Anti-Inflammatory Effect of Melittin on Porphyromonas Gingivalis LPS-Stimulated Human Keratinocytes

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
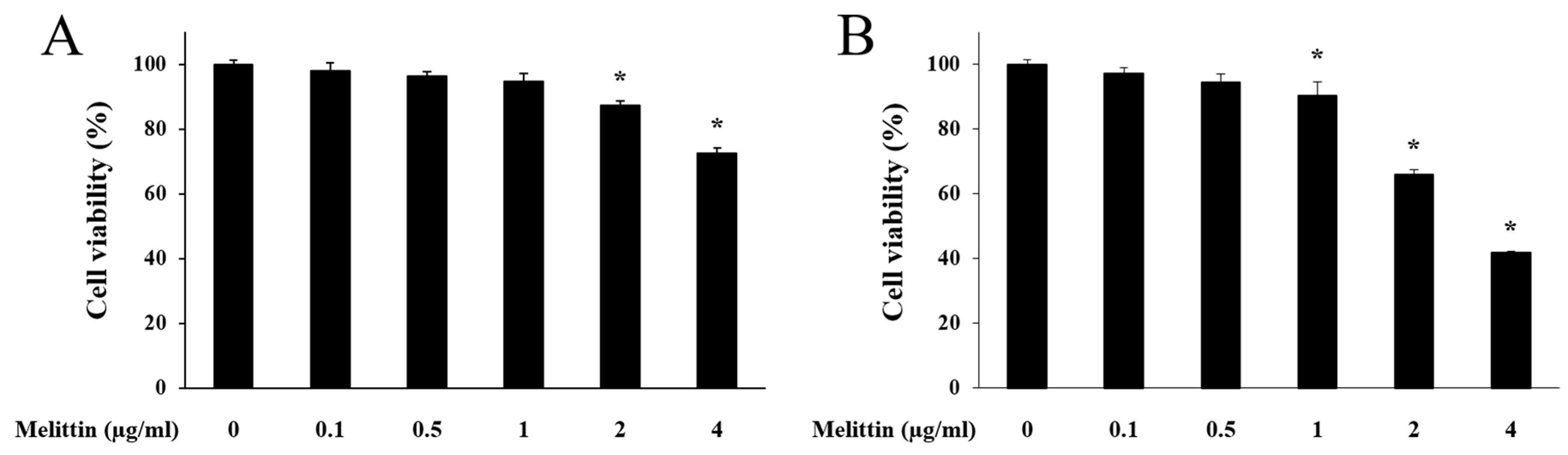
2.1. Effect of Melittin on HaCaT Cell Viability
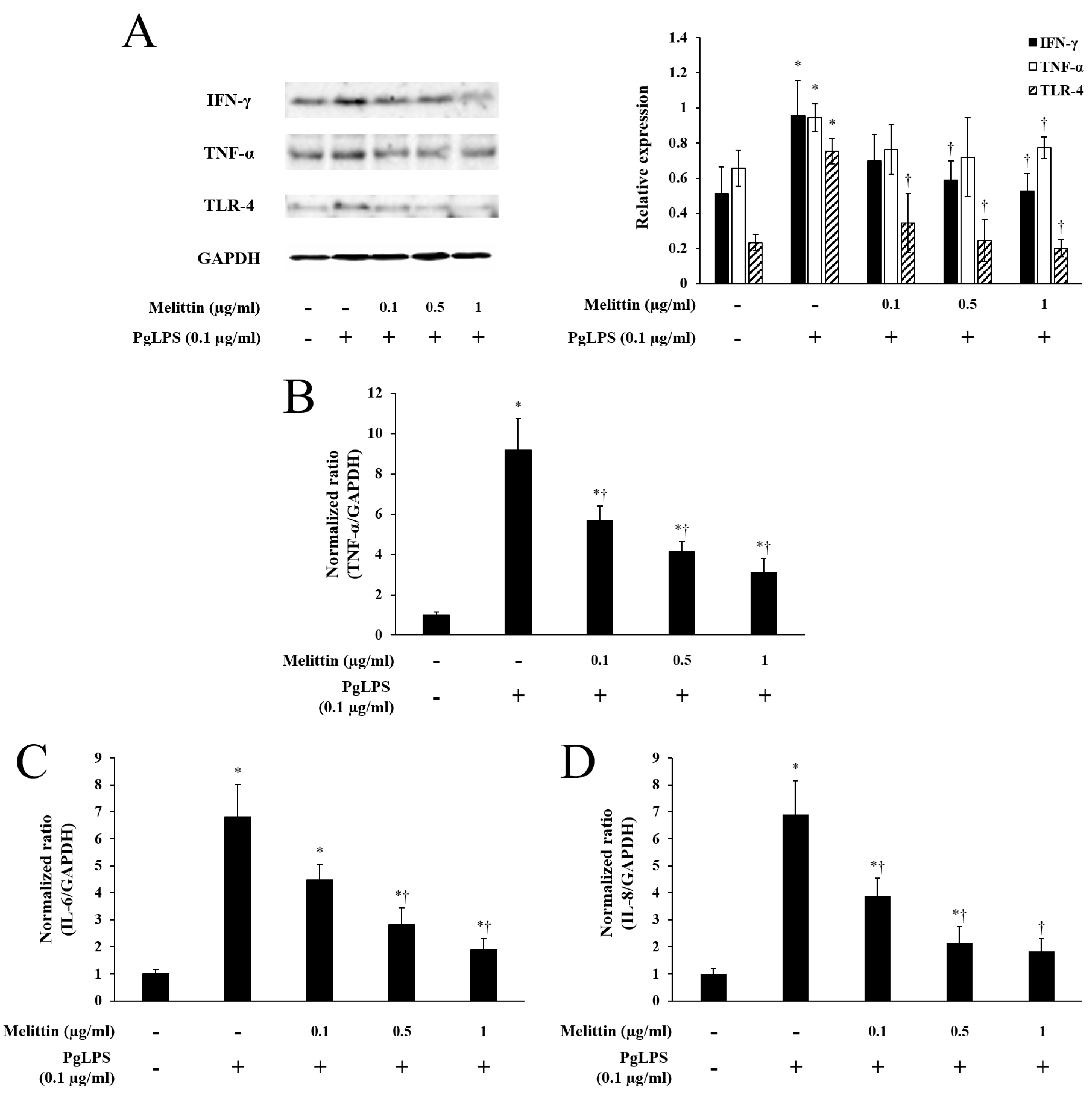
2.2. Melittin Inhibits PgLPS-Induced Expression of TLR-4 and Inflammatory Cytokines
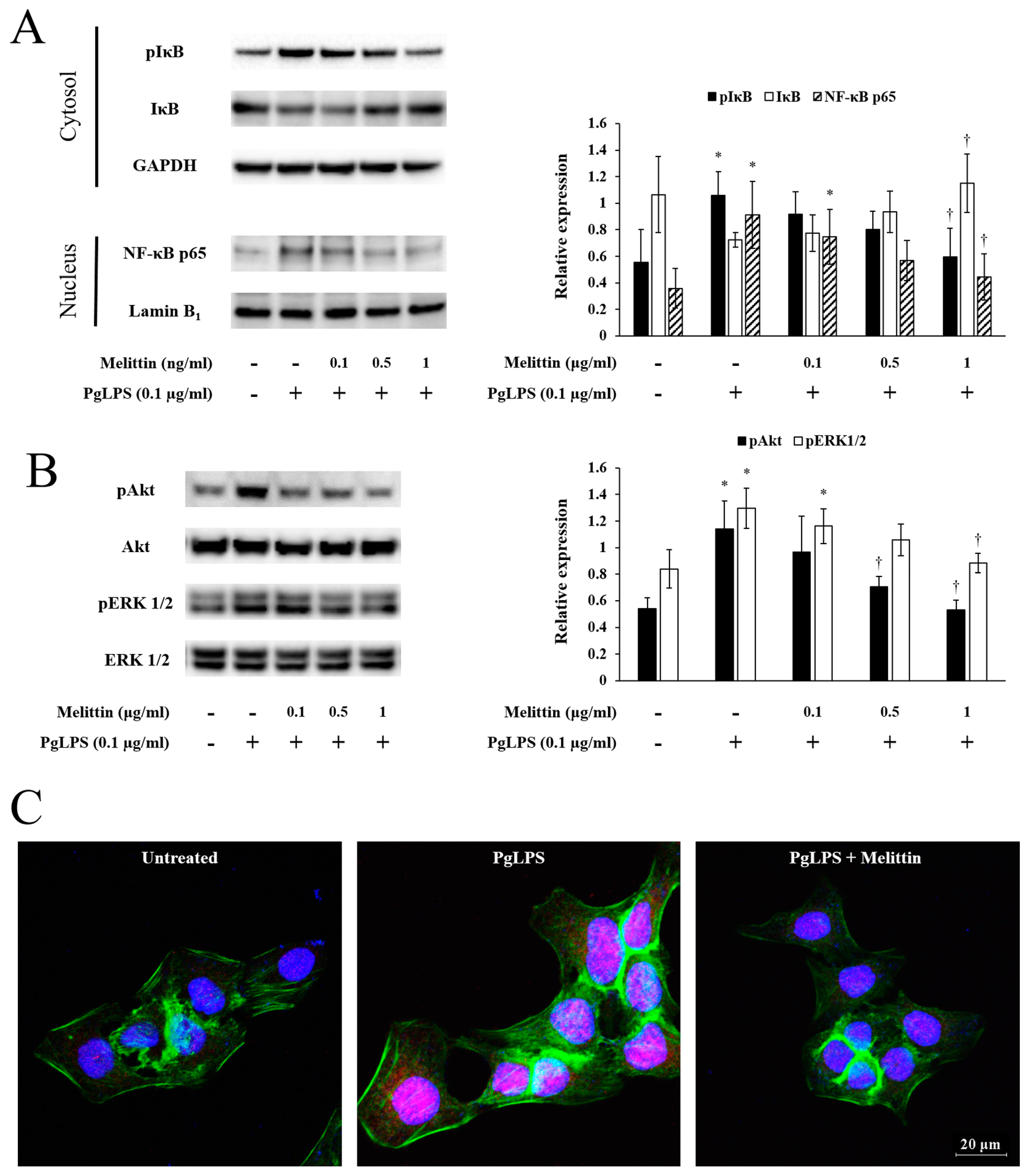
2.3. Melittin Inhibits PgLPS-Induced Activation of the NF-κB Signaling Pathway, Akt, and ERK
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Cell Viability Test
4.3. Quantitative Real-Time Polymerase Chain Reaction
4.4. Western Blot
4.5. Immunofluorescence Analysis
4.6. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kuboniwa, M.; Lamont, R.J. Subgingival biofilm formation. Periodontology 2000 2010, 52, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Silva, N.; Abusleme, L.; Bravo, D.; Dutzan, N.; Garcia-Sesnich, J.; Vernal, R.; Hernandez, M.; Gamonal, J. Host response mechanisms in periodontal diseases. J. Appl. Oral Sci. 2015, 23, 329–355. [Google Scholar] [CrossRef] [PubMed]
- How, K.Y.; Song, K.P.; Chan, K.G. Porphyromonas gingivalis: An overview of periodontopathic pathogen below the gum line. Front. Microbiol. 2016, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, A.; Zoheir, N.; Awang, R.A.; Culshaw, S.; Ramage, G.; Lappin, D.F.; Nile, C.J. The alpha 7 nicotinic receptor agonist pha-543613 hydrochloride inhibits porphyromonas gingivalis-induced expression of interleukin-8 by oral keratinocytes. Inflamm. Res. 2014, 63, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Sandros, J.; Karlsson, C.; Lappin, D.F.; Madianos, P.N.; Kinane, D.F.; Papapanou, P.N. Cytokine responses of oral epithelial cells to porphyromonas gingivalis infection. J. Dent. Res. 2000, 79, 1808–1814. [Google Scholar] [CrossRef] [PubMed]
- Ekhlassi, S.; Scruggs, L.Y.; Garza, T.; Montufar-Solis, D.; Moretti, A.J.; Klein, J.R. Porphyromonas gingivalis lipopolysaccharide induces tumor necrosis factor-alpha and interleukin-6 secretion, and ccl25 gene expression, in mouse primary gingival cell lines: Interleukin-6-driven activation of ccl2. J. Periodontal. Res. 2008, 43, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Arango Duque, G.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef] [PubMed]
