-3′, C-5′), 129.5 (C-2′, C-6′), 131.8 (C-1′), 155.5 (C-4′), 175.1 (C=O) ppm. MS/EI: m/z (% int. rel). 166 (M+).
3.6. Synthesis of Ethyl 3-(4-hydroxyphenyl)propanoate (10)
To a stirred solution of acid 9 (3 g, 18 mmol) in 30 mL of ethanol, under ice-cooling, was added thionyl chloride (1.1 eq, 1.44 mL, 19 mmol) dropwise over 20 min. After stirring the reaction mixture for 3 h at room temperature, methanol is distilled out and 25 mL of water is added. The separated ester is extracted with ethyl acetate and washed with 10 mL of saturated sodium bicarbonate solution. Evaporation of the ethyl acetate gave the ester in pure form. Yield 94%, yellow liquid; 1H-NMR (CDCl3) δ: 1.22 (d, 3H, CH3), 2.59 (t, 2H, CH2), 2.87 (t, 2H, CH2), 4.12, q, 2H, CH2), 6.74 (d, 2H, 2.09, 6.48, H-3A-H-5A), 7.03 (d, 2H, 6.54, H-2A, H-6A). 13C-NMR (CDCl3) δ: 14.2 (CH3), 30.2 (CH2), 36.3 (CH2), 60.6 (CH2), 115.4 (C-3A, C-5A), 129.4 (C-2A, C-6A), 154.3 (C-4A), 173.6 (C=O). MS/EI: m/z (% int. rel). 194 (M+) 49%, 142 (M − 74) 100%.
3.7. General Method for the Preparation of Compounds 11–14
A solution of ethyl 3-(4-hydroxyphenyl)propanoate 10 (0.5 g, 2.5 mmol) and K2CO3 (0.71 g, 5.1 mmol, 2 equiv) in appropriate polar aprotic solvent (5 mL acetonitrile or acetone) was heated at 40 °C for 30 min. Adequately substituted methylaryl halides (1.1 equiv.) were added dropwise. The mixture was heated to reflux for 8 h. Solvent was evaporated in vacuo. A mixture of ice-water (10 mL) was added and stirred for 15 min. The emulsion formed was extracted with ethyl acetate (3 × 15 mL). The solvent was removed in vacuo to give an oily product, which were purified by column chromatography.
Ethyl 3-[4-(biphenyl-3-ylmethoxy)phenyl] propanoate (11). Yield 65%, colorless liquid; 1H-NMR (CDCl3) δ: 1.21 (t, 3H, CH3), 2.58 (t, 2H, CH2), 2.88 (t, 2H, CH2), 4.11 (c, 2H, CH2′), 5.08 (s, 2H, CH2), 6.92 (d, 2H, J = 7.8, H-3A, H-5A), 7.11 (d, 2H, J = 8.1, H-2A, H-6A), 7.34 (t, 1H, J = 7.56, H-5B), 7.39 (d, 1H, 7.44, H-6B), 7.43–7.41 (m, 3H, H-3C, H-4C, H-5C), 7.54 (d, 1H, J = 7.68, H-4B), 7.58, (d, 2H, J = 7.68, H-2C, H-6C), 7.64 (s, 1H, H-2B) ppm. 13C-NMR (CDCl3) δ: 14.2 (CH3), 30.1 (CH2), 36.2 (CH2), 60.3 (CH2′), 70.1 (CH2), 114.9 (C-3A, C-5A), 126.2 (C-4B), 126.3 (C-2B), 126.7 (C-BB), 127.2 (C-2C, C-6C), 127.4 (C-5B), 128.9 (C-3C, C-5C), 129 (C-4C), 129.3 (C-2A, C-6A), 133.0 (C-1A), 137.7 (C-1C), 140.9 (C-1B), 141.5 (C-3B), 157.2 (C-4A), 172.9 (C=O) ppm. MS/EI: m/z (% int. rel). 360(M+) 31%, 167(M − 193) 100%.
