Role of Cationic Side Chains in the Antimicrobial Activity of C18G




Abstract
1. Introduction
2. Results
2.1. Peptide Sequences and Structure
2.2. Antibacterial Activity Assays
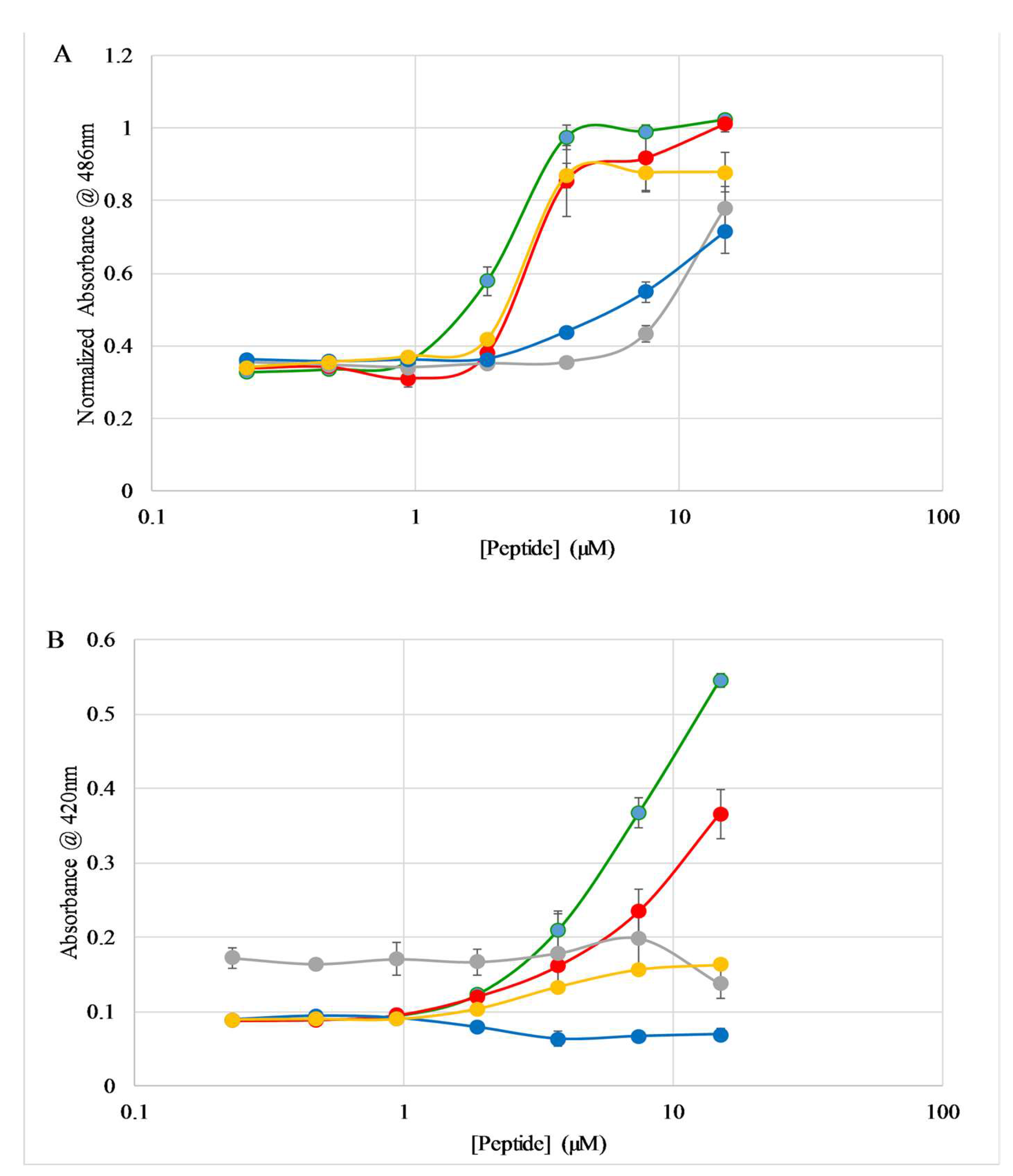
2.3. Binding to Lipid Vesicles
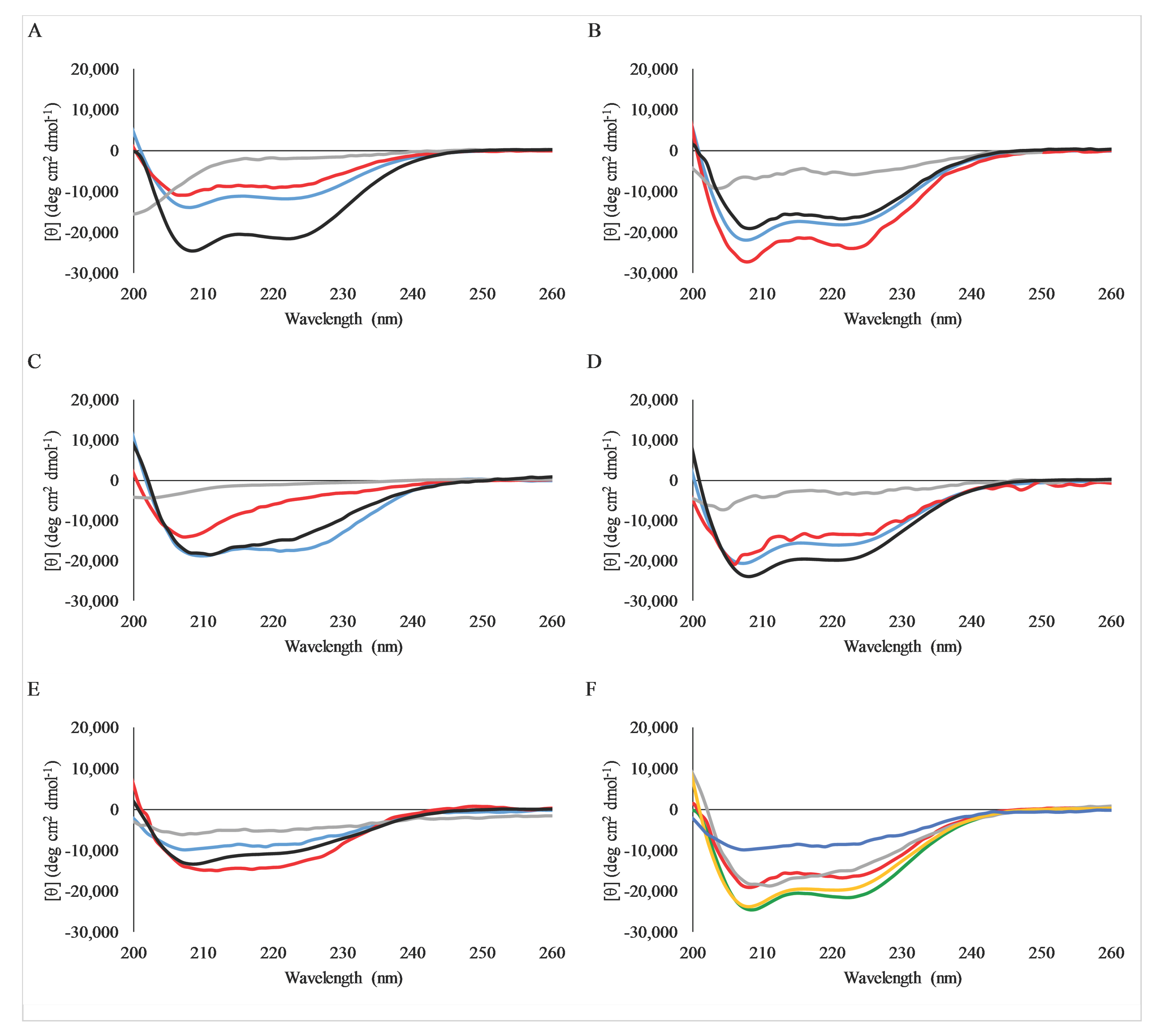
2.4. Secondary Structure Analysis by Circular Dichroism Spectroscopy
2.5. E. coli Membrane Permeabilization
2.6. S. aureus Membrane Permeabilization
2.7. Hemolysis
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Bacterial Culturing
4.3. Minimal Inhibitory Concentration/Minimal Bactericidal Concentration Assays
4.4. Fluorescence Spectroscopy
4.5. Circular Dichroism Spectroscopy
4.6. E. coli Outer Membrane Permeability Assay
4.7. E. coli Inner Membrane Permeability Assay
4.8. S. aureus Membrane Permeability Assay
4.9. Hemolysis Assay
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization (WHO). Global Action Plan on Antimicrobial Resistance; WHO: Geneva, Switzerland, 2015; p. 28. [Google Scholar]
- Centers for Disease Control and Prevention. Antibiotic Resistance Threats in the United States, 2013; Centers for Disease Control and Prevention: Druid Hills, GA, USA, 2017.