- Cekici, A.; Kantarci, A.; Hasturk, H.; Van Dyke, T.E. Inflammatory and immune pathways in the pathogenesis of periodontal disease. Periodontology 2000 2014, 64, 57–80. [Google Scholar] [CrossRef] [PubMed]
- Tribble, G.D.; Kerr, J.E.; Wang, B.Y. Genetic diversity in the oral pathogen porphyromonas gingivalis: Molecular mechanisms and biological consequences. Future Microbiol. 2013, 8, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Armitage, G.C. Classifying periodontal diseases—A long-standing dilemma. Periodontology 2000 2002, 30, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Colombo, A.P.; Bennet, S.; Cotton, S.L.; Goodson, J.M.; Kent, R.; Haffajee, A.D.; Socransky, S.S.; Hasturk, H.; Van Dyke, T.E.; Dewhirst, F.E.; et al. Impact of periodontal therapy on the subgingival microbiota of severe periodontitis: Comparison between good responders and individuals with refractory periodontitis using the human oral microbe identification microarray. J. Periodontol. 2012, 83, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Lerner, U.H.; Teng, Y.T. Cytokine responses against periodontal infection: Protective and destructive roles. Periodontology 2000 2010, 52, 163–206. [Google Scholar] [CrossRef] [PubMed]
- Cirano, F.R.; Casarin, R.C.; Ribeiro, F.V.; Casati, M.Z.; Pimentel, S.P.; Taiete, T.; Bernardi, M.M. Effect of resveratrol on periodontal pathogens during experimental periodontitis in rats. Braz. Oral Res. 2016, 30, e128. [Google Scholar] [CrossRef] [PubMed]
- Hasturk, H.; Kantarci, A.; Ebrahimi, N.; Andry, C.; Holick, M.; Jones, V.L.; Van Dyke, T.E. Topical h2 antagonist prevents periodontitis in a rabbit model. Infect. Immun. 2006, 74, 2402–2414. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Miao, B.; Zhu, J.; Wang, H.; Zhou, Z. Transplantation of periodontal ligament cell sheets expressing human betadefensin3 promotes antiinflammation in a canine model of periodontitis. Mol. Med. Rep. 2017, 16, 7459–7467. [Google Scholar] [CrossRef] [PubMed]