Ethyl 3-[4-(1-naphthylmethoxy)phenyl]propanoate (12). Yield 78%, colorless liquid; 1H-NMR (CDCl3) δ:1.23 (t, 3H, CH3), 2.59 (t, 2H, CH2) 2.90 (t, 2H, CH2) 4.12 (c, 2H, CH2′), 5.44 (s, 2H, CH2), 6.97 (d, 2H, 7.14, H-3A, H-5A) 7.14 (d, 2H, J = 7.68, H-2A, H-6A) 7.44 (t, 1H, J = 8.22, J = 8.28, H-3′) 7.52–7.49 (m, 2H, H-6′, H-7′) 7.57 (d, 1H, J = 6.9, H-2′) 7.83 (d, 1H, J = 8.22, H-4′) 7.85 (d, 1H, J = 7.5, H-5′) 8.03 (d, J = 7.86, H-8′) ppm. 13C-NMR (CDCl3) δ: 14.2 (CH3), 30.2 (CH2), 36.2 (CH2), 60.4 (CH2′), 68.7 (CH2), 114.9 (C-3A, C-5A), 123.7 (C-8′), 125.3 (C-3′), 125.9 (C-6′), 126.5 (C-7′), 128.7 (C-2′), 128.9 (C-5′), 129.3 (C-2A, C-6A), 131.5 (C-4′), 132.4 (C-8a), 133.1 (C-4a), 133.7 (C-1′), 157.4 (C-4a), 172.9 (C=O) ppm. MS/EI: m/z (% int. rel). 334 (M+) 11%, 141(M − 193) 100%.
Ethyl 3-{4-[(2′-cyanobiphenyl-4-yl)methoxy] phenyl}propanoate (13). Yield 64%, white crystals; 1H-NMR (DMSO-d6) δ: 1.14 (t, 3H, CH3), 2.56 (t, 2H, CH2) 2.78 (t, 2H, CH2) 4.03, (q, 2H, CH2′), 5.16 (s, 2H, CH2) 6.96 (d, 2H, J = 8.04, H-3A, H-5A) 7.15 (d, 2H, J = 8.04, H-2A, H-6A), 7.64-7.60 (m, 6H, H-2A, H-3A, H-5A, H-6A, H-4C, H-6C), 7.79 (t, 1H, J = 7.56, H-5C) 7.95 (d, 1H, J = 7.68, H-3C) ppm. 13C-NMR (DMSO-d6) δ:14.5 (CH3), 29.1 (CH2), 35.8 (CH2), 60.2 (CH2′), 69.1 (CH2), 110.6 (C-2C), 115 (C-3A, C-5A), 119 (CN), 128.7 (C-4C), 129.2 (C-2B, C-6B), 129.7 (C-2A, C-6A), 130.5 (C-6C), 133.2 (C-1A), 134 (C-5C), 134.3 (C-3C), 137.7 (C-4B), 138.3 (C-1B), 144.7 (C-1C), 157.2 (C-4A), 172.5 (C=O) ppm. MS/EI: m/z (% int. rel). 385 (M+) 11%, 192(M − 193) 100%.
Ethyl 3-[4-(quinolin-2-ylmethoxy)phenyl]propanoate (14). Yield 72%, yellow liquid; 1H-NMR (CDCl3) δ: 1.47 (t, 3H, CH3), 2.50 (t, 2H, CH2), 2.75 (t, 2H, CH2), 4.01 (c, 2H, CH2), 5.32 (s, 2H, CH2), 6.96 (d, 2H, J = 6.66, J = 8.67, H-3A, H-5A), 7.13 (d, 2H, J = 8.58, H-2A, H-6A), 7.62, (d, 1H, J = 7.35, H-6′), 7.65 (d, 1H, J = 8.49, H-3′), 7.79 (dd, 1H, J = 8.28, H-7′), 7.97 (dd, 1H, J = 8.85, H-5′), 8.01 (dd, 1H, J = 8.64, H-4′), 8.39 (d, 1H, 8.55, H-8′). 13C-NMR (CDCl3) δ: 14.0 (CH3), 29.4 (CH2), 35.4 (CH2), 59.7 (CH2′), 70.7 (CH2), 114.6 (C-3A, C-5A), 119.5 (C-3′), 127.1 (C-6′), 128.1 (C-4′), 128.5 (C-4a), 129.1 (C-7′), 129.8 (C-2A, C-6A), 130.5 (C-5′), 132.9 (C-1A), 137.9 (C-8′), 146.9 (C-8a), 158.7 (C-2′), 172.1 (C=O) ppm. MS/EI: m/z (% int. rel). 335 (M+) 10%.