- Yoneyama, H.; Katsumata, R. Antibiotic resistance in bacteria and its future for novel antibiotic development. Biosci. Biotechnol. Biochem. 2006, 70, 1060–1075. [Google Scholar] [CrossRef] [PubMed]
- Laxminarayan, R.; Duse, A.; Wattal, C.; Zaidi, A.K.; Wertheim, H.F.; Sumpradit, N.; Vlieghe, E.; Hara, G.L.; Gould, I.M.; Goossens, H.; et al. Antibiotic resistance-the need for global solutions. Lancet Infect. Dis. 2013, 13, 1057–1098. [Google Scholar] [CrossRef]
- Siala, W.; Mingeot-Leclercq, M.P.; Tulkens, P.M.; Hallin, M.; Denis, O.; Van Bambeke, F. Comparison of the antibiotic activities of daptomycin, vancomycin, and the investigational fluoroquinolone delafloxacin against biofilms from Staphylococcus aureus clinical isolates. Antimicrob. Agents Chemother. 2014, 58, 6385–6397. [Google Scholar] [CrossRef] [PubMed]
- Anderl, J.N.; Franklin, M.J.; Stewart, P.S. Role of antibiotic penetration limitation in Klebsiella pneumoniae biofilm resistance to ampicillin and ciprofloxacin. Antimicrob. Agents Chemother. 2000, 44, 1818–1824. [Google Scholar] [CrossRef] [PubMed]
- Lomovskaya, O.; Warren, M.S.; Lee, A.; Galazzo, J.; Fronko, R.; Lee, M.; Blais, J.; Cho, D.; Chamberland, S.; Renau, T.; et al. Identification and characterization of inhibitors of multidrug resistance efflux pumps in pseudomonas aeruginosa: Novel agents for combination therapy. Antimicrob. Agents Chemother. 2001, 45, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Wijnant, G.J.; Van Bocxlaer, K.; Yardley, V.; Murdan, S.; Croft, S.L. Efficacy of paromomycin-chloroquine combination therapy in experimental cutaneous leishmaniasis. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Mensa, B.; Kim, Y.H.; Choi, S.; Scott, R.; Caputo, G.A.; DeGrado, W.F. Antibacterial mechanism of action of arylamide foldamers. Antimicrob. Agents Chemother. 2011, 55, 5043–5053. [Google Scholar] [CrossRef] [PubMed]
- Mensa, B.; Howell, G.L.; Scott, R.; DeGrado, W.F. Comparative mechanistic studies of brilacidin, daptomycin, and the antimicrobial peptide LL16. Antimicrob. Agents Chemother. 2014, 58, 5136–5145. [Google Scholar] [CrossRef] [PubMed]
- Malik, I.T.; Brotz-Oesterhelt, H. Conformational control of the bacterial CLP protease by natural product antibiotics. Nat. Prod. Rep. 2017, 34, 815–831. [Google Scholar] [CrossRef] [PubMed]
- Townsley, L.; Shank, E.A. Natural-product antibiotics: Cues for modulating bacterial biofilm formation. Trends Microbiol. 2017, 25, 1016–1026. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Caputo, G.A.; Vemparala, S.; Kuroda, K. Synthetic random copolymers as a molecular platform to mimic host-defense antimicrobial peptides. Bioconj. Chem. 2017, 28, 1340–1350. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Wang, Y.; Wu, S.; Li, Y.; Li, J. Durable anti-superbug polymers: Covalent bonding of ionic liquid onto the polymer chains. Biomacromolecules 2017, 18, 4364–4372. [Google Scholar] [CrossRef] [PubMed]
- Goderecci, S.S.; Kaiser, E.; Yanakas, M.; Norris, Z.; Scaturro, J.; Oszust, R.; Medina, C.D.; Waechter, F.; Heon, M.; Krchnavek, R.R.; et al. Silver oxide coatings with high silver-ion elution rates and characterization of bactericidal activity. Molecules 2017, 22, E1487. [Google Scholar] [CrossRef] [PubMed]
- Thanh, N.H.; Goycoolea, F.M. Chitosan/Cyclodextrin/TPP nanoparticles loaded with quercetin as novel bacterial quorum sensing inhibitors. Molecules 2017, 22, E1975. [Google Scholar] [CrossRef] [PubMed]