- Habermann, E. Bee and wasp venoms. Science 1972, 177, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Billingham, M.E.; Morley, J.; Hanson, J.M.; Shipolini, R.A.; Vernon, C.A. Letter: An anti-inflammatory peptide from bee venom. Nature 1973, 245, 163–164. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.; Giralt, E. Three valuable peptides from bee and wasp venoms for therapeutic and biotechnological use: Melittin, apamin and mastoparan. Toxins 2015, 7, 1126–1150. [Google Scholar] [CrossRef] [PubMed]
- Raghuraman, H.; Chattopadhyay, A. Melittin: A membrane-active peptide with diverse functions. Biosci. Rep. 2007, 27, 189–223. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.T.; Cheng, H.H.; Huang, C.J.; Chang, H.C.; Chi, C.C.; Su, H.H.; Hsu, S.S.; Wang, J.L.; Chen, I.S.; Liu, S.I.; et al. Phospholipase a2-independent ca2+ entry and subsequent apoptosis induced by melittin in human mg63 osteosarcoma cells. Life Sci. 2007, 80, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, B.; Lu, S.Q.; Li, Y.; Su, Y.H.; Ling, C.Q. Effects of melittin on expressions of mitochondria membrane protein 7a6, cell apoptosis-related gene products fas and fas ligand in hepatocarcinoma cells. Chin. J. Integr. Med. 2007, 5, 559–563. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, K.H.; Lee, W.R.; Han, S.M.; Park, K.K. Protective effect of melittin on inflammation and apoptosis in acute liver failure. Apoptosis 2012, 17, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Park, J.H.; Kim, K.H.; Lee, W.R.; Kim, K.S.; Park, K.K. Melittin inhibits atherosclerosis in lps/high-fat treated mice through atheroprotective actions. J. Atheroscler. Thromb. 2011, 18, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.R.; Kim, K.H.; An, H.J.; Kim, J.Y.; Chang, Y.C.; Chung, H.; Park, Y.Y.; Lee, M.L.; Park, K.K. The protective effects of melittin on propionibacterium acnes-induced inflammatory responses in vitro and in vivo. J. Investig. Dermatol. 2014, 134, 1922–1930. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.H.; An, H.J.; Kim, J.Y.; Gwon, M.G.; Gu, H.; Park, J.B.; Sung, W.J.; Kwon, Y.C.; Park, K.D.; Han, S.M.; et al. Bee venom inhibits porphyromonas gingivalis lipopolysaccharides-induced pro-inflammatory cytokines through suppression of nf-kappab and ap-1 signaling pathways. Molecules 2016, 21. [Google Scholar] [CrossRef] [PubMed]