3.8. Synthesis of N-1,3-Benzothiazol-2-yl-2-chloroacetamide (15)
A mixture of 2-amino-1,3-benzothiazole (0.100 g), sodium bicarbonate (3 equiv) in acetone (10 mL), was stirred at 5 °C. After 30 min, chloroacetyl chloride (1.5 equiv.) was added dropwise using a drop funnel apparatus and stirred at room temperature for 8 h. Once the reaction was completed, the solvent was removed in vacuo, and the white precipitated was washed with cold water. Yield: 90%, m.p.: 156.5–159.3 °C; 1H-NMR (CDCl3) δ: 4.32 (s, CH2), 7.36 (t, H-6, J = 8.10 Hz), 7.48 (t, H-5, J = 8.28 Hz), 7.84 (q, H-4, H-7, J = 7.92 Hz); MS-EI: m/z 225.75 (M+) 40%, 149.61(M − 77) 100%.
3.9. Biological Assays
3.9.1. GPR40 Agonistic Activities of Compounds 1–5
This bioassay was previously described [2]. Briefly: About 4 × 104 h GPR40/HEK293 per well were seeded in 96-well plate. After culturing for 24 h, cells were loaded with 2 μmol/L Fluo-4 AM in Hanks’ balanced salt solution at 37 °C for 40 min. After thorough washing with 50 μL assay buffer, 25 μL assay buffer containing compound was dispensed into the well, using a FlexStation III microplate reader and intracellular calcium change was recorded with an excitation wavelength of 485 nm and emission wavelength of 525 nm.
3.9.2. Aldose Reductase (AKR1B1) Inhibition Assay
Human recombinant AKR1B1 was purified by electrophoretic homogeneity as previously described [24]. The enzyme activity was corroborated at 37 °C following the decrease of absorbance at 340 nm. The assay mixture (final volume of 0.7 mL) contained 0.25 M Na3PO4 buffer (pH 6.8), 4.67 mM d,l-glyceraldehyde, 0.11 mM NADPH, 0.38 M (NH4)2SO4 and 0.5 mM EDTA. Compounds 1–5, tested as AKR1B1 inhibitors were dissolved at appropriate concentrations in a mixture DMSO/methanol (40:60, v/v) and added to the assay mixture containing 5 mU of AKR1B1; the concentrations of DMSO and methanol in all the assays were kept constant at 0.2 and 0.3% (v/v), respectively. IC50 values was determined by nonlinear regression analysis. For each curve, five different concentrations of compounds were analyzed [25].
3.9.3. In Vitro PPARγ and GLUT-4 Assay
Both assays were achieved as described elsewhere [7,17,35,36,37]. Briefly: 3T3-L1 fibroblasts (9 × 105 cells per well) were cultured in 6-well plates in Dulbecco’s modified Eagle’s medium. After 2 days of confluence, the cells were differentiated to the adipocyte phenotype with 0.5 mM 3-isobutyl-1-methylxanthine, 0.25 μM dexamethasone acetate, and 0.8 μM insulin, for 48 h, followed by insulin alone for other 48 h. To determine the action of compounds on PPARγ and GLUT-4 expression, the cells were treated 24 h. RNA was isolated from cultured cells and two μg of total RNA were reverse-transcripted in a thermocycler. The enzyme was inactivated and finally samples were cooled. Then 1/10 volume of each reverse-transcripted reaction was amplified with SYBR Green master mix containing 0.5 mM of customized primers for PPAR-γ and GLUT-4. PCR was conducted and the threshold cycles (Ct) were measured in separate tubes and in duplicate. The ΔCt values were calculated in every sample for each gene of interest. Relative changes in the expression level of one specific gene (ΔΔCt) were calculated [37].