- Capilato, J.N.; Philippi, S.V.; Reardon, T.; McConnell, A.; Oliver, D.C.; Warren, A.; Adams, J.S.; Wu, C.; Perez, L.J. Development of a novel series of non-natural triaryl agonists and antagonists of the Pseudomonas aeruginosa LasR quorum sensing receptor. Bioorg. Med. Chem. 2017, 25, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Arranz-Trullen, J.; Lu, L.; Pulido, D.; Bhakta, S.; Boix, E. Host antimicrobial peptides: The promise of new treatment strategies against tuberculosis. Front. Immunol. 2017, 8, 1499. [Google Scholar] [CrossRef] [PubMed]
- Dhople, V.; Krukemeyer, A.; Ramamoorthy, A. The human beta-defensin-3, an antibacterial peptide with multiple biological functions. Biochim. Biophys. Acta 2006, 1758, 1499–1512. [Google Scholar] [CrossRef] [PubMed]
- Amiche, M.; Galanth, C. Dermaseptins as models for the elucidation of membrane-acting helical amphipathic antimicrobial peptides. Curr. Pharm. Biotechnol. 2011, 12, 1184–1193. [Google Scholar] [CrossRef] [PubMed]
- Mahalka, A.K.; Kinnunen, P.K. Binding of amphipathic alpha-helical antimicrobial peptides to lipid membranes: Lessons from temporins B and L. Biochim. Biophys. Acta 2009, 1788, 1600–1609. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Liang, X.; Liu, C.; Cheng, Y.; Zhou, L.; Wang, K.; Zhao, L. Influence of proline substitution on the bioactivity of mammalian-derived antimicrobial peptide NK-2. Probiotics Antimicrob. Proteins 2017. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Rangarajan, N.; Weisshaar, J.C. Lights, camera, action! Antimicrobial peptide mechanisms imaged in space and time. Trends Microb. 2016, 24, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.F.; Brezden, A.; Mohammad, H.; Chmielewski, J.; Seleem, M.N. A short D-enantiomeric antimicrobial peptide with potent immunomodulatory and antibiofilm activity against multidrug-resistant Pseudomonas aeruginosa and Acinetobacter baumannii. Sci. Rep. 2017, 7, 6953. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, U.; Sobczak, M.; Oledzka, E. Current state of a dual behaviour of antimicrobial peptides-therapeutic agents and promising delivery vectors. Chem. Biol. Drug Des. 2017, 90, 1079–1093. [Google Scholar] [CrossRef] [PubMed]
- Toke, O. Antimicrobial peptides: New candidates in the fight against bacterial infections. Biopolymers 2005, 80, 717–735. [Google Scholar] [CrossRef] [PubMed]
- Darveau, R.P.; Blake, J.; Seachord, C.L.; Cosand, W.L.; Cunningham, M.D.; Cassiano-Clough, L.; Maloney, G. Peptides related to the carboxyl terminus of human platelet factor iv with antibacterial activity. J. Clin. Investig. 1992, 90, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Peck-Miller, K.A.; Blake, J.; Cosand, W.L.; Darveau, R.P.; Fell, H.P. Structure-activity analysis of the antitumor and hemolytic properties of the amphiphilic alpha-helical peptide, c18g. Int. J. Pept. Protein Res. 1994, 44, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Bader, M.W.; Navarre, W.W.; Shiau, W.; Nikaido, H.; Frye, J.G.; McClelland, M.; Fang, F.C.; Miller, S.I. Regulation of salmonella typhimurium virulence gene expression by cationic antimicrobial peptides. Mol. Microbiol. 2003, 50, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Groisman, E.A. Acidic pH sensing in the bacterial cytoplasm is required for salmonella virulence. Mol. Microbiol. 2016, 101, 1024–1038. [Google Scholar] [CrossRef] [PubMed]
- Gunn, J.S. The salmonella PmrAB regulon: Lipopolysaccharide modifications, antimicrobial peptide resistance and more. Trends Microbiol. 2008, 16, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Saint Jean, K.D.; Henderson, K.D.; Chrom, C.L.; Abiuso, L.E.; Renn, L.M.; Caputo, G.A. Effects of hydrophobic amino acid substitutions on antimicrobial peptide behavior. Probiotics Antimicrob. Proteins 2017. [Google Scholar] [CrossRef] [PubMed]