- Tada, H.; Matsuyama, T.; Nishioka, T.; Hagiwara, M.; Kiyoura, Y.; Shimauchi, H.; Matsushita, K. Porphyromonas gingivalis gingipain-dependently enhances il-33 production in human gingival epithelial cells. PLoS ONE 2016, 11, e0152794. [Google Scholar] [CrossRef] [PubMed]
- De Camargo Pereira, G.; Guimaraes, G.N.; Planello, A.C.; Santamaria, M.P.; de Souza, A.P.; Line, S.R.; Marques, M.R. Porphyromonas gingivalis lps stimulation downregulates dnmt1, dnmt3a, and jmjd3 gene expression levels in human hacat keratinocytes. Clin. Oral Investig. 2013, 17, 1279–1285. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Taguchi, Y.; Tominaga, K.; Umeda, M.; Tanaka, A. Porphyromonas gingivalis lps inhibits osteoblastic differentiation and promotes pro-inflammatory cytokine production in human periodontal ligament stem cells. Arch. Oral Biol. 2014, 59, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Roberts, F.A.; McCaffery, K.A.; Michalek, S.M. Profile of cytokine mrna expression in chronic adult periodontitis. J. Dent. Res. 1997, 76, 1833–1839. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; van der Graaf, C.; Van der Meer, J.W.; Kullberg, B.J. Toll-like receptors and the host defense against microbial pathogens: Bringing specificity to the innate-immune system. J. Leukoc Biol. 2004, 75, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Aderem, A.; Ulevitch, R.J. Toll-like receptors in the induction of the innate immune response. Nature 2000, 406, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.T. Protective and destructive immunity in the periodontium: Part 1—Innate and humoral immunity and the periodontium. J. Dent. Res. 2006, 85, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Kawa, K.; Tsutsui, H.; Uchiyama, R.; Kato, J.; Matsui, K.; Iwakura, Y.; Matsumoto, T.; Nakanishi, K. Ifn-gamma is a master regulator of endotoxin shock syndrome in mice primed with heat-killed propionibacterium acnes. Int. Immunol. 2010, 22, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, N.; Gans, E.H. Tretinoin: A review of its anti-inflammatory properties in the treatment of acne. J. Clin. Aesthet. Dermatol. 2011, 4, 22–29. [Google Scholar] [PubMed]