3.10. In Vivo Assay
CD1 mice (20–25 g) were housed in groups of six (n = 6) under laboratory conditions. Before experimentation, all animals were fasted for 16 h with water ad libitum. All animal procedures were conducted in accordance with the Mexican Federal Regulations for Animal Experimentation and Care (SAGARPA, NOM-062-ZOO-1999), based on US National Institutes of Health Publication #85-23, revised 1985. Non-insulin-dependent diabetes mice model was induced in overnight fasted mice by a single i.p. injection of 100 mg/kg STZ, 15 min before the i.p. administration of 40 mg/kg of NA. Hyperglycaemia was established by the higher plasma glucose concentration, determined by glucometer after 2 weeks. The animals with glycaemia higher than 200 mg/dL were selected for the assay [38,39,40,41]. The diabetic mice were separated into three groups of six animals each (n = 6). Mice of experimental groups were administered with a suspension of the molecules 1–3 (100 mg/kg, prepared in 10% (v/v) tween 80). Control group mice were similarly treated with 10% tween 80. The hypoglycemic reference drug glibenclamide was used as positive control (5 mg/kg). The samples of blood were collected from the caudal vein at 0, 1, 3, 5 and 7 h after administration of each compound. Blood glucose levels were estimated using a commercial glucometer [41,42,43]. The percentage variation of glycemia for each group was calculated. Values are expressed as mean ± S.E.M. Statistical significance was estimated by ANOVA.
3.11. In Silico Docking Studies
The crystal structures of PPARγ, GPR40 and aldose reductase were recovered from the Protein Data Bank (PDB) with the ID: 1I7I, 4PHU and 1Z3N respectively. Docking calculations were performed using AutoDock, version 4.2. The program performs several runs (100) in each docking experiment. Each run provides one predicted binding mode. All water molecules and every co-crystall ligands were removed from the crystallographic structure and all hydrogen atoms were added. For all ligands and proteins, Gasteiger charges were assigned and non-polar hydrogen atoms were merged. All torsions could rotate during docking. The auxiliary program AutoGrid generated the grid maps. Each grid was centered at the crystallographic coordinates of the crystallographic compound with dimensions of 60 × 60 × 60 Å with points separated by 0.375 Å. After molecular docking, all solutions were clustered into groups with RMSD < 2.0 Å. Discovery Studio version 3.5, Pymol version 1.7.4 and MOE [44] were used for visualization.
Docking Validation
The docking protocols were validated by re-docking of cocrystal ligands into the active site of the structure of each protein target: PPARγ (Tesaglitazar), GPR40 (TAK-875) and aldose reductase (Lidorestat). The RMSD obtained in both cases was less than 2.0 Å in all cases. This value specifies that the parameters for docking simulations agree in reproducing the orientation and the conformation in the X-ray crystal coordinates of enzyme and receptors.
4. Conclusions
Five phenylpropanoic acid derivatives active on different relevant targets involved in the control of glucose level and in the cell damage linked to hyperglycaemic conditions, are presented as promising antidiabetic compounds. In fact, compounds 1 to 3 are able with different effectiveness to combine a significantly increase in the mRNA expression of PPARγ, GLUT-4, with a GPR40 agonist action and a well detectable inhibition of the AKR1B1 activity. Compounds 2 and 3 also revealed their antidiabetic effectiveness in vivo, showing a robust hypoglycemic efficacy similar to glibenclamide. The in vitro multitargeting profile and the in vivo hypoglycemic efficacy make some of them suitable leads in developing new chemical entities with polypharmacological ability for treatment of diabetes and its complications. To our knowledge, this is the first report describing a multiple combined action of a single molecule facing towards these four relevant targets involved into cellular glucose handling.