- Caputo, G.A.; London, E. Using a novel dual fluorescence quenching assay for measurement of tryptophan depth within lipid bilayers to determine hydrophobic alpha-helix locations within membranes. Biochemistry 2003, 42, 3265–3274. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, Z.; Picciano, A.L.; Gosavi, P.M.; Moroz, Y.S.; Angevine, C.E.; Chavis, A.E.; Reiner, J.E.; Korendovych, I.V.; Caputo, G.A. Functional characterization of a melittin analog containing a non-natural tryptophan analog. Biopolymers 2015, 104, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Caputo, G.A. Analyzing the effects of hydrophobic mismatch on transmembrane alpha-helices using tryptophan fluorescence spectroscopy. Methods Mol. Biol. 2013, 1063, 95–116. [Google Scholar] [PubMed]
- Bahar, A.A.; Ren, D. Antimicrobial peptides. Pharmaceuticals 2013, 6, 1543–1575. [Google Scholar] [CrossRef] [PubMed]
- Zelezetsky, I.; Tossi, A. Alpha-helical antimicrobial peptides—Using a sequence template to guide structure-activity relationship studies. Biochim. Biophys. Acta 2006, 1758, 1436–1449. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.P.; Girinath, P.; Ahmad, R. Effect of lysine side chain length on intra-helical glutamate-lysine ion pairing interactions. Biochemistry 2007, 46, 10528–10537. [Google Scholar] [CrossRef] [PubMed]
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic therapeutic peptides: Science and market. Drug Discov. Today 2010, 15, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Rabanal, F.; Grau-Campistany, A.; Vila-Farrés, X.; Gonzalez-Linares, J.; Borràs, M.; Vila, J.; Manresa, A.; Cajal, Y. A bioinspired peptide scaffold with high antibiotic activity and low in vivo toxicity. Sci. Rep. 2015, 5, 10558. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.P.; Wang, W.R.; Girinath, P.; Yang, P.A.; Ahmad, R.; Li, J.H.; Hart, P.; Kokona, B.; Fairman, R.; Kilpatrick, C.; et al. Effect of glutamate side chain length on intrahelical glutamate-lysine ion pairing interactions. Biochemistry 2012, 51, 7157–7172. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, K.; Caputo, G.A.; DeGrado, W.F. The role of hydrophobicity in the antimicrobial and hemolytic activities of polymethacrylate derivatives. Chemistry 2009, 15, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Palermo, E.F.; Vemparala, S.; Kuroda, K. Cationic spacer arm design strategy for control of antimicrobial activity and conformation of amphiphilic methacrylate random copolymers. Biomacromolecules 2012, 13, 1632–1641. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Isaacs, A.; Clements, D.; Liu, D.; Kim, H.; Scott, R.W.; Winkler, J.D.; DeGrado, W.F. De novo design and in vivo activity of conformationally restrained antimicrobial arylamide foldamers. Proc. Natl. Acad. Sci. USA 2009, 106, 6968–6973. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Choi, S.; Chen, B.; Doerksen, R.J.; Clements, D.J.; Winkler, J.D.; Klein, M.L.; DeGrado, W.F. Nontoxic membrane-active antimicrobial arylamide oligomers. Angew. Chem. 2004, 43, 1158–1162. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Koh, J.-J.; Liu, S.; Lakshminarayanan, R.; Verma, C.S.; Beuerman, R.W. Membrane active antimicrobial peptides: Translating mechanistic insights to design. Front. Neurosci. 2017, 11, 73. [Google Scholar] [CrossRef] [PubMed]