- Vowels, B.R.; Yang, S.; Leyden, J.J. Induction of proinflammatory cytokines by a soluble factor of propionibacterium acnes: Implications for chronic inflammatory acne. Infect. Immun. 1995, 63, 3158–3165. [Google Scholar] [PubMed]
- Basal, E.; Jain, A.; Kaushal, G.P. Antibody response to crude cell lysate of propionibacterium acnes and induction of pro-inflammatory cytokines in patients with acne and normal healthy subjects. J. Microbiol. 2004, 42, 117–125. [Google Scholar] [PubMed]
- Kim, J.; Ochoa, M.T.; Krutzik, S.R.; Takeuchi, O.; Uematsu, S.; Legaspi, A.J.; Brightbill, H.D.; Holland, D.; Cunliffe, W.J.; Akira, S.; et al. Activation of toll-like receptor 2 in acne triggers inflammatory cytokine responses. J. Immunol. 2002, 169, 1535–1541. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.A.; Smith, M.F., Jr.; Sanders, M.K.; Ernst, P.B. Reactive oxygen and nitrogen species differentially regulate toll-like receptor 4-mediated activation of nf-kappa b and interleukin-8 expression. Infect. Immun. 2004, 72, 2123–2130. [Google Scholar] [CrossRef] [PubMed]
- Miggin, S.M.; O’Neill, L.A. New insights into the regulation of tlr signaling. J. Leukoc Biol. 2006, 80, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Gaynor, R.B. Therapeutic potential of inhibition of the nf-kappab pathway in the treatment of inflammation and cancer. J. Clin. Investig. 2001, 107, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Napetschnig, J.; Wu, H. Molecular basis of nf-kappab signaling. Annu. Rev. Biophys. 2013, 42, 443–468. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, M.D.; Harrison, S.C. Structure of an ikappabalpha/nf-kappab complex. Cell 1998, 95, 749–758. [Google Scholar] [CrossRef]
- Grange, P.A.; Raingeaud, J.; Calvez, V.; Dupin, N. Nicotinamide inhibits propionibacterium acnes-induced il-8 production in keratinocytes through the nf-kappab and mapk pathways. J. Dermatol. Sci. 2009, 56, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Park, D.J.; Kim, Y.H.; Kim, Y.; Choi, Y.W.; Lee, S.J. Schisandra chinensis alpha-iso-cubebenol induces heme oxygenase-1 expression through pi3k/akt and nrf2 signaling and has anti-inflammatory activity in porphyromonas gingivalis lipopolysaccharide-stimulated macrophages. Int. Immunopharmacol. 