Supplementary Materials
Spectra are available online.
Acknowledgments
The work was funded by the Consejo Nacional de Ciencia y Tecnología (CONACyT), grant No. 253814 (Ciencia Básica 2015) given to Gabriel Navarrete-Vazquez. Blanca Colín-Lozano received a CONACYT fellowship (378373) to complete Doctoral studies. This article is dedicated to all the people, mainly Pharmacists, who helped in the medication management and classification after the recent Mexico’s earthquake.
Author Contributions
Blanca Colín-Lozano: performed the chemical synthesis of all compounds, did the acquisition of antidiabetic in vitro and in vivo data, and analyzed the chemical and biological results. Samuel Estrada-Soto, Fabiola Chavez Silva and Litzia Cerón Romero: Carried out and interpreted the antidiabetic assays. Abraham Gutierrez-Hernandez: performed and analyzed the in silico refined molecular docking in to the PPARγ, GPR40 and AKR1B1. Abraham Giacoman-Martinez and Julio Cesar Almanza-Perez: Conducted and interpreted the expression of PPARγ and GLUT-4 experiments. Emanuel Hernández-Núñez: performed and interpreted the spectroscopic analysis using nuclear magnetic resonance in 1 and 2 dimensions. He also wrote the Supplementary Information. Zhilong Wang and Xin Xie: performed and supervised the in vitro GPR40 experiments and edited the manuscript. Mario Capiello, Francesco Balestri and Umberto Mura: performed the in vitro aldose reductase inhibition assay, interpreted the data for SAR analysis, contributed with reagents and analysis tools, drafted some parts of the manuscript and did a critical revision. Gabriel Navarrete-Vázquez: Developed the concept, designed the compounds, analyzed and interpreted data, prepared and wrote the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Schwartz, S.S.; Epstein, S.; Corkey, B.E.; Grant, S.F.; Gavin, J.R., 3rd; Aguilar, R.B. The time is right for a new classification system for diabetes: Rationale and implications of the β-cell-centric classification schema. Diabetes Care 2016, 39, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Guo, B.; Chu, W.J.; Xie, X.; Yang, Y.S.; Zhou, X.L. Design, synthesis and evaluation of potent G-protein coupled receptor 40 agonists. Chin. Chem. Lett. 2016, 27, 159–162. [Google Scholar] [CrossRef]
- Edfalk, S.; Steneberg, P.; Edlund, H. GPR40 is expressed in enteroendocrine cells and mediates free fatty acid stimulation of incretin secretion. Diabetes 2008, 57, 2280–2287. [Google Scholar] [CrossRef] [PubMed]
- Mancini, A.D.; Poitout, V. The fatty acid receptor FFA1/GPR40 a decade later: How much do we know? Trends Endocrinol. Metab. 2013, 24, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Morgan, N.G.; Dhayal, S. G-protein coupled receptors mediating long chain fatty acid signalling in the pancreatic beta-cell. Biochem. Pharmacol. 2009, 78, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Li, H.; Long, Y.Q. GPR40 agonists for the treatment of type 2 diabetes mellitus: The biological characteristics and the chemical space. Bioorg. Med. Chem. Lett. 2016, 26, 5603–5612. [Google Scholar] [CrossRef] [PubMed]