- Cutrona, K.J.; Kaufman, B.A.; Figueroa, D.M.; Elmore, D.E. Role of arginine and lysine in the antimicrobial mechanism of histone-derived antimicrobial peptides. FEBS Lett. 2015, 589, 3915–3920. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, M.; Hällbrink, M.; Prochiantz, A.; Langel, Ü. Cell-penetrating peptides. Trends Pharmacol. Sci. 2000, 21, 99–103. [Google Scholar] [CrossRef]
- Schmidt, N.; Mishra, A.; Lai, G.H.; Wong, G.C. Arginine-rich cell-penetrating peptides. FEBS Lett. 2010, 584, 1806–1813. [Google Scholar] [CrossRef] [PubMed]
- Marquette, A.; Mason, A.J.; Bechinger, B. Aggregation and membrane permeabilizing properties of designed histidine-containing cationic linear peptide antibiotics. J. Pept. Sci. 2008, 14, 488–495. [Google Scholar] [CrossRef] [PubMed]
- McDonald, M.; Mannion, M.; Pike, D.; Lewis, K.; Flynn, A.; Brannan, A.M.; Browne, M.J.; Jackman, D.; Madera, L.; Power Coombs, M.R.; et al. Structure–function relationships in histidine-rich antimicrobial peptides from atlantic cod. Biochim. Biophys. Acta 2015, 1848, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Kacprzyk, L.; Rydengård, V.; Mörgelin, M.; Davoudi, M.; Pasupuleti, M.; Malmsten, M.; Schmidtchen, A. Antimicrobial activity of histidine-rich peptides is dependent on acidic conditions. Biochim. Biophys. Acta 2007, 1768, 2667–2680. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Hu, K.; Hu, G.; Shi, D.; Jiang, Y.; Hui, L.; Zhu, R.; Xie, Y.; Yang, L. Long hydrophilic-and-cationic polymers: A different pathway toward preferential activity against bacterial over mammalian membranes. Biomacromolecules 2014, 15, 3267–3277. [Google Scholar] [CrossRef] [PubMed]
- Palermo, E.F.; Sovadinova, I.; Kuroda, K. Structural determinants of antimicrobial activity and biocompatibility in membrane-disrupting methacrylamide random copolymers. Biomacromolecules 2009, 10, 3098–3107. [Google Scholar] [CrossRef] [PubMed]
- Dathe, M.; Schumann, M.; Wieprecht, T.; Winkler, A.; Beyermann, M.; Krause, E.; Matsuzaki, K.; Murase, O.; Bienert, M. Peptide helicity and membrane surface charge modulate the balance of electrostatic and hydrophobic interactions with lipid bilayers and biological membranes. Biochemistry 1996, 35, 12612–12622. [Google Scholar] [CrossRef] [PubMed]
- Tachi, T.; Epand, R.F.; Epand, R.M.; Matsuzaki, K. Position-dependent hydrophobicity of the antimicrobial magainin peptide affects the mode of peptide−lipid interactions and selective toxicity. Biochemistry 2002, 41, 10723–10731. [Google Scholar] [CrossRef] [PubMed]
- Hicks, R.P.; Bhonsle, J.B.; Venugopal, D.; Koser, B.W.; Magill, A.J. De novo design of selective antibiotic peptides by incorporation of unnatural amino acids. J. Med. Chem. 2007, 50, 3026–3036. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Guarnieri, M.T.; Vasil, A.I.; Vasil, M.L.; Mant, C.T.; Hodges, R.S. Role of peptide hydrophobicity in the mechanism of action of alpha-helical antimicrobial peptides. Antimicrob. Agents Chemother. 2007, 51, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- Hawrani, A.; Howe, R.A.; Walsh, T.R.; Dempsey, C.E. Origin of low mammalian cell toxicity in a class of highly active antimicrobial amphipathic helical peptides. J. Biol. Chem. 2008, 283, 18636–18645. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Shi, Z.; Baumgart, T. Regulation of membrane-shape transitions induced by i-bar domains. Biophys. J. 2015, 109, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Krishnakumar, S.S.; Franco, D.; Paul, A.V.; London, E.; Wimmer, E. Membrane topography of the hydrophobic anchor sequence of poliovirus 3A and 3AB proteins and the functional effect of 3A/3AB membrane association upon RNA replication. Biochemistry 2007, 46, 5185–5199. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, G.E.; Baleja, J.D. Membrane-binding peptide from the C2 domain of factor viii forms an amphipathic structure as determined by NMR spectroscopy. Biochemistry 1995, 34, 3022–3031. [Google Scholar] [CrossRef] [PubMed]