2011, 11, 1907–1915. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.O.; Park, S.Y.; Lee, K.J.; Heo, M.S.; Kim, K.C.; Kim, M.O.; Lee, J.D.; Choi, Y.H.; Kim, G.Y. Bee venom and melittin reduce proinflammatory mediators in lipopolysaccharide-stimulated bv2 microglia. Int. Immunopharmacol. 2007, 7, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, D.H.; Baek, S.H.; Lee, H.J.; Kim, M.R.; Kwon, H.J.; Lee, C.H. Rengyolone inhibits inducible nitric oxide synthase expression and nitric oxide production by down-regulation of nf-kappab and p38 map kinase activity in lps-stimulated raw 264.7 cells. Biochem. Pharmacol. 2006, 71, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.L.; Kim, C.K.; Kim, T.J.; Sun, J.; Rim, D.; Kim, Y.J.; Ko, S.B.; Jang, H.; Yoon, B.W. Anti-inflammatory effects of fimasartan via akt, erk, and nfkappab pathways on astrocytes stimulated by hemolysate. Inflamm. Res. 2016, 65, 115–123. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, W.-H.; An, H.-J.; Kim, J.-Y.; Gwon, M.-G.; Gu, H.; Jeon, M.; Kim, M.-K.; Han, S.-M.; Park, K.-K. Anti-Inflammatory Effect of Melittin on Porphyromonas Gingivalis LPS-Stimulated Human Keratinocytes. Molecules 2018, 23, 332. https://doi.org/10.3390/molecules23020332
Kim W-H, An H-J, Kim J-Y, Gwon M-G, Gu H, Jeon M, Kim M-K, Han S-M, Park K-K. Anti-Inflammatory Effect of Melittin on Porphyromonas Gingivalis LPS-Stimulated Human Keratinocytes. Molecules. 2018; 23(2):332. https://doi.org/10.3390/molecules23020332
Chicago/Turabian StyleKim, Woon-Hae, Hyun-Jin An, Jung-Yeon Kim, Mi-Gyeong Gwon, Hyemin Gu, Minji Jeon, Min-Kyung Kim, Sang-Mi Han, and Kwan-Kyu Park. 2018. "Anti-Inflammatory Effect of Melittin on Porphyromonas Gingivalis LPS-Stimulated Human Keratinocytes" Molecules 23, no. 2: 332. https://doi.org/10.3390/molecules23020332
APA StyleKim, W.-H., An, H.-J., Kim, J.-Y., Gwon, M.-G., Gu, H., Jeon, M., Kim, M.-K., Han, S.-M., & Park, K.-K. (2018). Anti-Inflammatory Effect of Melittin on Porphyromonas Gingivalis LPS-Stimulated Human Keratinocytes. Molecules, 23(2), 332. https://doi.org/10.3390/molecules23020332