- Navarrete-Vázquez, G.; Torres-Gómez, H.; Hidalgo-Figueroa, S.; Ramírez- Espinosa, J.J.; Estrada-Soto, S.; Medina-Franco, J.L.; León-Rivera, I.; Alarcón-Aguilar, F.J.; Almanza-Pérez, J.C. Synthesis, in vitro and in silico studies of a PPARγ and GLUT-4 modulator with hypoglycemic effect. Bioorg. Med. Chem. Lett. 2014, 24, 4575–4579. [Google Scholar] [CrossRef] [PubMed]
- Meinke, P.T.; Wood, H.B.; Szewczyk, J.W. Nuclear hormone receptor modulators for the treatment of diabetes and dyslipidemia. Annu. Rep. Med. Chem. 2006, 41, 99–126. [Google Scholar]
- Nevin, D.K.; Lloyd, D.G.; Fayne, D. Rational targeting of peroxisome proliferating activated receptor subtypes. Curr. Med. Chem. 2011, 18, 5598–5623. [Google Scholar] [CrossRef] [PubMed]
- Sternbach, D.D. Modulators of the peroxisome proliferator-activated receptors (PPARs). Annu. Rep. Med. Chem. 2003, 38, 71–80. [Google Scholar]
- Yasmin, S.; Jayaprakash, V. Thiazolidinediones and PPAR orchestra as antidiabetic agents: From past to present. Eur. J. Med. Chem. 2016, 126, 879–893. [Google Scholar] [CrossRef] [PubMed]
- Balestri, F.; Cappiello, M.; Moschini, R.; Rotondo, R.; Abate, M.; Del Corso, A.; Mura, U. Modulation of aldose reductase activity by aldose hemiacetals. Biochim. Biophys. Acta 2015, 1850, 2329–2339. [Google Scholar] [CrossRef] [PubMed]
- Del Corso, A.; Cappiello, M.; Mura, U. From a dull enzyme to something else: Facts and perspectives regarding aldose reductase. Curr. Med. Chem. 2008, 15, 1452–1461. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.; Chung, S.S. Contributions of polyol pathway to oxidative stress in diabetic cataract. FASEB J. 1999, 13, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Hamada, Y.; Araki, N.; Koh, N.; Nakamura, J.; Horiuchi, S.; Hotta, N. Rapid formation of advanced glycation end products by intermediate metabolites of glycolytic pathway and polyol pathway. Biochem. Biophys. Res. Commun. 1996, 228, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, C.P.; Peat, A.J.; McKeown, S.C.; Corbett, D.F.; Goetz, A.S.; Littleton, T.R.; McCoy, D.C.; Kenakin, T.P.; Andrews, J.L.; Ammala, C.; et al. Pharmacological regulation of insulin secretion in MIN6 cells through the fatty acid receptor GPR40: Identification of agonist and antagonist small molecules. Br. J. Pharmacol. 2006, 148, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo-Figueroa, S.; Ramírez-Espinosa, J.J.; Estrada-Soto, S.; Almanza-Pérez, J.C.; Román-Ramos, R.; Alarcón-Aguilar, J.F.; Hernández-Rosado, J.V.; Moreno-Díaz, H.; Díaz-Coutiño, D.; Navarrete-Vázquez, G. Discovery of thiazolidine-2,4-dione/biphenylcarbonitrile hybrid as dual PPAR α/γ modulator with antidiabetic effect: In vitro, in silico and in vivo approaches. Chem. Biol. Drug Des. 2013, 81, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Awad, K.S.; Elinoff, J.M.; Dougherty, E.J.; Ferreyra, G.A.; Wang, J.Y.; Cai, R.; Sun, J.; Ptasinska, A.; Danner, R.L. G protein-coupled receptor 40 (GPR40) and peroxisome proliferator-activated receptor γ (PPARγ): An integrated two-receptor signaling pathway. J. Biol. Chem. 2015, 290, 19544–19557. [Google Scholar] [CrossRef] [PubMed]