- Burman, L.G.; Nordstrom, K.; Boman, H.G. Resistance of Escherichia coli to Penicillins. V. Physiological comparison of two isogenic strains, one with chromosomally and one with episomally mediated ampicillin resistance. J. Bacteriol. 1968, 96, 438–446. [Google Scholar] [PubMed]
- Wiegand, I.; Hilpert, K.; Hancock, R.E. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Nanda, V.; Cristian, L.; Toptygin, D.; Brand, L.; Degrado, W.F. Nanosecond dynamics of InfluenzaA/M2TM and an amantadine resistant mutant probed by time-dependent red shifts of a native tryptophan. Chem. Phys. 2013, 422. [Google Scholar] [CrossRef] [PubMed]
- Pitcher, W.H., 3rd; Huestis, W.H. Preparation and analysis of small unilamellar phospholipid vesicles of a uniform size. Biochem. Biophys. Res. Commun. 2002, 296, 1352–1355. [Google Scholar] [CrossRef]
- Lapinski, M.M.; Castro-Forero, A.; Greiner, A.J.; Ofoli, R.Y.; Blanchard, G.J. Comparison of liposomes formed by sonication and extrusion: Rotational and translational diffusion of an embedded chromophore. Langmuir 2007, 23, 11677–11683. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the peptides may be available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence | Net Charge a | MW (Da) | MW (Da) Found |
|---|---|---|---|---|
| C18G | ALWKKLLKKLLKSAKKLG | +8 | 2065.7 | 2065.4 |
| C18G-Arg | ALWRRLLRRLLRSARRLG | +8 | 2261.8 | 2261.5 |
| C18G-His | ALWHHLLHHLLHSAHHLG | +1 b | 2128.5 | 2128.2 |
| C18G-Orn | ALWOOLLOOLLOSAOOLG | +8 | 1967.7 | 1967.3 |
| C18G-Dap | ALWXXLLXXLLXSAXXLG | +8 | 1771.7 | 1771.1 |
| E. coli | P. aeruginosa | A. baumannii | S. aureus | B. subtilis | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MIC | MIC | MBC | MIC | MBC | MBC | MIC | MBC | MIC | MBC | |
| C18G | 1.875 | 7.5 | 7.5 | 15 | 1.875 | n.d. | 1.875 | 3.75 | 1.875 | 1.875 |
| C18G-Arg | 1.875 | 7.5 | 7.5 | >15 | 3.75 | 7.5 | 3.75 | 3.75 | 1.875 | 1.875 |
| C18G-His | 3.75 | >15 | >15 | >15 | >15 | >15 | 15 | 15 | 1.875 | >15 |
| C18G-Orn | 1.875 | 3.75 | 3.75 | 3.75 | 1.875 | 1.875 | 1.875 | 1.875 | 3.75 | 7.5 |
| C18G-Dap | 3.75 | 7.5 | 7.5 | 7.5 | 3.75 | 3.75 | 7.5 | 7.5 | 1.875 | 1.875 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kohn, E.M.; Shirley, D.J.; Arotsky, L.; Picciano, A.M.; Ridgway, Z.; Urban, M.W.; Carone, B.R.; Caputo, G.A. Role of Cationic Side Chains in the Antimicrobial Activity of C18G. Molecules 2018, 23, 329. https://doi.org/10.3390/molecules23020329
Kohn EM, Shirley DJ, Arotsky L, Picciano AM, Ridgway Z, Urban MW, Carone BR, Caputo GA. Role of Cationic Side Chains in the Antimicrobial Activity of C18G. Molecules. 2018; 23(2):329. https://doi.org/10.3390/molecules23020329
Chicago/Turabian StyleKohn, Eric M., David J. Shirley, Lubov Arotsky, Angela M. Picciano, Zachary Ridgway, Michael W. Urban, Benjamin R. Carone, and Gregory A. Caputo. 2018. "Role of Cationic Side Chains in the Antimicrobial Activity of C18G" Molecules 23, no. 2: 329. https://doi.org/10.3390/molecules23020329
APA StyleKohn, E. M., Shirley, D. J., Arotsky, L., Picciano, A. M., Ridgway, Z., Urban, M. W., Carone, B. R., & Caputo, G. A. (2018). Role of Cationic Side Chains in the Antimicrobial Activity of C18G. Molecules, 23(2), 329. https://doi.org/10.3390/molecules23020329