- Reddy, T.N.; Ravinder, M.; Bagul, P.; Ravikanti, K.; Bagul, C.; Nanubolu, J.B.; Srinivas, K.; Banerjee, S.K.; Rao, V.J. Synthesis and biological evaluation of new epalrestat analogues as aldose reductase inhibitors (ARIs). Eur. J. Med. Chem. 2014, 71, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qiang, X.; Luo, L.; Yang, X.; Xiao, G.; Zheng, Y.; Cao, Z.; Sang, Z.; Su, F.; Deng, Y. Multitarget drug design strategy against Alzheimer’s disease: Homoisoflavonoid Mannich base derivatives serve as acetylcholinesterase and monoamine oxidase B dual inhibitors with multifunctional properties. Bioorg. Med. Chem. 2017, 25, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Kitamura, S.; Negoro, N.; Suzuki, M.; Tsujihata, Y.; Suzuki, N.; Santou, T.; Kanzaki, N.; Harada, M.; Tanaka, Y.; et al. Design, synthesis, and biological activity of potent and orally available G protein-coupled receptor 40 agonists. J. Med. Chem. 2011, 54, 1365–1378. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, E.; Due-Hansen, M.E.; Urban, C.; Merten, N.; Pfleiderer, M.; Karlsen, K.K.; Rasmussen, S.S.; Steensgaard, M.; Hamacher, A.; Schmidt, J.; et al. Structure-activity study of dihydrocinnamic acids and discovery of the potent FFA1 (GPR40) agonist TUG-469. ACS Med. Chem. Lett. 2010, 1, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Negoro, N.; Sasaki, S.; Mikami, S.; Ito, M.; Tsujihata, Y.; Ito, R.; Suzuki, M.; Takeuchi, K.; Suzuki, N.; Miyazaki, J.; et al. Optimization of (2,3-dihydro-1-benzofuran-3-yl)acetic acids: Discovery of a non-free fatty acid-like, highly bioavailable G protein-coupled receptor 40/free fatty acid receptor 1 agonist as a glucose-dependent insulinotropic agent. J. Med. Chem. 2012, 55, 3960–3974. [Google Scholar] [CrossRef] [PubMed]
- Balestri, F.; Cappiello, M.; Moschini, R.; Rotondo, R.; Buggiani, I.; Pelosi, P.; Mura, U.; Del Corso, A. L-Idose: An attractive substrate alternative to D-glucose for measuring aldose reductase activity. Biochem. Biophys. Res. Commun. 2015, 456, 891–895. [Google Scholar] [CrossRef] [PubMed]
- Balestri, F.; Rotondo, R.; Moschini, R.; Pellegrino, M.; Cappiello, M.; Barracco, V.; Misuri, L.; Sorce, C.; Andreucci, A.; Del-Corso, A.; et al. Zolfino landrace (Phaseolus vulgaris L.) from Pratomagno: General and specific features of a functional food. Food. Nutr. Res. 2016, 60, 31792. [Google Scholar] [CrossRef] [PubMed]
- Tsao, T.S.; Li, J.; Chang, K.S.; Stenbit, A.E.; Galuska, D.; Anderson, J.E.; Zierath, J.R.; McCarter, R.J.; Charron, M.J. Metabolic adaptations in skeletal muscle overexpressing GLUT4: Effects on muscle and physical activity. FASEB J. 2001, 15, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Dia-DB, A Web Server for the Prediction of Diabetes Drugs. Available online: http://bio-hpc.ucam.edu/dia-db/ (accessed on 5 January 2017).
- Sánchez-Pérez, A.; Muñoz, A.; Peña-García, J.; den-Haan, H.; Bekas, N.; Katsikoudi, A.; Tzakos, A.G.; Pérez-Sánchez, H. DIA-DB: A Web-Accessible Database for the Prediction of Diabetes Drugs. Bioinform. Biomed. Eng. 2015, 9044, 655–663. [Google Scholar]
- Guasch, L.; Sala, E.; Valls, C.; Blay, M.; Mulero, M.; Arola, L.; Pujadas, G.; Garcia-Vallvé, S. Structural insights for the design of new PPARgamma partial agonists with high binding affinity and low transactivation activity. J. Comput. Aided Mol. Des. 2011, 25, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Capelli, D.; Cerchia, C.; Montanari, R.; Loiodice, F.; Tortorella, P.; Laghezza, A.; Cervoni, L.; Pochetti, G.; Lavecchia, A. Structural basis for PPAR partial or full activation revealed by a novel ligand binding mode. Sci. Rep. 2016, 6, 34792. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Chen, Y.; Pan, Y.; Skiles, G.L.; Shou, M. Prediction of human drug-drug interactions from time-dependent inactivation of CYP3A4 in primary hepatocytes using a population-based simulator. Drug Metabol. Dispos. 2009, 37, 2330–2339. [Google Scholar] [CrossRef] [PubMed]
- Taboureau, O.; Jørgensen, F.S. In silico predictions of hERG channel blockers in drug discovery: From ligand-based and target-based approaches to systems chemical biology. Comb. Chem. High Throughput Screen. 2011, 14, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.-U. Polypharmacology—Foe or Friend? J. Med. Chem. 2013, 56, 8955–8971. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Lucio, O.; Naveja, J.J.; Vite-Caritino, H.; Prieto-Martínez, F.D.; Medina-Franco, J.L. One Drug for Multiple Targets: A Computational Perspective. J. Mex. Chem. Soc. 2016, 60, 168–181. [Google Scholar] [CrossRef]
- García-Macedo, R.; Sánchez-Muñoz, F.; Almanza-Pérez, J.C.; Duran-Reyes, G.; Alarcón-Aguilar, F.; Cruz, M. Glycine increases mRNA adiponectin and diminishes pro-inflammatory adipokines expression in 3T3-L1 cells. Eur. J. Pharmacol. 2008, 587, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Almanza-Pérez, J.C.; Alarcón-Aguilar, F.J.; Blancas-Flores, G.; Campos-Sepúlveda, A.E.; Román-Ramos, R.; García-Macedo, R.; Cruz, M. Glycine regulates inflammatory markers modifying the energetic balance through PPAR and UCP-2. Biomed. Pharmacother. 2010, 64, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo-Figueroa, S.; Navarrete-Vázquez, G.; Estrada-Soto, S.; Giles-Rivas, D.; Alarcón-Aguilar, F.J.; León-Rivera, I.; Giacoman-Martínez, A.; Miranda-Pérez, E.; Almanza-Pérez, J.C. Discovery of new dual PPARγ-GPR40 agonists with robust antidiabetic activity: Design, synthesis and in combo drug evaluation. Biomed. Pharmacother. 2017, 90, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Verspohl, E.J. Recommended testing in diabetes research. Planta Medica 2002, 68, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Goyal, S.N.; Reddy, N.M.; Patil, K.R.; Nakhate, K.T.; Ojha, S.; Patil, C.R.; Agrawal, Y.O. Challenges and issues with streptozotocin-induced diabetes—A clinically relevant animal model to understand the diabetes pathogenesis and evaluate therapeutics. Chem. Biol. Interact. 2016, 244, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Kojima, R.; Ito, M. Strain differences in the diabetogenic activity of streptozotocin in mice. Biol. Pharm. Bull. 2006, 6, 1110–1119. [Google Scholar] [CrossRef]
- Radenkovíc, M.; Stojanović, M.; Prostran, M. Experimental diabetes induced by alloxan and streptozotocin: The current state of the art. J. Pharm. Toxicol. Methods 2016, 78, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Andrade, R.; Rodríguez-López, V.; Garduño-Ramírez, M.; Castillo-España, P.; Estrada-Soto, S. Anti-diabetic effect on alloxanized and normoglycemic rats and some pharmacological evaluations of Tournefortia hartwegiana. J. Ethnopharmacol. 2005, 101, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Lara, E.; Martínez-Conde, C.; Rosales-Ortega, E.; Ramírez-Espinosa, J.J.; Rivera-Leyva, J.C.; Centurión, D.; Carvajal, K.; Ortega-Cuellar, D.; Estrada-Soto, S.; Navarrete-Vázquez, G. Synthesis and in vitro AMPK activation of cycloalkyl/alkarylbiguanides with robust in vivo antihyperglycemic action. J. Chem. 2017, 2017, 1212609. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE), 2016.08; Chemical Computing group ULC, 1010 Sherbooke St. West, Site #910, Montreal, QC, Canada, H3A 2R7, 2018. Available online: http://www.chemcomp.com (accessed on 30 September 2017).
Sample Availability: Samples of the compounds are available from the authors. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).